The familial Alzheimer's disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons (original) (raw)
Abstract
Alzheimer's disease (AD) is a complex neurodegenerative disorder characterized by extracellular plaques containing amyloid β (Aβ)-protein and intracellular tangles containing hyperphosphorylated Tau protein. Here, we describe the generation of inducible pluripotent stem cell lines from patients harboring the London familial AD (fAD) amyloid precursor protein (APP) mutation (V717I). We examine AD-relevant phenotypes following directed differentiation to forebrain neuronal fates vulnerable in AD. We observe that over differentiation time to mature neuronal fates, APP expression and levels of Aβ increase dramatically. In both immature and mature neuronal fates, the APPV717I mutation affects both β- and γ-secretase cleavage of APP. Although the mutation lies near the γ-secretase cleavage site in the transmembrane domain of APP, we find that β-secretase cleavage of APP is elevated leading to generation of increased levels of both APPsβ and Aβ. Furthermore, we find that this mutation alters the initial cleavage site of γ-secretase, resulting in an increased generation of both Aβ42 and Aβ38. In addition to altered APP processing, an increase in levels of total and phosphorylated Tau is observed in neurons with the APPV717I mutation. We show that treatment with Aβ-specific antibodies early in culture reverses the phenotype of increased total Tau levels, implicating altered Aβ production in fAD neurons in this phenotype. These studies use human neurons to reveal previously unrecognized effects of the most common fAD APP mutation and provide a model system for testing therapeutic strategies in the cell types most relevant to disease processes.
INTRODUCTION
Alzheimer's disease (AD) is a common and devastating dementia that is pathologically defined by the accumulation of extracellular amyloid β (Aβ)-containing amyloid plaques and intraneuronal hyperphosphorylated Tau protein aggregates associated with neuronal loss in the cerebral cortex. Over 200 known missense mutations in amyloid precursor protein (APP) or the presenilin-1 and -2 genes (PSEN1/2) can cause dominantly inherited, early-onset forms of AD, termed familial AD (fAD) (reviewed in 1). The catalytic site of γ-secretase activity resides within PSEN (2), and APP is cleaved within its transmembrane domain by the γ-secretase complex to generate Aβ species primarily of 38, 40 or 42 amino acid lengths (3). The fAD mutations in APP or PSEN have been shown to either increase Aβ production generally or to increase the ratio of Aβ42 to Aβ40 peptides (reviewed in 1,4). These genotype-to-phenotype relationships provide strong evidence that Aβ42 plays a causal role in at least some cases of AD.
APPV717I was the first mutation linked to fAD (5) and is the most common fAD APP mutation (6). Residue 717 resides in the transmembrane domain of APP, near the γ-secretase cleavage site. Previous studies have shown that overexpression of APP cDNA with the V717I mutation results in an increase in the ratio of Aβ42/40 generated in cell lines (7) and mouse primary neurons (8). Brain lysates from transgenic mice expressing human APPV717I also showed an increased Aβ42/40 ratio (9,10). In most studies, the increased ratio of Aβ42/40 is mainly attributable to an increase in Aβ42 with no change or a slight decrease in Aβ40 levels. Importantly, both plasma and lysates of brains of patients carrying APPV717I have shown elevated Aβ42 levels relative to total Aβ, confirming the effect of this mutation on Aβ42 levels in the subjects of interest (11,12).
Rapid advancements in stem cell biology in recent years have provided neuroscientists with a unique opportunity to examine the effects of genetic alterations in disease-relevant human cell types. Previously, analyses of risk genes for neurological diseases were primarily limited to research on postmortem brains, mouse models and heterologous cell lines. With the advent of induced pluripotent stem cell (iPSC) technology (13–17), it is now possible to study genetic risk factors in neurons derived from primary cells of affected subjects (18). Two recent studies showed that neurons derived from iPSCs generated from subjects with APP duplication (including from a Down's syndrome line) secreted higher levels of Aβ and developed increased levels of Tau phosphorylated at Thr231 (19,20). A third study showed that a unique mutation in APP (E693delta) decreased overall levels of Aβ, but increased the accumulation of intracellular Aβ oligomers (21). In another study, iPSC lines were derived from two fAD subjects, one harboring a mutation in PSEN1 and another in PSEN2 (13). This study showed that each mutation increased secretion of Aβ42 and that γ-secretase inhibitors and modulators effectively decreased Aβ generation (13). These first efforts utilizing iPSCs to study AD provided an important proof-of-principle regarding the utility of such cells to model biochemical processes relevant to AD.
Here, we establish a model of AD using iPSCs from patients harboring a dominant, fully penetrant fAD mutation in APP (V717I). In neurons of forebrain fate derived from iPSCs, we confirm the previous finding from other model systems that the V717I mutation leads to increased Aβ42 levels. We show for the first time that this mutation alters (i) APP subcellular localization, (ii) Aβ38 and APPsβ generation and (iii) Tau expression and phosphorylation. Furthermore, we demonstrate that the increase in Tau can be rescued by treatment with anti-Aβ antibodies, providing direct evidence linking disease-relevant changes in Aβ to aberrant Tau metabolism. Aβ vaccines are a promising therapeutic option in early AD, which multiple pharmaceutical companies are actively pursuing. These rescue studies suggest that iPSC-derived neuronal cultures may be a useful model to test and compare Aβ antibodies, as well as alternate therapeutic strategies, for efficacy in the cell types of interest.
RESULTS
Generation and differentiation of iPSC lines with the London (V717I) APP mutation
Skin biopsies were obtained from a father and daughter each harboring a mutation in APP (V717I) (Fig. 1A). The father was 57 years old and diagnosed with AD while the daughter was asymptomatic at age 33 (Fig. 1A). Fibroblasts from the biopsy were reprogrammed using lentiviruses encoding Oct4, SOX2, cMYC and KLF4, and three iPSC clones from each subject were established and characterized by the Harvard Stem Cell Institute (HSCI) iPSC core facility (Supplementary Material, Fig. S1). All clones maintained stem cell morphology, expressed the pluripotency-associated genes Oct4, NANOG, SSEA3, SSEA4, TRA-1-60 and Alkaline Phosphatase, repressed retroviral transgenes and could be differentiated into cells of ectodermal, mesodermal and endodermal lineages in vitro (Supplementary Material, Fig. S1A–C). All clones from the daughter (fAD2) displayed a normal euploid karyotype (Supplementary Material, Fig. S1D), while all clones from the father (fAD1) displayed a normal chromosome number, but a balanced (t(1;12)(q42.1;q15)) translocation in all cells from four clonal lines (Supplementary Material, Fig. S1E). The fibroblasts obtained from this subject displayed the same abnormal karyotype, suggesting that this was a preexisting abnormality that did not arise during the reprogramming process.
Figure 1.

Characterization of the neuronal differentiation capacity of familial AD iPSC lines harboring the APP V717I mutation. Human iPSC lines were derived from a father and daughter with an fAD mutation (APP V717I). (A) Table summarizing human iPS lines used in this study. ND, not determined. (B) Schematic outlining α-, β- and γ-secretase cleavage sites in APP. Residue in red is V717, those in blue encode wild-type Aβ42. (C) Control and fAD lines were differentiated to neuronal fates using an embryoid aggregate protocol. After 40 days of differentiation, cells were fixed and immunostained for general neuronal markers such as MAP2, Tau and TuJ1, a marker of lower layer cortical neurons (Tbr1), a marker of upper layer cortical neurons (Cux1) and/or synaptic markers (PSD95, SYP, VGLUT1). Data shown are representative images from control and fAD lines. Scale bars = 50 μm. Magnified views of dotted boxes are shown as insets or adjacent to each image. (D) A representative single-unit waveform (∼15 μV, 3 ms), extracted from a voltage trace using extracellular whole well MEA (Axion Biosystems) recordings, is shown for both control and fAD lines. (E, F) After 40 days of differentiation, cells were lysed, RNA extracted and expression of 150 genes analyzed using the NanoString platform. Expression of general neuronal markers are shown in (E), cell fate-specific markers are shown in (F). For control, n = 20 and fAD, n = 10. Data were normalized to a panel of seven HK genes. AU, arbitrary units. Error bars represent SEM.
Because variability exists in the differentiation efficiency among pluripotent stem cell lines, we first compared the capacity of each line to differentiate to neuronal fates. In order to direct the differentiation of these cells to forebrain neuronal fates, we utilized an embryoid body-based protocol (22) (with modifications described in Materials and Methods). Using this method, cultures highly enriched in neurons were generated, with over 90% of the cells expressing MAP2 in each well (Fig. 1C). Initial characterizations of each clone for differentiation capacity informed the selection of two clones from each subject to be further analyzed. In parallel, differentiation experiments were performed with iPSC lines generated from healthy donors, which were obtained from the HSCI iPSC Core (18,23) and from the UCONN Stem Cell Core (Fig. 1A). No significant differences were observed in differentiation capacity between control and fAD cell lines, as assayed by immunostaining for the general neuronal markers MAP2, Tau and TuJ1, markers of upper (Cux1) and lower (Tbr1) layer cortical neurons and synaptic markers synaptophysin (SYP), PSD95 and VGLUT1 (Fig. 1C). Recordings from cultures of differentiated neurons showed spontaneous activity using a microelectrode array (MEA) platform in all cell lines tested. Representative waveforms from control and fAD lines, isolated from voltage traces, are shown in Figure 1D. Representative extracellular voltage traces and raster plots of spiking events are shown in Supplementary Material, Figure S2. No obvious quantitative differences were observed between the fAD and control neurons. To provide a quantitative analysis of differentiation capacity over multiple clones and rounds of differentiation, NanoString analyses were performed with a custom designed probe set measuring 150 genes. Quantitative comparison of control and fAD lines showed no significant differences in expression of general neuronal markers (Fig. 1E) or cell fate-specific markers (Fig. 1F).
Cleavage of APP by α-, β- and γ-secretases in fAD and control iPSC-derived neurons
Multiple studies in non-neuronal cell lines and transgenic mice overexpressing the APP V717I mutation report that this mutation alters γ-secretase cleavage of APP to generate higher levels of Aβ42 (1,3,5,7,8,11,12,24). Here, we aimed to examine the effects of this mutation expressed from the endogenous APP gene in human neuronal cells. At 40 days of neuronal differentiation, conditioned media were collected 48 h after application for analysis of secreted APP cleavage products, and cells remaining in the well were lysed to collect RNA for expression analyses. NanoString analyses showed no significant differences in RNA expression between control and fAD lines for APP splice variants and APP family members (APLP1 and APLP2) (Fig. 2A). Western blot analyses suggested that APP holoprotein levels were increased ∼1.4-fold in fAD-derived neuronal cultures (Fig. 2C; Supplementary Material, Fig. S3A and data not shown). No significant changes were observed in expression of genes encoding α-, β-secretases or components of γ-secretase (Fig. 2B).
Figure 2.
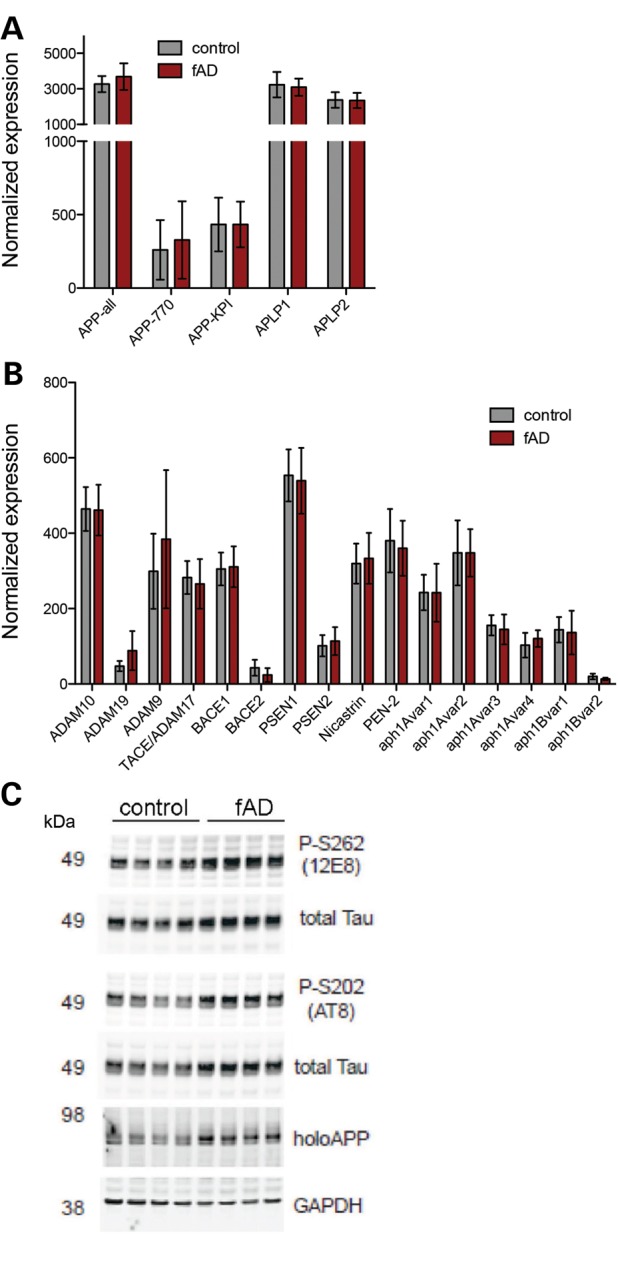
APP protein levels are altered and levels of secretases are unchanged in neurons with APPV717I mutation. Control and fAD APP V717I lines were differentiated to neuronal fates. (A, B) After 40 days, cells were lysed, RNA extracted and expression of 150 genes analyzed using the NanoString platform. Expression of APP family members are shown in (A) and components of α-, β- and γ-secretases in (B). Each bar represents data from nine independent wells collected from three rounds of differentiation. Data from four different iPSC lines are represented. Error bars represent SEM. (C) Representative Western blot analysis of selected genes following 100 days of differentiation.
Conditioned media from days 40 to 50 of differentiation from control and APPV717I lines were analyzed to examine Aβ38, 40 and 42 levels using a multiplex ELISA. Cells remaining in the well were lysed and RNA collected to assay differentiation efficiency by qPCR and/or NanoString (as in Figs 1E, F and 2A, B). Only those experiments with efficient neuronal differentiation (as assayed by morphology and expression of neuronal markers such as MAP2, Tau, Tbr1 and Cux1) were further analyzed. Differentiation efficiency was not significantly different between the control and fAD lines used here. In agreement with data from previous studies using other experimental paradigms, neurons derived from each line harboring this APP fAD mutation secreted Aβ with a higher ratio of 42/40 than neurons from control lines (control 0.25 SD ±0.05; fAD 0.39 SD ±0.10; Fig. 3B). Notably, the ratios observed here appear to be physiologically relevant, as published results examining Aβ in TBS-extracted human brain lysates showed similar ratios (0.25–0.42) (25). Furthermore, we observed an increased ratio with the APPV717I mutation (1.6-fold) that is highly similar to the ratio increase observed in the plasma of human subjects with the same mutation (1.7-fold) (12). Here, data from each Aβ species show that this ratio change was primarily due to a 2-fold increase in production of Aβ42 (Fig. 3C, D, F and G; Supplementary Material, Fig. S4A and B). Aβ40 levels showed a trend towards increased levels, but this did not achieve significance. Of note, the intra- and inter-clonal variability in Aβ secretion was quite low between wells and between experiments in both fAD and control cell lines (Fig. 3A). The variability observed was due in part to slight technical differences between rounds of differentiation, with differences between ELISA plates also contributing to the mild variability observed (Supplementary Material, Fig. S4E and F). Interestingly, human neurons harboring the APPV717I mutation also secreted higher levels of Aβ38 (Fig. 3E and H; Supplementary Material, Fig. S4C). Accordingly, the calculated Aβ38/40 ratio was also significantly higher with the fAD mutation (data not shown). Aβ levels also were measured in the lysates of a subset of samples. However, Aβ38 and Aβ42 were not detectable in the lysates, which contain both intracellular and cell-associated Aβ (Supplementary Material, Fig. S4D).
Figure 3.

FAD mutation (APPV717I) in forebrain neuronal cells leads to increased Aβ42 and Aβ38 production. Control and fAD iPSC lines were differentiated for 40–50 days to neuronal fates. Media conditioned on the cells for the final 48 h were collected, and Aβ 38, 40 and 42 were detected in a single well using a multiplex ELISA (MesoScale Discoveries). Following collection of media, cells were lysed and RNA collected for parallel analyses of markers of differentiation. Data are shown for Aβ42/40 ratio in individual clones (A) or pooled as control and APPV717I (fAD) (B–H). For (B)–(H), Aβ data were generated from the two control and four fAD lines shown in (A) and averaged over 13 rounds of differentiation (YZ1 n = 45, YK26 n = 24, 1a n = 33, 1b n = 16, 2a n = 29, 2b n = 41). One-way ANOVA performed with Tukey's multiple comparisons test, **P < 0.01; ***P < 0.001. In (A), black asterisks show significance versus YZ1 and green asterisks show significance versus YK26. In (F)–(H), day 40 neurons were treated with vehicle or DAPT (5 μm) for the final 48 h of differentiation (n = 4–5 for each condition). Two-tailed _t_-tests were performed, **P < 0.01; ***P < 0.001. Error bars = SEM. Data normalized to RNA in (C)–(E) and total protein in F–H.
Overall levels of Aβ were increased in fAD neurons (Fig. 3). Prior to cleavage by γ-secretase, APP must first be cleaved by α- or β-secretase to release the large N-terminal fragment of APP, termed APPsα or APPsβ, respectively (26). Cleavage by β-secretase prior to γ-secretase generates Aβ, while α-cleavage precludes Aβ generation. Because of the importance of these cleavage events, we next examined whether the APPV717I fAD mutation affects α- and/or β-secretase cleavage of APP in a neuronal context. Surprisingly, fAD neurons secreted a lower ratio of APPsα to APPsβ (Fig. 4A), due to a 1.4-fold increase in the production of APPsβ (Fig. 4B and C). The shift in the ratio of APPsα to APPsβ production was confirmed by Western blot for a limited subset of samples (Supplementary Material, Fig. S3B and C). This effect did not appear to be due to increased expression of β-secretase, as both NanoString and Western blot analyses showed no differences in RNA or protein levels of the genes encoding β-secretase activity (BACE 1 and 2) (Fig. 2A and B and data not shown). In order to test whether γ-secretase activity is necessary for the enhancement of β-secretase cleavage of APP by V717I, differentiated neuronal cells from control and fAD mutant lines were treated with a low dose (5 μm) of a potent γ-secretase inhibitor (DAPT) for 48 h. As expected, this treatment efficiently inhibited the production of Aβ38, 40 and 42 in both control and fAD neurons (Fig. 3F–H). Interestingly, inhibition of γ-secretase potently blocked the effect of V717I in increasing β-secretase cleavage of APP (Fig. 4E and F). Further, γ-secretase inhibition increased APPsα generation relative to APPsβ generation in control neurons (Fig. 4D–F). The inhibitor effect on this ratio was enhanced in neurons harboring the APPV717I mutation (Fig. 4D).
Figure 4.

APP V717I mutation in forebrain neuronal cells leads to increased cleavage of APP at the β-secretase site. Control and fAD iPSC lines were differentiated for 40–50 days to neuronal fates. Media conditioned for the final 48 h were collected, and APPsα and APPsβ detected in a single well using a duplex ELISA (MesoScale Discovery). Following collection of media, cells were lysed and RNA collected for parallel analyses of markers of differentiation. (A) Ratio of APPsα/β in each line analyzed. Green asterisks represent significance relative to YZ1, red asterisks relative to YK26. (B) APPsα or (C) APPsβ levels normalized to total RNA from control and fAD neurons are shown, pooled by APP genotype. Data in (A) are combined from six differentiation rounds, for YZ1 n = 19, YK26 n = 11, 1A n = 8, 1B n = 15, 2a n = 26, and 2b n = 28; for (B) and (C) n = 20 for controls and n = 38 for fAD. One-way ANOVA performed with Tukey's multiple comparisons test, *P < 0.05; **P < 0.01; ***P < 0.001. (D–F) Cells differentiated to neuronal fates for 50 days were treated with 5 μm DAPT or vehicle (DMSO) for the last 48 h of culture prior to lysis. Media were collected and APPsα and APPsβ measured using multiplex ELISA. In (D), ‘pre’ conditions show data from the media collected from the same wells 48 h prior to treatments. Green asterisks in (D) show significance relative to control cells treated with DMSO, and the purple asterisks in (D,F) show significance relative to fAD DMSO. Representative data from a single round of differentiation are shown, n = 3–5. (G, H) Neurons differentiated from control and fAD lines were immunostained and imaged using confocal microscopy. Data shown are representative images from control and fAD lines for APP, EEA-1 and MAP2 staining (G). Scale bars = 20 μm. Magnified views of dotted boxes within the middle panel are shown in the last panel. (H) APP/EEA-1 co-localization was measured using Zen Black software from Zeiss. Two lines for each conditioned were used. Number of cells counted: for control, n = 126 and for fAD, n = 115. Error bars represent SEM, ***P < 0.001.
The effect of APPV717I on β- and γ-secretase cleavage was confirmed in human embryonic kidney (HEK) 293 cells following transient transfection with cDNAs encoding either human WT or V717I APP (Supplementary Material, Figs S5 and S6). Treatments using γ-secretase inhibitors (DAPT, L685,458, compound E) and a β-secretase inhibitor (C3) were performed in these cells to further probe the interaction between β- and γ-secretase activities. All tested γ-secretase inhibitors significantly lowered Aβ38, 40 and 42 (Supplementary Material, Fig. S6A–C). The inhibitor panel also recapitulated the increase in β-secretase cleavage of APP observed in treatment of iPSC-derived neurons (Supplementary Material, Fig. S6D). APPV717I expressing HEK cells had significantly higher β-CTF (C-terminal fragments) levels than APPWT-expressing HEK cells (Supplementary, Material, Fig. S5J and K), and β-CTFs were further increased in both APPWT and APPV717I expressing HEK cells in the presence of DAPT. As expected, the β-secretase inhibitor C3 increased the ratio of APPsα to APPsβ in both APPWT and APPV717I expressing HEK cells (Supplementary Material, Fig. S6F). However, β-secretase inhibition did not affect the Aβ42/40 ratio, which is determined by γ-secretase cleavage events (Supplementary Material, Fig. S6G). Thus, while γ-secretase inhibition affects β-secretase cleavage, β-secretase inhibition does not affect the site of γ-secretase cleavage. Taken together, these results suggest that β- and γ-secretase cleavages of APP are tightly linked processes, and that a single point mutation of APP can influence both cleavage events.
Since the APP V717I mutation induced increased β-secretase cleavage of APP, and β-secretase is most active in acidic compartments such as in the endosomal pathway, we examined whether APP subcellular localization was affected by the mutation. Using immunocytochemistry and confocal microscopy, we investigated the co-localization coefficient between APP and the endosomal marker EEA1 in control and fAD differentiated forebrain neurons (Fig. 4G). FAD neurons had a significantly higher co-localization coefficient of APP with EEA1, indicating that APP subcellular localization is affected by the V717I mutation (Fig. 4H).
Changes in APP processing across differentiation from stem cell to neuron
We next addressed whether the effects observed on APP cleavage varied as a function of cell fate. RNA and conditioned media were collected from control and fAD iPSCs at multiple time points during differentiation from stem cell to neuron, in order to assess how differentiation alters the secretion of APPsα, APPsβ and Aβ. As expected, over differentiation time, cells changed morphologically and lost expression of pluripotency markers (Oct4), first turning on neuronal precursor markers (CyclinD1 and Nestin, not shown), and then markers of mature neurons (MAP2, Tau, VGLUT1, GAD1) (Fig. 5A and B). Over differentiation time from day 0 to day 100, Aβ secretion increased markedly in both control and fAD lines, and the fAD-dependent increase in Aβ38 and 42 was observed consistently and significantly after day 40 (Fig. 5C and E). Accordingly, we observed higher Aβ42/40 ratios in fAD versus control lines over the differentiation timecourse, with statistical significance obtained beginning at day 24 (Fig. 5F). Furthermore, as cells became more neuronal in their RNA and protein expression profiles, there was a consistent and steady decrease in the ratio of APPsα to APPsβ secreted by these cells (Fig. 5I). While both APPsα and APPsβ increase over differentiation [in part due to an increase in APP expression (Fig. 5J)], there is a greater increase in APPsβ due to a robust increase in expression of BACE1 with neuronal differentiation (Fig. 5G, H and J). The effect of the APPV717I mutation on significantly elevating the β-secretase cleavage of APP was observed at all time points examined (Fig. 5H).
Figure 5.

Examination of APP cleavage products generated over differentiation time to mature neuronal fates. Control and APP V717I (fAD) iPSC lines were differentiated over 100 days to neuronal fates. (A) At multiple time points, cells were fixed and immunostained for a pluripotency marker (Oct4), a neuronal marker (MAP2) and a nuclear marker (TOPRO3). (B) Alternatively, cells were lysed following collection of media and RNA purified for qPCR analysis. Media were analyzed by ELISA to measure levels of Aβ (C–F) and/or APPsα and APPsβ (G–I). (J) qPCR analysis of APP and BACE mRNAs across the differentiation time course, normalized to GAPDH expression. For data in (B)–(J), error bars, SEM. For each comparison, a two-tailed _t_-test was performed, *P < 0.05; **P < 0.01; ***P < 0.001. For d9, d17, d24, n = 2–4, for d40, n = 70–100, for d60, d80, n = 5–10. Data normalized to total RNA in (C)–(E), (G) and (H).
Aβ-specific antibodies engage Aβ and rescue the Tau increase observed in fAD neurons
To determine whether iPSC-derived human neurons can reflect putative downstream effects of the APPV717I mutation observed in AD patients, we quantified the levels and phosphorylation state of Tau. In both early and late cultures (day 35 and 100, respectively), APPV717I neurons exhibited a 2-fold increase in Tau protein levels (Fig. 2C and 6A, C; Supplementary Material, Fig. S7C and D). There was no significant difference in phospho-Tau levels relative to total Tau levels between control and APPV717I neurons at the day 35 time point (Fig. 6B). However, at day 100 APPV717I neurons exhibited higher levels of phospho-Tau at amino acid S262 (Figs 2C and 6D). When normalized to total Tau, pS262 was significantly elevated beyond the increase of total Tau expression observed (Figs 2C and 6D).
Figure 6.

Tau protein levels are increased in fAD neurons directed to a forebrain fate, which is reversed by treatment with Aβ-specific antibodies. Total Tau (A) and phospho-Tau (B) levels from control and fAD neurons differentiated for 35 days were determined by Western blotting and densitometry. Total Tau (C) and phospho-Tau (D) levels from control and fAD iPSCs differentiated for 100 days were determined by Western blotting and densitometry. (E–K) Control and fAD neural progenitors (days 18–20) were treated with the Aβ-specific antibodies 3D6 (F, H, J) or AW7 (G, I, K) and compared with isotype-specific or preimmune serum, respectively, for 15 days. (E) Western blots of media probed for Aβ after pull down with protein Agarose-A/G beads. (F, G) Aβ ELISA data are shown from conditioned media following pull down of Aβ with either 3D6 or AW7. Treatment with a monoclonal Aβ antibody (3D6) was compared with its isotype control (IgG) (H, J) or else treatment with a polyclonal antibody (AW7) was compared with treatment with its preimmune serum (I, K). ELISA data for total Tau (H, I) and quantification by densitometry from Western blot (J, K) is shown for 3D6 and AW7 experiments, respectively. Fresh neural differentiation media with antibody was applied every 3 days. *P < 0.05; **P < 0.01; ***P < 0.001. PI, preimmune; ND, not detected; error bars, SEM.
In order to determine if the Tau phenotype was caused by differences in secreted Aβ in fAD neurons, we specifically targeted Aβ using anti-Aβ antibodies. Neurons directed to a forebrain neuronal fate were treated with either 3D6 (27), an anti-Aβ monoclonal antibody (Fig. 6H and J) or else AW7 (28), an anti-Aβ polyclonal antibody (Fig. 6I and K). Both antibodies bound and sequestered Aβ, as evidenced by Western blot and depletion of Aβ from the media following pull down of antibody-bound Aβ (Fig. 6E–G). Very low or no Aβ levels were detected by ELISA in the media following pull down, suggesting that very little unbound Aβ of all three species analyzed remained in media with antibody treatment. Treatment with both Aβ-specific antibodies prevented the increase in total Tau in APPV717I neurons, as quantified by both Tau ELISA (Fig. 6H and I) and Western blot (Fig. 6J and K). This observation supports the hypothesis that increased Tau levels in fAD neurons are a downstream result of the Aβ generated. Antibody treatment did not alter the APPsα/APPsβ ratio (data not shown), supporting the hypothesis that the mechanism causing increased APPsβ in fAD neurons is Aβ independent. Of note, the timing of antibody treatment was critical for rescuing the Tau phenotype: treating with antibodies later in differentiation, or for shorter periods of time, did not allow for significant rescue of Tau levels (Supplementary Material, Fig. S7, and data not shown). The proportional increase in phospho-Tau observed early in differentiation also is rescued by antibody treatment (data not shown). However, due to the lack of efficacy of antibody treatment initiated late in differentiation, ability of antibodies to rescue the elevated phospho-Tau levels observed at day 100 was not tested.
DISCUSSION
Aβ homeostasis plays a central role in the pathogenesis of AD (29). Aβ is generated physiologically by sequential cleavages of APP by β- and γ-secretase. Cleavage by γ-secretase occurs at multiple sites and results in the generation of a variety of Aβ species varying in length between 36 and 43 residues. Aβ40 is the most abundant, followed by Aβ42 and 38 (26,29). Although Aβ42 is a minor form, the two extra amino acids (isoleucine and alanine) make this peptide more hydrophobic and prone to self-aggregation. It has previously been shown that fAD mutations in APP, PSEN1 or PSEN2 each act in part to either increase total Aβ levels or, more commonly, to increase the amount of Aβ42 relative to Aβ40 generated (reviewed in 3). Here, we demonstrate that human neurons derived from iPSC lines established from subjects harboring one such mutation (V717I) generate significantly more Aβ42. The fold-increase in Aβ42/40 ratio reported here (1.6-fold) is highly similar to that observed in plasma from subjects with the V717I mutation (1.7-fold) (12). We extend these findings to show that Aβ38 also is elevated, in accordance with the helical model of γ-secretase processivity within the transmembrane domain (30). This model of APP cleavage describes stepwise cleavages of APP by γ-secretase, beginning with epsilon cleavage near the transmembrane-cytoplasmic interface to release the intracellular domain of APP (AICD) as well as Aβ of 48 or 49 residues (31). These longer Aβ-like species are then cleaved every 3–4 amino acids along the transmembrane domain to generate smaller species, such that Aβ49 is cleaved to generate Aβ peptides of 46, 43 and 40 amino acids in length, whereas Aβ48 similarly is cleaved to generate Aβ peptides of 45, 42 and 38 amino acids (26,32). In agreement with this processivity model, we observe an increase in both Aβ42 and 38 caused by the APPV717I mutation, suggesting that this mutation may primarily act to alter the initial epsilon site of cleavage within APP. Indeed, changes in epsilon site cleavage with other fAD mutations have been previously reported (33).
In addition to effects on the sites of γ-secretase cleavage within APP, we also describe an unexpected effect of this fAD mutation on β-secretase cleavage of APP. β-secretase activity is encoded by the genes BACE1 and BACE2 (34–36), and expression of these genes is high in the CNS (36,37,40). Human iPSCs provide a model system to examine the activity of these enzymes in human neuronal development. Here, we show a dramatic increase in β-secretase cleavage of APP as cells differentiate to neuronal fates. Moreover, although the V717I mutation occurs near the site of the γ-secretase cleavages in the transmembrane domain of APP, a significant effect of this mutation on β-secretase cleavage of APP was observed at all differentiation time points examined. The increase was not due to indirect effects on BACE expression, as both RNA and protein levels of BACE were unchanged in the fAD neurons. BACE is an aspartyl protease that has optimal activity in an acidic environment, such as in the endosomal pathway (38,39,41,42). One possible explanation is that the V717I mutation affects the subcellular localization of APP to compartments containing active BACE, and this in turn results in increased β-secretase cleavage of APP in fAD neurons. Indeed, we observed altered subcellular localization of APP to EEA1-positive sites within neurons derived from APPV717I carriers. In addition, APPV717I may affect the position of APP within the membrane, which in turn affects both the site of epsilon cleavage of APP and the position of APP relative to the active site of BACE, which is a membrane-anchored protease that cleaves its substrates just outside of their transmembrane domains. To examine whether the β- and γ-secretase effects of the APP mutation are linked, we asked whether γ-secretase inhibition could rescue the effect of the APPV717I mutation on β-cleavage. Indeed, γ-secretase inhibition prevented the effect of the V717I mutation in elevating β-secretase processing of APP. This was observed in both iPSC-derived human neurons and HEK cells, confirming the observation and suggesting that these enzymatic activities are functionally interdependent. This result is in agreement with recent evidence that these proteases may exist in a previously unrecognized complex and affect the activities of one another (A. C. Chen and D. J. Selkoe, personal communication). Further studies are therefore warranted to examine whether other fAD APP and PSEN1/2 mutations affect β-secretase cleavage of APP, in addition to their known effects on γ-secretase processivity.
The experimental paradigm outlined here allows us to follow changes in APP expression and processing over a human neuronal developmental timeline. Using our differentiation protocol, stem cells are directed to neuronal fates over the course of several months. Between days 17 and 24, cells express markers of neuronal precursor cells and are actively dividing. At day 24, cells are cultured as a monolayer on Matrigel in differentiation media, and between this day and ∼day 40, general neuronal markers are upregulated including those for cytoskeletal proteins important in neurite outgrowth such as Tau, MAP2 and βIII-tubulin (TuJ1). Between days 30 and 50, spontaneous electrophysiological activity first appears in our cultures, coinciding with an upregulation of synaptic markers such as PSD95, SYP and synapsin. Between days 50 and 100, the dynamic changes in neuronal gene expression that are present during differentiation are no longer observed, and cultures have more stable expression of neuronal and synaptic markers. Over differentiation time, we observe a consistent increase in APP expression at both the RNA and protein levels, with a corresponding increase in both APPsα and APPsβ secretion. The increase in the percentage of APPsβ generated as cells differentiate to mature neuronal fates likely reflects the time-dependent increase in BACE1 levels. Similarly, there is a dramatic increase in Aβ secretion over differentiation time until day 60, at which point the levels plateau to day 100. At all observed neuronal time points, fAD neurons secrete more Aβ38 and 42, and a higher ratio of Aβ42/40 than control neurons. Interestingly, between days 40 and 100, the ratio of Aβ42/40 secreted from fAD neurons increases but remains relatively constant in control neurons.
The accumulation of Aβ in all AD brains, as well as the dominant effects of APP, PSEN1 and PSEN2 mutations in causing an accelerated but otherwise typical AD phenotype, point to Aβ being critical to pathogenesis. Accumulation of intraneuronal hyperphosphorylated Tau is a key feature observed in the AD brain. Multiple lines of evidence suggest that this pathological hallmark of AD can arise as a downstream result of accumulation of extracellular Aβ, but this hypothesis remains controversial (20,43–45). Accumulation of intracellular Aβ also has been implicated in causing endosomal defects that can lead to pathology (20,21, and others). However, in the fAD APP London V717I neurons described here, levels of intracellular and membrane-associated Aβ are substantially lower than the Aβ that accumulates extracellularly in the media (Supplementary Material, Fig. S4D).
We observe a significant increase in Tau protein levels in neurons of forebrain fate derived from APP V717I iPSCs. This increase is observed as early as day 26 (data not shown) and continues through the latest time point examined (day 100). Phospho-Tau levels also are increased in fAD neurons at day 100, as well as at day 40. However, at day 40 this phospho-Tau elevation is proportional to the increase in total Tau. Thus, we observe an increase in total Tau levels prior to a further increase in phospho-Tau levels. In order to address whether the phenotype of increased Tau expression in fAD neurons is due to altered Aβ generation, we performed rescue experiments with antibodies specific to Aβ. Both a polyclonal and a monoclonal Aβ-antibody were able to reverse the increased total and phospho-Tau protein levels observed in fAD neurons at early time points. We propose that treatment with these antibodies in the media binds to extracellular Aβ and prevents a non-autonomous effect of Aβ on Tau. This rescue was observed when neurons were treated at early time points (days 18–20), but not when neurons were treated at later time points of maturity (d40+), suggesting that intervening earlier in this culture system is more effective than at later time points. This may be important when considering using AD culture models for screening and testing disease-modifying therapies.
Postmortem studies of AD brain have shown that total Tau levels are elevated 8-fold, and that most of this Tau is phosphorylated (46), in agreement with our observations. A large number of studies have reported an increase in the levels of phospho-Tau in postmortem brain from fAD and sporadic AD subjects. However, surprisingly little is known about the overall levels of soluble Tau in fAD or even sporadic AD brain, especially at the preclinical stages of the disease, which is most relevant to our studies. Elevated Tau in the CSF is an early biomarker for AD, but this elevation is thought to be due to a combination of release from neurons following degeneration and increased active Tau secretion (reviewed in 47). Future studies should address whether early increases in total Tau levels are specific to neurons derived from our APP V717I cultures, or if this phenotype extends to other fAD cases and/or sporadic AD cases.
These data indicate that increased Tau levels can result directly from the effects of altered Aβ generation induced by the fAD mutation V717I in a relatively simple neuronal cell culture system, thus connecting the two major abnormalities of AD pathogenesis. Notably, cultures of both control and fAD neurons remain healthy at day 100 of differentiation, with no obvious cell death observed (data not shown). However, in contrast to other studies, which have shown an increase in cell death of primary rodent neurons in response to exogenously added Aβ, in this study the extracellular Aβ is cleared from the cultures every 2–3 days with full media changes. The use of this system provides the opportunity to further probe the initiating events in AD pathogenesis, namely, the generation of pathological Aβ and the accumulation of phosphorylated Tau, in the absence of frank cell loss, which is not observed until late stages of the disease.
The findings presented here provide several unexpected insights into the effects of APP fAD mutations on processing by the β- and γ-secretases. Furthermore, we provide evidence that altered cleavage of APP leads to increased Tau expression, which can be reversed by treatment with Aβ-specific antibodies. Taken together, this study demonstrates the utility of iPSCs from human donors to model AD-relevant phenotypes, with related therapeutic implications.
MATERIALS AND METHODS
Patients and fibroblast derivation and iPSC generation
iPSCs were generated in collaboration with the HSCI. Skin punch biopsies were taken from a father and daughter pair, each with the APP V717I mutation, after informed consent and in accordance with Institutional Review Board approval. iPSCs were reprogrammed as described (18). cDNAs for Oct4, Sox2, Klf4 and Myc were cloned into pMIG vectors and packaged into VSVG-pseudotyped retroviruses. Fibroblasts were transduced with retroviruses. Valproic acid (Sigma, 50 μm) was added for 7 days, beginning on day 2 after transfection. iPSC colonies appeared after ∼3 weeks and were picked based on morphology and GFP silencing. Colonies were transferred to six-well plates containing irradiated mouse embryonic fibroblasts (MEFs) (Globalstem). ROCK inhibitor (Y27632) (Millipore) was used at 10 μm to increase cell survival. Each picked colony was one line. For passaging, cells were dissociated with collagenase (Stemcell Technologies). For a 10 cm culture plate, between 5 and 50 colonies emerged.
Karyotype analysis and characterization
iPS clones were karyotyped by Cell Line Genetics. For pluripotency assays, iPSCs were dissociated from the plate with collagenase and then resuspended in ultra low-attachment plates and fed with iPSC media. Cells were re-plated onto gelatin (Millipore) after 1 week with DMEM (Invitrogen) 10% FBS (Sigma). Cells were harvested for gDNA extraction after 1 week of plating. Copy number analyses were routinely performed using the NanoString nCounter CNV CodeSets, in order to ensure a normal chromosome number across passages.
iPS cell culture
iPSCs were cultured in iPSC media with bFGF (Millipore) added fresh daily at 10 μg/ml. Cells were maintained at 37°C/5% CO2 and were split as necessary based on colony growth (5–6 days). Differentiating colonies were removed from the plate prior to splitting. iPSCs were maintained on an irradiated MEF feeder layer at 1.7–2.0 × 105 cells/well. For passaging, cells were dissociated with collagenase.
Neuronal differentiation
For the induction of forebrain neurons, iPSCs were differentiated using an embryoid body-based protocol (22) that was further optimized, as described here. iPSC colonies were dissociated from MEFs at day 1 and cultured as aggregates 4 days in suspension with iPSC media. Aggregates were switched to Neural Induction media (N2) at day 5. Aggregates were plated on Matrigel-coated (BD Biosciences) culture dishes at day 7, forming primitive neuroepithelial (NE) structures over 10 days with Neural Induction media (N2). By day 17 definitive NE structures were present, and neural progenitors further cultured in suspension using Neural Induction (N2/B27) media. Neural rosettes were either selected manually or with STEMDiff Neural Rosette Selection reagent (Stemcell Technologies). The second culture in suspension aimed to purify neuronal progenitors, by clearing improperly differentiating cells. Cells were dissociated to single cells with accutase (Invitrogen) and plated on Matrigel for final differentiation at day 24 in Neural Differentiation media. Matrigel was used per the manufacturer's instructions. A full media change was performed every 2–3 days for the duration of the experiment.
Medias
iPSC Medium consisted of 400 ml (DMEM/F12, Invitrogen), 100 ml Knockout Serum Replacement (Invitrogen), 5 ml MEM-NEAA (Invitrogen), 5 ml of penicillin/streptomycin/glutamine (Invitrogen) and 500 μl 2-mercaptoethanol (100×) (Invitrogen). Neural Induction Medium (N2) consisted of 490 ml DMEM/F12, 5 ml N2 supplement (Invitrogen), 5 ml MEM-NEAA, 2 mg/ml Heparin (Sigma-Aldrich). Neural Induction Medium (N2/B27) consisted of 480 ml DMEM/F12, 5 ml N2 supplement, 10 ml B27 supplement (Invitrogen), 5 ml MEM-NEAA, 2 mg/ml Heparin, with cAMP (1 μm, Sigma) and IGF-1 (10 ng/ml, Peprotech) added to the medium. Neural Differentiation Medium consisted of 480 ml of Neurobasal medium (Invitrogen), 5 ml N2 supplement, 10 ml B27 supplement, 5 ml of MEM-NEAA, with the addition of fresh cAMP (1 mm), BDNF, GDNF and IGF1 (PeproTech, 10 ng/ml) to the medium.
Microelectrode array recordings
Sterile 12-well MEA plates were coated with sterile-filtered poly-d-lysine (PDL) (Sigma) for 1 h at room temperature. After aspiration of the PDL solution, wells were washed 3× with sterile milliQ water. Plates were allowed to dry overnight in the tissue culture hood. Seventy-five microliters of sterile Matrigel solution were coated directly over the electrode grid for 1 h at 37°C. To prevent drying of the Matrigel, sterile water was added to the area surrounding the electrodes. Neural aggregates were single-cell dissociated at day 24 of differentiation using accutase, and ∼75 000 cells were plated in each well. Neurons were co-cultured with ∼75 000 human astrocytes (Sciencell) in a volume of 75–100 μl. Cells were allowed to attach over the course of 30 min. Following attachment, 500 μl of a 1.5:1 ratio of neural differentiation to astrocyte media was added to the wells. Media was switched to neural differentiation media thereafter, and changed every 2–3 days.
MEA recordings were performed using the Maestro system from Axion Biosystems. Recordings were done in 12-well plates, with each well consisting of 64 electrodes in an 8 × 8 grid, for a plate total of 768. Nano-porous platinum electrodes are 30 μm in diameter and 200 µm apart. Using the AxIS software (Axion Biosystems), data were acquired using a sampling rate of 12.5 kHz and filtered using a 200–2500 Hz Butterworth band-pass filter. A detection threshold was set to ±5.5 × SD of the baseline electrode noise. Filtering and plotting of trace and waveform data were done using MATLAB with custom scripts (The MathWorks, Natick, MA, USA). Spike raster plots were analyzed using Neuroexplorer (NEX Technologies). At least two independent recordings were performed, from five different differentiation experiments.
Primers
- Actin: forward, ggacttcgagcaagagatgg; reverse, agcactgtgttggcgtacag
- Dnmt3b: forward, ataagtcgaaggtgcgtcgt; reverse, ggcaacatctgaagccattt
- hTERT: forward, tgtgcaccaacatctacaag; reverse, gcgttcttggctttcaggat
- Nanog: forward, tccaacatcctgaacctcag; reverse, gactggatgttctgggtctg
- Oct4: forward (transgene), gtggaggaagctgacaacaa; reverse (endogenous), caggttttctttccctagct
- RexI: forward, tggacacgtctgtgctcttc; reverse, gtcttggcgtcttctcgaac
- Sox2: forward, ttgtcggagacggagaagcg; reverse, tgaccaccgaacccatggag
- β-III Tubulin: forward, cagatgttcgatgccaagaa; reverse, tgctgttcttgctctggatg
- NCAM: forward, atggaaactctattaaagtgaacctg; reverse, tagacctcatactcagcattccagt
- Pax6: forward, tctaatcgaagggccaaatg; reverse, tgtgagggctgtgtctgttc
- AFP: forward, agcttggtggtggatgaaac; reverse, ccctcttcagcaaagcagac
- GATA4: forward, ctagaccgtgggttttgcat; reverse, tgggttaagtgcccctgtag
- Flk1: forward, agtgatcggaaatgacactgga; reverse, gcacaaagtgacacgttgagat
- GATA2: forward, gcaacccctactatgccaacc; reverse, cagtggcgtcttggagaag
- PECAM: forward, cccagcccaggatttcttat; reverse, accgcaggatcatttgagtt
- VECAD: forward, cagcccaaagtgtgtgagaa; reverse, tgtgatgttggccgtgttat
qPCR
qPCR was performed using Fast SYBR Green Master Mix (Applied Biosystems) and run on a ViiA 7 System (Applied Biosystems). Samples were assayed in three technical replicates. Data were analyzed using the ΔΔ_C_T method and expression was normalized to GAPDH expression (48). RNA was purified from individual samples and processed through a PureLink RNA Mini Kit (Ambion), followed by reverse transcription using SuperScript II (Invitrogen). Primer efficiency was calculated for each pair of primers and the slope of the dilution line was found to be within the appropriate range. Dissociation curves also showed single peak traces, indicating template-specific products.
Nanostring analysis
To analyze gene expression for a large number of genes from an individual sample, we utilized a custom probe set designed by NanoString Technologies (nCounter Gene Expression Assay). The assays were performed using the NanoString protocols, 12 samples per run. The first step hybridization reactions were carried out with 100–200 ng RNA. Posthybridization samples were processed with the nCounter Prep-station. Following run completion, the cartridge was scanned using the nCounter Digital Analyzer, at max resolution (∼1000 images/sample). Data were analyzed using the nSolver Analysis Software and normalized to a set of seven house-keeping (HK) genes or to the total gene set, as noted. HK genes = GAPDH, GUSB, HPRT1, LDHA, POLR2A, RPL13a and RPL27.
Immunocytochemistry and microscopy
Cultures were fixed with 4% paraformaldehyde (Sigma), followed by membrane permeabilization with 0.1% Triton X-100 and then staining with primary and secondary antibodies (see Western blots and antibodies). Imaging was performed using a Zeiss LSM710 confocal microscope and images were acquired using ZEN black software. ZEN black software was used to pseudo-color images and add scale bars.
Western blots and antibodies
Lysates and conditioned media were electrophoresed on 4–12% Bis–Tris gels (Invitrogen) and transferred to nitrocellulose. Lysates were prepared with standard buffer containing 1% NP40, 0.5 m EDTA, 5 m NaCl, 1 m Tris and cOmplete protease inhibitors and phosSTOP (Roche). Western blotting and immunostaining were performed with antibodies from Abcam: MAP2 (1:5000), Oct4 (1:1000), Tbr1 (1:200), Cux1 (1:100), SYP (1:250), PSD95 (1:250) VGLUT1 (1:500), Nanog (1:50). From Millipore: SSEA3 (1:200), SSEA4 (1:200), TRA-1-60 (1:200), GAPDH (1:2000), BACE (1:500), EEA1 (1:500). From Sigma: TuJ1 (1:1000). From Dako: Tau (K9JA) (1:200). From Covance: Aβ (6E10) (1:1000). From Selkoe Lab (Brigham and Women's Hospital): APP (C7,9) (1:1000), APPs (4F2, 3F3) (1:1000, 1:200). From Malinow Lab, UCSD: pS262 (12E8) (1:2000). From Pierce: pS202 (AT8) (1:200). Secondaries were from Jackson ImmunoResearch: anti-chicken Cy2/Cy3/Cy5, anti-rabbit Cy2/Cy3, anti-mouse Cy2/Cy3, anti-rat Cy2/Cy3 (1:1000). TOPRO3, DAPI 1:1000 Invitrogen.
APP/EEA1 colocalization experiment
Control and fAD day 50–60 neurons were immunostained for APP and EEA1 (see Immunocytochemistry and microscopy methods). Zeiss ZEN black software was used to analyze the colocalization coefficient. MAP2+ neurons were outlined for pixel measurement. The colocalization coefficient was calculated by comparing colocalized pixels of APP and EEA1 in the outlined area, to the total number of pixels. Values ranged from 0 to 1, with 0 being no colocalization and 1 being all pixels colocalized.
Aβ, sAPPα/β and Tau ELISAs
Neuronal cells were plated in 96-well plates at various time points up to 100 days. ELISA assays was carried out using the reagents, protocols and imager manufactured by MesoScale Diagnostics, LLC. Media were collected after 48–72 h and analyzed using the 6E10 Abeta Triplex, and sAPPα/sAPPβ ELISA assays (specific to human). Lysates were prepared as noted above and analyzed using the Phospho(Thr231)/Total Tau ELISA assay. Data were normalized to either total RNA or intracellular protein values, as noted.
Inhibitor treatments
Neuronal cells were plated at day 24 and allowed to differentiate until days 40–50. Additionally, for some inhibitor experiments, transiently transfected HEK cells were cultured. Conditioned media were collected 48 h prior to treatment and saved for ELISA assays. Cultures were then treated with either DAPT (5 μm, Sigma), Compound E (50 nm, Millipore), L-685,458 (1.5 μm, Sigma), C3 (0.3 μm, Calbiochem) or DMSO (Sigma) for 48 h, followed by media collection and harvesting for protein. For protein, cells were lysed with standard buffer containing 1% NP40, 0.5 m EDTA, 5 m NaCl, 1 m Tris and cOmplete protease inhibitors and phosSTOP.
Transfections
HEK293 cells were transiently transfected with cDNA encoding either WT human APP695 or with the V717I mutation, using Fugene HD (Promega). Twenty-four hours after transfection, media were changed and cells were treated with either vehicle or inhibitors. Forty-eight hours after transfection, conditioned media were collected and cells lysed in 1% NP40 STEN buffer.
Tau rescue experiments
Neurons were directed to a forebrain neuronal fate and treated with Aβ-specific antibodies: either AW7 (28) or else 3D6 (27) at days 18–20 of differentiation. Cultures were treated for 15 days, with media changes occurring every 3 days. At the end of treatment, conditioned media were collected and cells were lysed for protein (see above). Lysates were utilized for western blotting and the Tau-specific ELISA, while conditioned media was probed for binding of the antibodies to Aβ.
Statistics
Data were analyzed using GraphPad PRISM 5 software. Values are expressed as either ±SD or ±SEM as indicated by figure legend text. Statistical significance was tested by either an unpaired Student's _t_-test (two-tailed) or one-way ANOVA with a Tukey's post-test. Statistically significant differences were determined by _P_-values of <0.05.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
FUNDING
This work was supported by funding from the Harvard Stem Cell Institute, the Massachusetts Alzheimer's Disease Research Center, the BWH Neurosciences Institute and the National Institute on Aging [grant number R21AG042776(TLY-P)].
Supplementary Material
Supplementary Data
ACKNOWLEDGEMENTS
We thank Bianca DiChiaro and Dr Tilman Kispersky for technical assistance and Dr Matt LaVoie, Dr Phil De Jager and members of the LaVoie, Selkoe and Young-Pearse laboratories for helpful discussions. We are grateful to Drs Peter Seubert and Dale Schenk for providing the 3D6 antibody. Familial AD iPSC lines were derived at the HSCI iPS Core facility under the direction of Dr Laurence Daheron.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Bertram L., Lill C.M., Tanzi R.E. The genetics of Alzheimer disease: back to the future. Neuron. 2010;68:270–281. doi: 10.1016/j.neuron.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 2.Wolfe M.S., Xia W., Ostaszewski B.L., Diehl T.S., Kimberly W.T., Selkoe D.J. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- 3.Selkoe D.J., Podlisny M.B. Deciphering the genetic basis of Alzheimer's disease. Annu. Rev. Genomics Hum. Genet. 2002;3:67–99. doi: 10.1146/annurev.genom.3.022502.103022. [DOI] [PubMed] [Google Scholar]
- 4.Bertram L., Tanzi R.E. The genetics of Alzheimer's disease. Prog. Mol. Biol. Transl. Sci. 2012;107:79–100. doi: 10.1016/B978-0-12-385883-2.00008-4. [DOI] [PubMed] [Google Scholar]
- 5.Goate A., Chartier-Harlin M.C., Mullan M., Brown J., Crawford F., Fidani L., Giuffra L., Haynes A., Irving N., James L. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 6.Cruts M., Theuns J., Van Broeckhoven C. Locus-specific mutation databases for neurodegenerative brain diseases. Hum. Mutat. 2012;33:1340–1344. doi: 10.1002/humu.22117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suzuki N., Cheung T.T., Cai X.D., Odaka A., Otvos L., Jr, Eckman C., Golde T.E., Younkin S.G. An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science. 1994;264:1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 8.De Jonghe C., Esselens C., Kumar-Singh S., Craessaerts K., Serneels S., Checler F., Annaert W., Van Broeckhoven C., De Strooper B. Pathogenic APP mutations near the gamma-secretase cleavage site differentially affect Abeta secretion and APP C-terminal fragment stability. Hum. Mol. Genet. 2001;10:1665–1671. doi: 10.1093/hmg/10.16.1665. [DOI] [PubMed] [Google Scholar]
- 9.Moechars D., Dewachter I., Lorent K., Reversé D., Baekelandt V., Naidu A., Tesseur I., Spittaels K., Haute C.V., Checler F., et al. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J. Biol. Chem. 1999;274:6483–6492. doi: 10.1074/jbc.274.10.6483. [DOI] [PubMed] [Google Scholar]
- 10.Dewachter I., van Dorpe J., Spittaels K., Tesseur I., Van Den Haute C., Moechars D., Van Leuven F. Modeling Alzheimer's disease in transgenic mice: effect of age and of presenilin1 on amyloid biochemistry and pathology in APP/London mice. Exp. Gerontol. 2000;35:831–841. doi: 10.1016/s0531-5565(00)00149-2. [DOI] [PubMed] [Google Scholar]
- 11.Tamaoka A., Odaka A., Ishibashi Y., Usami M., Sahara N., Suzuki N., Nukina N., Mizusawa H., Shoji S., Kanazawa I. APP717 missense mutation affects the ratio of amyloid beta protein species (a beta 1-42/43 and a beta 1-40) in familial Alzheimer's disease brain. J. Biol. Chem. 1994;269:32721–32724. [PubMed] [Google Scholar]
- 12.Scheuner D., Eckman C., Jensen M., Song X., Citron M., Suzuki N., Bird T.D., Hardy J., Hutton M., Kukull W., et al. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat. Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 13.Yagi T., Ito D., Okada Y., Akamatsu W., Nihei Y., Yoshizaki T., Yamanaka S., Okano H., Suzuki N. Modeling familial Alzheimer's disease with induced pluripotent stem cells. Hum. Mol. Genet. 2011;20:4530–4539. doi: 10.1093/hmg/ddr394. [DOI] [PubMed] [Google Scholar]
- 14.Ooi L., Sidhu K., Poljak A., Sutherland G., O'Connor M.D., Sachdev P., Münch G. Induced pluripotent stem cells as tools for disease modelling and drug discovery in Alzheimer's disease. J. Neural Transm. 2013;120:103–111. doi: 10.1007/s00702-012-0839-2. [DOI] [PubMed] [Google Scholar]
- 15.Malgrange B., Borgs L., Grobarczyk B., Purnelle A., Ernst P., Moonen G., Nguyen L. Using human pluripotent stem cells to untangle neurodegenerative disease mechanisms. Cell. Mol. Life Sci. 2011;68:635–649. doi: 10.1007/s00018-010-0557-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 17.Park I.-H., Daley G.Q. Human iPS cell derivation/reprogramming. Curr. Protoc. Stem Cell Biol. 2009 doi: 10.1002/9780470151808.sc04a01s8. Chapter 4, Unit 4A.1. [DOI] [PubMed] [Google Scholar]
- 18.Park I.-H., Arora N., Huo H., Maherali N., Ahfeldt T., Shimamura A., Lensch M.W., Cowan C., Hochedlinger K., Daley G.Q. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi Y., Kirwan P., Smith J., MacLean G., Orkin S.H., Livesey F.J. A human stem cell model of early Alzheimer's disease pathology in Down syndrome. Sci. Transl. Med. 2012;4:124–129. doi: 10.1126/scitranslmed.3003771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Israel M.A., Yuan S.H., Bardy C., Reyna S.M., Mu Y., Herrera C., Hefferan M.P., Van Gorp S., Nazor K.L., Boscolo F.S., et al. Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells. Nature. 2012;482:216–220. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kondo T., Asai M., Tsukita K., Kutoku Y., Ohsawa Y., Sunada Y., Imamura K., Egawa N., Yahata N., Okita K., et al. Modeling Alzheimer's disease with iPSCs reveals stress phenotypes associated with intracellular Aβ and differential drug responsiveness. Cell Stem Cell. 2013;12:487–496. doi: 10.1016/j.stem.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 22.Zeng H., Guo M., Martins-Taylor K., Wang X., Zhang Z., Park J.W., Zhan S., Kronenberg M.S., Lichtler A., Liu H.-X., et al. Specification of region-specific neurons including forebrain glutamatergic neurons from human induced pluripotent stem cells. PLoS ONE. 2010;5:e11853. doi: 10.1371/journal.pone.0011853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boulting G.L., Kiskinis E., Croft G.F., Amoroso M.W., Oakley D.H., Wainger B.J., Williams D.J., Kahler D.J., Yamaki M., Davidow L., et al. A functionally characterized test set of human induced pluripotent stem cells. Nat. Biotechnol. 2011;29:279–286. doi: 10.1038/nbt.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Theuns J., Marjaux E., Vandenbulcke M., Van Laere K., Kumar-Singh S., Bormans G., Brouwers N., Van den Broeck M., Vennekens K., Corsmit E., et al. Alzheimer dementia caused by a novel mutation located in the APP C-terminal intracytosolic fragment. Hum. Mutat. 2006;27:888–896. doi: 10.1002/humu.20402. [DOI] [PubMed] [Google Scholar]
- 25.Moore B.D., Chakrabarty P., Levites Y., Kukar T.L., Baine A.-M., Moroni T., Ladd T.B., Das P., Dickson D.W., Golde T.E. Overlapping profiles of Aβ peptides in the Alzheimer's disease and pathological aging brains. Alzheimers Res. Ther. 2012;4:18. doi: 10.1186/alzrt121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haass C., Kaether C., Thinakaran G., Sisodia S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012;2:a006270. doi: 10.1101/cshperspect.a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson-Wood K., Lee M., Motter R., Hu K., Gordon G., Barbour R., Khan K., Gordon M., Tan H., Games D., et al. Amyloid precursor protein processing and A beta42 deposition in a transgenic mouse model of Alzheimer disease. Proc. Natl. Acad. Sci. USA. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McDonald J.M., Cairns N.J., Taylor-Reinwald L., Holtzman D., Walsh D.M. The levels of water-soluble and triton-soluble Aβ are increased in Alzheimer's disease brain. Brain Res. 2012;1450:138–147. doi: 10.1016/j.brainres.2012.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suh Y.-H., Checler F. Amyloid precursor protein, presenilins, and alpha-synuclein: molecular pathogenesis and pharmacological applications in Alzheimer's disease. Pharmacol. Rev. 2002;54:469–525. doi: 10.1124/pr.54.3.469. [DOI] [PubMed] [Google Scholar]
- 30.Lichtenthaler S.F., Wang R., Grimm H., Uljon S.N., Masters C.L., Beyreuther K. Mechanism of the cleavage specificity of Alzheimer's disease γ-secretase identified by phenylalanine-scanning mutagenesis of the transmembrane domain of the amyloid precursor protein. Proc. Natl. Acad. Sci. 1999;96:3053–3058. doi: 10.1073/pnas.96.6.3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qi-Takahara Y., Morishima-Kawashima M., Tanimura Y., Dolios G., Hirotani N., Horikoshi Y., Kametani F., Maeda M., Saido T.C., Wang R., et al. Longer forms of amyloid beta protein: implications for the mechanism of intramembrane cleavage by gamma-secretase. J. Neurosci. 2005;25:436–445. doi: 10.1523/JNEUROSCI.1575-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Strooper B. Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process. Physiol. Rev. 2010;90:465–494. doi: 10.1152/physrev.00023.2009. [DOI] [PubMed] [Google Scholar]
- 33.Chávez-Gutiérrez L., Bammens L., Benilova I., Vandersteen A., Benurwar M., Borgers M., Lismont S., Zhou L., Van Cleynenbreugel S., Esselmann H., et al. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012;31:2261–2274. doi: 10.1038/emboj.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahmed R.R., Holler C.J., Webb R.L., Li F., Beckett T.L., Murphy M.P. BACE1 and BACE2 enzymatic activities in Alzheimer's disease. J. Neurochem. 2010;112:1045–1053. doi: 10.1111/j.1471-4159.2009.06528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cai H., Wang Y., McCarthy D., Wen H., Borchelt D.R., Price D.L., Wong P.C. BACE1 is the major β-secretase for generation of Aβ peptides by neurons. Nat. Neurosci. 2001;4:233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- 36.Vassar R., Bennett B.D., Babu-Khan S., Kahn S., Mendiaz E.A., Denis P., Teplow D.B., Ross S., Amarante P., Loeloff R., et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 37.Bennett B.D., Babu-Khan S., Loeloff R., Louis J.-C., Curran E., Citron M., Vassar R. Expression analysis of BACE2 in brain and peripheral tissues. J. Biol. Chem. 2000;275:20647–20651. doi: 10.1074/jbc.M002688200. [DOI] [PubMed] [Google Scholar]
- 38.Haass C., Selkoe D.J. Cellular processing of beta-amyloid precursor protein and the genesis of amyloid beta-peptide. Cell. 1993;75:1039–1042. doi: 10.1016/0092-8674(93)90312-e. [DOI] [PubMed] [Google Scholar]
- 39.Das U., Scott D.A., Ganguly A., Koo E.H., Tang Y., Roy S. Activity-induced convergence of APP and BACE-1 in acidic microdomains via an endocytosis-dependent pathway. Neuron. 2013;79:447–460. doi: 10.1016/j.neuron.2013.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marcinkiewicz M., Seidah N.G. Coordinated expression of beta-amyloid precursor protein and the putative beta-secretase BACE and alpha-secretase ADAM10 in mouse and human brain. J. Neurochem. 2000;75:2133–2143. doi: 10.1046/j.1471-4159.2000.0752133.x. [DOI] [PubMed] [Google Scholar]
- 41.Vassar R., Kovacs D.M., Yan R., Wong P.C. The β-secretase enzyme BACE in health and Alzheimer's disease: regulation, cell biology, function, and therapeutic potential. J. Neurosci. 2009;29:12787–12794. doi: 10.1523/JNEUROSCI.3657-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knops J., Suomensaari S., Lee M., McConlogue L., Seubert P., Sinha S. Cell-type and amyloid precursor protein-type specific inhibition of A beta release by bafilomycin A1, a selective inhibitor of vacuolar ATPases. J. Biol. Chem. 1995;270:2419–2422. doi: 10.1074/jbc.270.6.2419. [DOI] [PubMed] [Google Scholar]
- 43.Roberson E.D., Scearce-Levie K., Palop J.J., Yan F., Cheng I.H., Wu T., Gerstein H., Yu G.-Q., Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 44.Jin M., Shepardson N., Yang T., Chen G., Walsh D., Selkoe D.J. Soluble amyloid {beta}-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. USA. 2011 doi: 10.1073/pnas.1017033108. 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lewis J., Dickson D.W., Lin W.L., Chisholm L., Corral A., Jones G., Yen S.H., Sahara N., Skipper L., Yager D., et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 46.Khatoon S., Grundke-Iqbal I., Iqbal K. Brain levels of microtubule-associated protein tau are elevated in Alzheimer's disease: a radioimmuno-slot-blot assay for nanograms of the protein. J. Neurochem. 1992;59:750–753. doi: 10.1111/j.1471-4159.1992.tb09432.x. [DOI] [PubMed] [Google Scholar]
- 47.Musiek E.S., Holtzman D.M. Origins of Alzheimer's disease: reconciling cerebrospinal fluid biomarker and neuropathology data regarding the temporal sequence of amyloid-beta and tau involvement. Curr. Opin. Neurol. 2012;25:715–720. doi: 10.1097/WCO.0b013e32835a30f4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data