Open questions for Alzheimer’s disease immunotherapy (original) (raw)
Abstract
Perhaps more definitively than any other class of novel Alzheimer’s disease (AD) therapy, pre-clinical studies in mouse models of amyloid β (Aβ) deposition have established the disease-modifying potential of anti-Aβ immunotherapy. Despite disappointing results to date from anti-Aβ immunotherapy therapeutic trials, there is continued hope that such immunotherapies, especially if used in the preclinical stages, could prove to be the first disease-modifying therapies available for AD. The general optimism that Aβ-targeting and emerging tau-targeting immunotherapies may prove to be disease modifying is tempered by many unanswered questions regarding these therapeutic approaches, including but not limited to i) lack of precise understanding of mechanisms of action, ii) the factors that regulate antibody exposure in the brain, iii) the optimal target epitope, and iv) the mechanisms underlying side effects. In this review I discuss how answering these and other questions could increase the likelihood of therapeutic success. As passive immunotherapies are also likely to be extremely expensive, I also raise questions relating to cost-benefit of biologic-based therapies for AD that could limit future impact of these therapies by limiting access due to economic constraints.
Introduction
Over the past several years, data from human trials testing the efficacy of anti-amyloid β (anti-Aβ) immunotherapies and intravenous immunoglobulin in symptomatic Alzheimer’s disease (AD) patients have been disappointing, although this is perhaps not unexpected. Yet despite these clinical setbacks, development and clinical testing of immunotherapies for AD remain the most active areas of both clinical and pre-clinical development [1]. For over a decade, the main target of immunotherapies has been Aβ, but in the past few years anti-tau immunotherapies have emerged and are rapidly advancing to the clinic. Despite the huge investments, both in therapeutic development and clinical testing, there remain many fundamental gaps in our knowledge regarding how immunotherapies for AD work and how to optimize them [2]. In this review, I address some of these gaps in our knowledge and discuss how filling them in will likely result in therapeutics more likely to have significant clinical efficacy.
Is brain exposure the key?
The issue of how a small amount of anti-Aβ monoclonal antibody (mAb) present in the brain following peripheral dosing can have a therapeutic effect on plaque pathology has posed a dilemma for the field. It is well established that steady state central nervous system (CNS) levels of a peripherally administered anti-Aβ mAb are approximately 0.1% of the levels found in the plasma [3-5]. Although it remains remotely plausible that anti-Aβ therapy promotes efflux of Aβ or an Aβ aggregate from the brain to the plasma via a peripheral sink [6], a growing body of evidence suggests that mAb exposure in the brain is critical for efficacy [2]. If this proves to be the case, then increasing total mAb CNS exposure can have a huge positive impact on efficacy. Indeed, given a set of anti-Aβ mAbs with similar pharmacokinetic properties, one would predict that those that can be dosed at higher levels would be more efficacious. Alternatively, efforts to increase brain uptake (for example, by hijacking transferrin or insulin receptor-mediated transcytosis machinery [7,8]) might also be worth the extensive antibody engineering required to achieve modest, but nevertheless significant, increases in brain exposure [5]. In support of this concept, two preclinical studies, one testing mAb infusion via mini pumps into the ventricles and another testing the effects of direct transgenic expression in the brain of an anti-Aβ mAb, both demonstrate enhanced efficacy relative to peripheral mAb administration [9,10]. Although some in the field remain skeptical about a central mechanism of action of anti-Aβ antibodies in the brain, there are numerous examples of peripherally produced natural antibodies that cause neurological syndromes by targeting a CNS protein [11,12]. Thus, for remaining skeptics I would simply state that if a peripherally produced antibody can cause CNS disease, then a peripherally injected antibody that targets a pathologic target should also be capable of having a therapeutic effect.
A more general review of the literature reveals that there is a paucity of data regarding antibody exposure in the CNS. Based on findings that centrally administered antibodies are rapidly exported to the periphery, it nevertheless appears likely that there is cycling of the mAb between the CNS and plasma compartments [3-5]. Thus, the 0.1% of antibody should not be viewed as in a static steady state, but rather a dynamic equilibrium in which the mAb rapidly enters the brain and subsequently is rapidly exported from the brain. As shown in Figure 1, if the cycling time is rapid (for example, 1 hour) one can estimate that CNS exposures of a human therapeutic dose of anti-Aβ could influence Aβ through stoichiometric binding. Given the limited data available, it would seem that a renewed effort to understand mAb efflux from the brain is warranted. If mAb cycling times are fast and the influx and efflux mechanisms are distinct, it may be possible to increase CNS mAb exposure by identifying and then manipulating these mechanisms. Alternatively, perhaps we should collectively consider direct infusion of the mAb into the brain [9]. Indeed, given the costs of mAb production and the amounts required in current trials (typically 2 to 3 g per patient), direct infusion might require dramatically less mAb to achieve equivalent efficacy. Although it would be more invasive, direct infusion might be more cost-effective. Furthermore, direct infusion of the mAb might also be used as proof of concept studies in small human trials to establish efficacy with no uncertainty regarding sufficient brain exposure. Two caveats with respect to possible clinical trials of direct infusion studies would be the unknowns regarding how the antibodies distribute in the brain following infusion and how site of infusion might influence that distribution.
Figure 1.
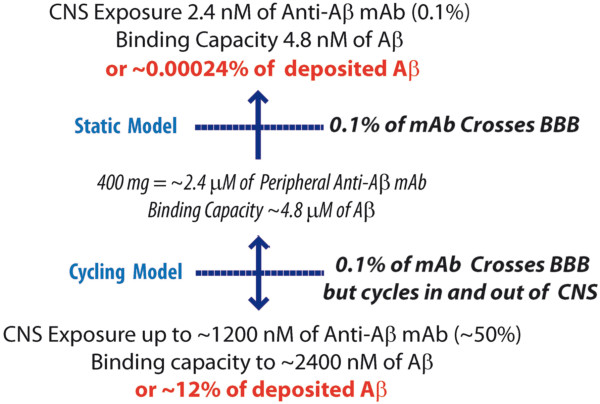
Comparisons of central nervous system (CNS) monoclonal antibody (mAb) exposure in a static influx model versus a cycling influx and efflux model. Based on estimates that ~20 μM of amyloid β (Aβ) (~100 mg) are deposited in the Alzheimer’s disease brain, the potential target engagement in each model is shown. The parameters used correspond to human studies using a 400 mg dose of anti-Aβ mAb. A method to estimate exposure based on a trapezoidal method for estimating the area under the curve was used with a theoretical cycling time of 1 hour (complete exchange) and antibody half-life of 21 days. In the cycling model, the estimate of how much Aβ could be targeted in the brain is almost certainly an overestimate as the model does not take into account the efficiency of antibody binding within the brain and the extent to which binding of plasma Aβ or other peripheral sources could decrease the amount of free mAb entering the brain. The issue of how much binding of plasma/peripheral Aβ might decrease free mAb exposure in the brain is complex and will be related to the target epitope and antibody affinity. Given an estimate for the daily turnover of Aβ in plasma of ~50 nmol, if the mAb bound all plasma Aβ produced in a day and the binding was essentially irreversible (as has been observed for several anti-Aβ mAbs), then that binding would be predicted to reduce the exposure of free antibody in the brain by ~50%. BBB, blood-brain barrier.
The neonatal Fc receptor (FcRn) is a major mediator of immunoglobulin (Ig)G transcytosis and recycling of IgG that is initially taken up by cells through fluid phase endocytosis (reviewed in [13]). Although FcRn has been reported to mediate efflux of IgG from the CNS to the blood [14] and also play a role in IgG assisted clearance of Aβ [15], other data suggest that FcRn and other FcR-mediated mechanisms of efflux may be more complicated [16]. Indeed, studies in FcRn-deficient mice have demonstrated that the brain levels of IgG are similar to wild-type mice following intravenous administration of IgG [16]. Thus, it is clear that additional studies on both antibody influx into and efflux out of the CNS are needed to better understand the mechanism that would regulate antibody exposure in the brain [13]. Other key gaps in our knowledge are whether the mAb influx into the brain from the periphery results in homogenous mAb distribution and whether reported dysfunction of the blood-brain barrier in AD would alter the normal distribution. In this regard, it would be interesting to evaluate whether antibody transport within the brain and efflux from the brain are mediated by the newly described drainage pathway for cerebrospinal fluid [17-19]. This brain-wide clearance pathway, which has been termed the glymphatic system, has been shown to facilitate clearance of solutes from the brain, with cerebrospinal fluid entering along periarteriolar channels where there is solute exchange with interstitial fluid, and then exiting via para-venous pathways [18]. The glymphatic pathway may also be of interest regarding the potential for redistribution of parenchymal amyloid deposits to the vascular deposits as a result of immunotherapy [20].
How do target epitope and binding affinity influence potential efficacy in humans?
Another critical unanswered issue is how the Aβ target epitope and binding affinity influence efficacy in humans [2,21]. Based on preclinical studies showing enhanced efficacy of mAbs that bind to Aβ, the vast majority of mAbs that have advanced to trials have been selected to bind both monomeric and aggregated forms of Aβ. In many cases, either simply because of increased avidity, recognition of a conformational epitope, or some combination of these properties, these antibodies often appear to have higher affinity for aggregated Aβ, and, for the most part, are reported to bind near the amino terminus of Aβ [1]. Solanezumab is the exception; it binds the mid-domain of monomeric, but not aggregated, Aβ with extremely high affinity [22,23]. Although, the human data to date are quite limited, in contrast to what might be predicted based on preclinical studies, solanezumab is the only mAb for which there is evidence of a hint of clinical efficacy in phase 3.
As the notion of targeting pathological Aβ aggregates, either fibrils or oligomers, makes a great deal of conceptual sense in terms of both avoiding targeting presumably non-toxic, non-aggregated forms of Aβ that could have some physiological role and also potentially increasing exposure of free mAb to the CNS by avoiding binding of plasma Aβ, the current negative bapinezumab data and suggestive solanezumab data present the field with somewhat of a dilemma. Moreover, the clinical data raise larger questions of whether we really understand how target epitope and affinity can be optimized to enhance efficacy. For example, do we want an Aβ-targeting antibody with extremely high affinity that will bind plaques in the brain and stay bound until degraded? Or do we want an antibody that binds soluble Aβ or soluble Aβ aggregates with modest affinity so that the antibody can carry them to the periphery where they could dissociate and be degraded? Or do we want to target specific modified epitopes of Aβ that are preferentially found in aggregated forms such as pE3-Aβ or nitrosylated forms of Aβ [24-27]? Unfortunately, given the differences between mouse models of Aβ deposition and the limited data on detailed binding constants that are available for many of the mAbs as well as the lack of comparative binding data [26], these questions may ultimately only be answered by the data that emerge from ongoing human trials, which is a very expensive and inefficient path forward.
In addition to having different biologic activities, binding affinity may skew interpretation of antibody target engagement studies in humans. Antibodies that bind Aβ with high affinity tend to raise plasma Aβ to the greatest extent, probably by preventing the rapid clearance of plasma Aβ, which normally has a half-life of approximately 10 minutes [3,28]. Although some of the mAb-bound Aβ may have come from mAb binding in the brain and then the complex being transported to the plasma, it is challenging to distinguish such brain-derived complexes from complexes that form when the antibody binds Aβ in blood. Furthermore, if an antibody has modest affinity for monomeric Aβ and exhibits a relatively rapid off-rate, then it may be difficult to see engagement of monomeric Aβ as assessed by rise in plasma Aβ; though the antibody binds Aβ, the complex is not stable and thus Aβ will dissociate and be rapidly degraded. In any case, more information on how affinity and other binding properties determine not only clinical efficacy but also Aβ biomarker changes will help us better understand how these anti-Aβ mAbs are acting in humans and what properties are most predictive of various clinical outcomes.
What is the role of antibody effector function?
Preclinical studies demonstrate that, depending on the timing of the intervention, antibody effector functions mediated by the Fc region may not be required for efficacy [3,29-31]. In prevention studies in amyloid precursor protein (APP) mice, recombinant antibodies lacking effector functions can be shown to be effective. In contrast, there is some evidence in therapeutic studies targeting Aβ in mice with pre-existing amyloid deposits that antibody effector functions may facilitate or even be required for reduction of deposited Aβ [27]. Anti-Aβ antibodies likely attenuate amyloid deposition through multiple non-exclusive mechanisms that include direct binding and subsequent export from the brain, inhibition of aggregation (even at substoichiometric levels), and enhancing microglial phagocytosis and degradation. Varying conclusions derived from these and other preclinical studies likely reflect the complex actions that antibodies have on CNS amyloid and how those actions are, in part, determined by the pre-existing amyloid load at the time the treatment is initiated [2,21].
As the Fc regions can bind FcR on immune cells (presumably microglial cells in the brain), it is possible that this engagement enhances Aβ phagocytosis and also elicits signaling that could indirectly enhance Aβ clearance. Again, in humans, there is insufficient data to understand the importance of IgG isotype and effector functions, but preliminary reports suggest that utilization of different IgG isotypes or engineered isotypes may have clinical significance. Indeed, crenezumab, which uses an IgG4 backbone with mutations that reduce affinity for FcR [32], appeared in the initial phase I study to avoid amyloid-related imaging abnormalities (ARIAs) even at higher doses than have been tolerated for other anti-Aβ mAbs. Although on the surface this may seem desirable, if Aβ removal is key this could inadvertently impair clearance by limiting glial activation. For example, in early phase human studies of gantenerumab, a fully human anti-Aβ IgG1, there was evidence that the regions of the brain showing radiographic abnormalities following antibody administration also showed the highest reduction in signal on subsequent amyloid PIB (C11-Pittsburgh Compound B) scans [33,34]. Other modifications, such as deglycosylation, that reduce affinity for FcγR and impair ability to bind complement can be shown in mice to reduce potential vascular side effects of anti-Aβ mAbs [35]. However, deglycosylated antibodies that decrease effector function or Fab fragments and single chain antibodies that have no effector functions have not advanced to human studies.
What causes amyloid-related imaging abnormality?
ARIA is an acronym that refers to both vasogenic edema (ARIA-e) and microhemorrhage (ARIA-H) observed by magnetic resonance imaging (MRI) in patients receiving anti-Aβ immunotherapies [36]. Although cortical microhemorrhage is frequently observed during the natural history of AD and is thought to be in part related to amyloid angiopathy, vasogenic edema is rarely observed; however, passive immunotherapy with select anti-Aβ mAbs (for example, bapinezumab) results in increased ARIAs that appear to be more frequent in APOE4 carriers and increases in frequency with increasing dose of mAb [37]. In most cases, ARIAs produce no detectable clinical symptoms, but in some cases are associated with acute worsening of cognitive function. Although the prevailing mechanistic theory relates to mobilization of Aβ by the mAb leading to local edema and hemorrhage, no data clearly demonstrate the mechanisms underlying ARIAs. Indeed, it is just as plausible that anti-Aβ mAb engages amyloid in vessels leading to focal immune activation and that the inflammation clears the amyloid indirectly. Given the prevalence of ARIAs in the bapinezumab trial and their clear association with escalating dose, an enhanced understanding of ARIAs is needed. Indeed, given the costs associated with MRI and the dose-limiting effect, ARIAs pose a significant obstacle for development of certain mAbs. Certainly, a better understanding of the mechanism underlying ARIAs would streamline mAb development and perhaps lead to a more optimal immunotherapy.
Why so little news on second generation active vaccines targeting amyloid β?
Three active anti-Aβ vaccines are in phase II trials for AD, but, except for CAD106 (Novartis/Cytos), almost no data have been released regarding their ability to induce anti-Aβ immune responses, avoid side effects observed with the AN-1792 vaccine, and to alter relevant biomarkers [38]. Although a cautious approach is justified given that the vaccines are targeting a self-epitope and thus can induce autoimmune disease, it is puzzling as to why there are so few data, let alone word of mouth insight, regarding how the testing of these vaccine candidates is proceeding. Although most of the second generation anti-Aβ vaccines are designed to maximize humoral anti-Aβ responses and minimize harmful T-cell responses, it should be noted that the mechanism responsible for the meningoencephalitic reaction in a subset of patients receiving the AN1792 vaccine remains uncertain [39,40]. It has been inferred that the likely cause was harmful T-cell response, but the data supporting this inference are only circumstantial. Moreover, given the rather uneven distribution of the apparent clearance of Aβ in the brain in a handful of subjects who had a postmortem brain autopsy, one has to speculate whether T-cell or other cellular immune mechanisms played a role in clearance [41,42]. Indeed, most T-cell-related disease of the brain is patchy in nature, and it is hard to envisage how there could be extensive focal clearance mediated solely by a peripherally produced mAb.
Anti-tau immunotherapy?
Multiple reports now demonstrate the therapeutic potential of active and passive immunotherapies for tau, at least in terms of ability to reduce pathological tau burden in mouse models (reviewed in [21,43,44]). Coupled with reports demonstrating that tau is secreted and that extracellular pathological forms of tau can induce intracellular tau pathology in culture and in mice, there is a burgeoning effort to move both active and passive tau immunotherapies towards the clinic [45-47]. Notably, almost all of the gaps in our knowledge regarding CNS exposure, effector functions, and target epitope discussed above with respect to anti-Aβ immunotherapies apply to tau targeted therapies. Furthermore, though many in the field now accept the potential of tau immunotherapy based on the premise that extracellular tau may be the target, as supported by data from a recent study showing antibodies that block spread of tau seeding in culture also effectively attenuate tau pathology _in vivo_[48], additional mechanisms should be considered. For example, neurons do express FcR, and therefore could bind and even internalize mAbs [49,50], and, in contrast to the recent report that supports extracellular targeting of tau as a primary mechanism of tau antibodies [48], another recent report provides further evidence that tau antibodies can enter neurons and target intracellular tau [51]. In addition, a recent study has shown that an intracellular protein called TRIM21, which contains a high-affinity Fc-binding domain, can recognize low levels of antibody bound to cargo, ubiquitinate that cargo, and thereby target it for degradation by the proteosome [52]. Thus, it is possible that these mechanisms, or others that are largely under the radar, may contribute to the efficacy of anti-tau immunotherapies. As with anti-Aβ immunotherapies, it is likely that a better understanding of the mechanism will ultimately result in more efficacious and safer immunotherapy.
What do the failed immunotherapy trials tell us about targeting amyloid β in symptomatic Alzheimer’s disease patients?
The amyloid or Aβ aggregate hypothesis only predicts that preventing Aβ aggregation and accumulation in the brain will prevent the development of AD [53]. It does not predict that clearing deposits in symptomatic patients will have clinical benefit. Furthermore, although slowing ongoing deposition or clearing pre-existing deposits in preclinical stages of AD might be predicted to have some clinical benefit, there is a reasonable possibility that Aβ aggregates trigger downstream events that contribute to neurodegeneration that subsequently becomes self-sustaining. If this is the case, then even clearance of Aβ in preclinical AD may have limited efficacy. Studies of postmortem brains from patients previously enrolled in the AN1792 vaccine trial certainly provide some support for the assertion that regional clearance of Aβ is not associated with clinical benefit in patients with AD [41]. In this context, the recent failures of anti-Aβ mAbs to show significant and consistent efficacy are, in fact, likely and not unexpected outcomes.
Another pressing question regarding these trials relates to biologic effects of the mAbs in the brain. Although increased incidence of ARIAs and suggestive evidence that there may be slight reductions in amyloid loads based on serial amyloid scans support target engagement in the brain, the consequences of such engagement are poorly understood [54]. Though practically challenging, efforts to systematically obtain postmortem brains from subjects in these trials would be of major utility for the field. Given the differences between human and mouse brain and the ongoing uncertainties regarding mechanisms of action, rigorous postmortem analyses could provide unique insights into Aβ immunotherapies that might be used to guide future efforts designed to optimize them.
Can we afford suboptimal passive immunotherapies?
A final question that relates to public health policy is whether we can afford passive immunotherapy for AD that has limited clinical benefit? Given the likely costs of a biologic therapy and the ancillary testing (for example, amyloid scans and MRIs) that may be required to prescribe and monitor an approved passive immunotherapy, it is highly likely that the yearly costs of passive immunotherapy for AD will exceed 25,000to25,000 to 25,000to30,000. It is unclear whether any country’s health system can afford such therapy if it has a very modest effect on disease course. (Indeed, it is not even clear that we can afford it even if it has a more robust clinical effect.) Furthermore, given set-costs associated with manufacture of antibodies in quantities that would be needed to treat a prevalent disease and the uncertain road map for developing less-expensive generic biosimilars, it is unlikely that costs for such therapy would decline in the foreseeable future. This general issue of cost versus benefit of any novel therapy for most diseases is under increasing scrutiny in many countries and decisions whether to pay or not can have huge socioeconomic implications. Clearly, any convincing evidence that a passive immunotherapy had clinical benefit will be welcome news for the field, but given the changing climate the field may be well-served by more openly discussing the issue of whether society can afford this type of therapy, especially if it has only limited clinical benefit.
Conclusions
Despite intensive study for over a decade, many aspects of immunotherapy for AD remain enigmatic. Future studies designed to answer questions raised in this review, such as those relating to mechanism of antibody action and factors regulating CNS antibody exposure, could play major roles in guiding development of more optimal therapies. Given the challenges of developing active vaccines that potentially target self-epitopes and thus could induce autoimmune disease, passive immunotherapies, which appear to be relatively safe and have more certainty regarding target engagement, are clearly ideal ways to move forward to evaluate potential efficacy in AD. Given their expense, however, treatment with passive immunotherapies may not represent an ideal long-term public health solution to the AD epidemic. In contrast, from a public health perspective, vaccines would almost certainly be a cost-effective solution; thus, efforts to develop effective and sufficiently safe vaccines need to be supported.
More generally, there has been a general lack of appreciation for how successful development of AD vaccines and passive immunotherapies could result in a paradigm shift regarding immunotherapies for many CNS disorders. Largely based on the dogma that only a little antibody gets into the brain, until Schenk and colleagues demonstrated the potential utility of this approach in AD animal models [55,56], there was essentially no interest in development of antibody-based therapies against CNS targets. Given the ability to develop immunologic reagents with incredible specificity for a given target, if any form of AD immunotherapy proves efficacious, it is quite possible that in the future we may see antibodies and vaccines used for not only other neurodegenerative disease but even many other neurologic and psychiatric conditions.
Abbreviations
AD: Alzheimer’s disease; ARIA: Amyloid-related imaging abnormality; Aβ: Amyloid β; CNS: Central nervous system; FcRn: Neonatal Fc receptor; Ig: Immunoglobulin; mAb: Monoclonal antibody; MRI: Magnetic resonance imaging.
Competing interests
TEG is co-Editor-in-Chief of Alzheimer’s Research & Therapy and receives an annual honorarium. TEG is an inventor on patents relating to anti-amyloid vaccines and antibodies.
References
- Prins ND, Scheltens P. Treating Alzheimer’s disease with monoclonal antibodies; current status and outlook to the future. Alzheimers Res Ther. 2013;6:56. doi: 10.1186/alzrt220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golde TE, Das P, Levites Y. Quantitative and mechanistic studies of abeta immunotherapy. CNS Neurol Disord Drug Targets. 2009;6:31–49. doi: 10.2174/187152709787601830. [DOI] [PubMed] [Google Scholar]
- Levites Y, Smithson LA, Price RW, Dakin RS, Yuan B, Sierks MR, Kim J, McGowan E, Reed DK, Rosenberry TL, Das P, Golde TE. Insights into the mechanisms of action of anti-Abeta antibodies in Alzheimer’s disease mouse models. FASEB J. 2006;6:2576–2578. doi: 10.1096/fj.06-6463fje. [DOI] [PubMed] [Google Scholar]
- Atwal JK, Chen Y, Chiu C, Mortensen DL, Meilandt WJ, Liu Y, Heise CE, Hoyte K, Luk W, Lu Y, Peng K, Wu P, Rouge L, Zhang Y, Lazarus RA, Scearce-Levie K, Wang W, Wu Y, Tessier-Lavigne M, Watts RJ. A therapeutic antibody targeting BACE1 inhibits amyloid-beta production in vivo. Sci Transl Med. 2011;6:84ra43. doi: 10.1126/scitranslmed.3002254. [DOI] [PubMed] [Google Scholar]
- Yu YJ, Zhang Y, Kenrick M, Hoyte K, Luk W, Lu Y, Atwal J, Elliott JM, Prabhu S, Watts RJ, Dennis MS. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci Transl Med. 2011;6:84ra44. doi: 10.1126/scitranslmed.3002230. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM. Brain to plasma amyloid-beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer’s disease. Science. 2002;6:2264–2267. doi: 10.1126/science.1067568. [DOI] [PubMed] [Google Scholar]
- Bickel U, Yoshikawa T, Pardridge WM. Delivery of peptides and proteins through the blood-brain barrier. Adv Drug Deliv Rev. 2001;6:247–279. doi: 10.1016/S0169-409X(00)00139-3. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Engelhardt B, Lesley J, Bickel U, Pardridge WM. Targeting rat anti-mouse transferrin receptor monoclonal antibodies through blood-brain barrier in mouse. J Pharmacol Exp Ther. 2000;6:1048–1052. [PubMed] [Google Scholar]
- Thakker DR, Weatherspoon MR, Harrison J, Keene TE, Lane DS, Kaemmerer WF, Stewart GR, Shafer LL. Intracerebroventricular amyloid-beta antibodies reduce cerebral amyloid angiopathy and associated micro-hemorrhages in aged Tg2576 mice. Proc Natl Acad Sci U S A. 2009;6:4501–4506. doi: 10.1073/pnas.0813404106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paganetti P, Reichwald J, Bleckmann D, Abramowski D, Ammaturo D, Barske C, Danner S, Molinari M, Müller M, Papin S, Rabe S, Schmid P, Staufenbiel M. Transgenic expression of beta1 antibody in brain neurons impairs age-dependent amyloid deposition in APP23 mice. Neurobiol Aging. 2013;6:2866–2878. doi: 10.1016/j.neurobiolaging.2013.06.013. [DOI] [PubMed] [Google Scholar]
- Irani SR, Vincent A. NMDA receptor antibody encephalitis. Curr Neurol Neurosci Rep. 2011;6:298–304. doi: 10.1007/s11910-011-0186-y. [DOI] [PubMed] [Google Scholar]
- Panzer J, Dalmau J. Movement disorders in paraneoplastic and autoimmune disease. Curr Opin Neurol. 2011;6:346–353. doi: 10.1097/WCO.0b013e328347b307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strazielle N, Ghersi-Egea JF. Physiology of blood-brain interfaces in relation to brain disposition of small compounds and macromolecules. Mol Pharm. 2013;6:1473–1491. doi: 10.1021/mp300518e. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Pardridge WM. Mediated efflux of IgG molecules from brain to blood across the blood-brain barrier. J Neuroimmunol. 2001;6:168–172. doi: 10.1016/S0165-5728(01)00242-9. [DOI] [PubMed] [Google Scholar]
- Deane R, Sagare A, Hamm K, Parisi M, LaRue B, Guo H, Wu Z, Holtzman DM, Zlokovic BV. IgG-assisted age-dependent clearance of Alzheimer’s amyloid beta peptide by the blood-brain barrier neonatal Fc receptor. J Neurosci. 2005;6:11495–11503. doi: 10.1523/JNEUROSCI.3697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abuqayyas L, Balthasar JP. Investigation of the role of FcgammaR and FcRn in mAb distribution to the brain. Mol Pharm. 2013;6:1505–1513. doi: 10.1021/mp300214k. [DOI] [PubMed] [Google Scholar]
- Iliff JJ, Lee H, Yu M, Feng T, Logan J, Nedergaard M, Benveniste H. Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J Clin Invest. 2013;6:1299–1309. doi: 10.1172/JCI67677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med. 2012;6:147ra111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Kress BT, Weber HJ, Thiyagarajan M, Wang B, Deane R, Benveniste H, Iliff JJ, Nedergaard M. Evaluating glymphatic pathway function utilizing clinically relevant intrathecal infusion of CSF tracer. J Transl Med. 2013;6:107. doi: 10.1186/1479-5876-11-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller RO, Preston SD, Subash M, Carare RO. Cerebral amyloid angiopathy in the aetiology and immunotherapy of Alzheimer disease. Alzheimers Res Ther. 2009;6:6. doi: 10.1186/alzrt6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YJ, Watts RJ. Developing therapeutic antibodies for neurodegenerative disease. Neurotherapeutics. 2013;6:459–472. doi: 10.1007/s13311-013-0187-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seubert P, Barbour R, Khan K, Motter R, Tang P, Kholodenko D, Kling K, Schenk D, Johnson-Wood K, Schroeter S, Gill D, Jacobsen JS, Pangalos M, Basi G, Games D. Antibody capture of soluble Abeta does not reduce cortical Abeta amyloidosis in the PDAPP mouse. Neurodegener Dis. 2008;6:65–71. doi: 10.1159/000112834. [DOI] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer’s disease model. Nat Neurosci. 2002;6:452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- Jawhar S, Wirths O, Bayer TA. Pyroglutamate amyloid-beta (Abeta): a hatchet man in Alzheimer disease. J Biol Chem. 2011;6:38825–38832. doi: 10.1074/jbc.R111.288308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer MP, Hermes M, Delekarte A, Hammerschmidt T, Kumar S, Terwel D, Walter J, Pape HC, König S, Roeber S, Jessen F, Klockgether T, Korte M, Heneka MT. Nitration of tyrosine 10 critically enhances amyloid beta aggregation and plaque formation. Neuron. 2011;6:833–844. doi: 10.1016/j.neuron.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Frost JL, Le KX, Cynis H, Ekpo E, Kleinschmidt M, Palmour RM, Ervin FR, Snigdha S, Cotman CW, Saido TC, Vassar RJ, St George-Hyslop P, Ikezu T, Schilling S, Demuth HU, Lemere CA. Pyroglutamate-3 amyloid-beta deposition in the brains of humans, non-human primates, canines, and Alzheimer disease-like transgenic mouse models. Am J Pathol. 2013;6:369–381. doi: 10.1016/j.ajpath.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demattos RB, Lu J, Tang Y, Racke MM, Delong CA, Tzaferis JA, Hole JT, Forster BM, McDonnell PC, Liu F, Kinley RD, Jordan WH, Hutton ML. A plaque-specific antibody clears existing beta-amyloid plaques in Alzheimer’s disease mice. Neuron. 2012;6:908–920. doi: 10.1016/j.neuron.2012.10.029. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2001;6:8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levites Y, Jansen K, Smithson LA, Dakin R, Holloway VM, Das P, Golde TE. Intracranial adeno-associated virus-mediated delivery of anti-pan amyloid beta, amyloid beta40, and amyloid beta42 single-chain variable fragments attenuates plaque pathology in amyloid precursor protein mice. J Neurosci. 2006;6:11923–11928. doi: 10.1523/JNEUROSCI.2795-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das P, Howard V, Loosbrock N, Dickson D, Murphy MP, Golde TE. Amyloid-beta immunization effectively reduces amyloid deposition in FcRgamma-/- knock-out mice. J Neurosci. 2003;6:8532–8538. doi: 10.1523/JNEUROSCI.23-24-08532.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacskai BJ, Kajdasz ST, McLellan ME, Games D, Seubert P, Schenk D, Hyman BT. Non-Fc-mediated mechanisms are involved in clearance of amyloid-beta in vivo by immunotherapy. J Neurosci. 2002;6:7873–7878. doi: 10.1523/JNEUROSCI.22-18-07873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adolfsson O, Pihlgren M, Toni N, Varisco Y, Buccarello AL, Antoniello K, Lohmann S, Piorkowska K, Gafner V, Atwal JK, Maloney J, Chen M, Gogineni A, Weimer RM, Mortensen DL, Friesenhahn M, Ho C, Paul R, Pfeifer A, Muhs A, Watts RJ. An effector-reduced anti-beta-amyloid (Abeta) antibody with unique abeta binding properties promotes neuroprotection and glial engulfment of Abeta. J Neurosci. 2012;6:9677–9689. doi: 10.1523/JNEUROSCI.4742-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowitzki S, Deptula D, Thurfjell L, Barkhof F, Bohrmann B, Brooks DJ, Klunk WE, Ashford E, Yoo K, Xu ZX, Loetscher H, Santarelli L. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch Neurol. 2012;6:198–207. doi: 10.1001/archneurol.2011.1538. [DOI] [PubMed] [Google Scholar]
- Bohrmann B, Baumann K, Benz J, Gerber F, Huber W, Knoflach F, Messer J, Oroszlan K, Rauchenberger R, Richter WF, Rothe C, Urban M, Bardroff M, Winter M, Nordstedt C, Loetscher H. Gantenerumab: a novel human anti-Abeta antibody demonstrates sustained cerebral amyloid-beta binding and elicits cell-mediated removal of human amyloid-beta. J Alzheimers Dis. 2012;6:49–69. doi: 10.3233/JAD-2011-110977. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Alamed J, Gottschall PE, Grimm J, Rosenthal A, Pons J, Ronan V, Symmonds K, Gordon MN, Morgan D. Deglycosylated anti-amyloid-beta antibodies eliminate cognitive deficits and reduce parenchymal amyloid with minimal vascular consequences in aged amyloid precursor protein transgenic mice. J Neurosci. 2006;6:5340–5346. doi: 10.1523/JNEUROSCI.0695-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Jack CR Jr, Black SE, Frosch MP, Greenberg SM, Hyman BT, Scheltens P, Carrillo MC, Thies W, Bednar MM, Black RS, Brashear HR, Grundman M, Siemers ER, Feldman HH, Schindler RJ. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement. 2011;6:367–385. doi: 10.1016/j.jalz.2011.05.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling R, Salloway S, Brooks DJ, Tampieri D, Barakos J, Fox NC, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Lieberburg I, Arrighi HM, Morris KA, Lu Y, Liu E, Gregg KM, Brashear HR, Kinney GG, Black R, Grundman M. Amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol. 2012;6:241–249. doi: 10.1016/S1474-4422(12)70015-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winblad B, Andreasen N, Minthon L, Floesser A, Imbert G, Dumortier T, Maguire RP, Blennow K, Lundmark J, Staufenbiel M, Orgogozo JM, Graf A. Safety, tolerability, and antibody response of active Abeta immunotherapy with CAD106 in patients with Alzheimer’s disease: randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol. 2012;6:597–604. doi: 10.1016/S1474-4422(12)70140-0. [DOI] [PubMed] [Google Scholar]
- Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Rovira MB, Forette F, Orgogozo JM. AN1792(QS-21)-201 Study Team. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;6:1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;6:46–54. doi: 10.1212/01.WNL.0000073623.84147.A8. [DOI] [PubMed] [Google Scholar]
- Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;6:216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- Boche D, Donald J, Love S, Harris S, Neal JW, Holmes C, Nicoll JA. Reduction of aggregated Tau in neuronal processes but not in the cell bodies after Abeta42 immunisation in Alzheimer’s disease. Acta Neuropathol. 2010;6:13–20. doi: 10.1007/s00401-010-0705-y. [DOI] [PubMed] [Google Scholar]
- Sigurdsson EM. Immunotherapy targeting pathological tau protein in Alzheimer’s disease and related tauopathies. J Alzheimers Dis. 2008;6:157–168. doi: 10.3233/jad-2008-15202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Sigurdsson EM. Immunotherapy for tauopathies. J Mol Neurosci. 2011;6:690–695. doi: 10.1007/s12031-011-9576-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Cirrito JR, Stewart FR, Jiang H, Finn MB, Holmes BB, Binder LI, Mandelkow EM, Diamond MI, Lee VM, Holtzman DM. In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci. 2011;6:13110–13117. doi: 10.1523/JNEUROSCI.2569-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012;6:e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires-Jones TL, Hyman BT. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron. 2012;6:685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, Maloney SE, Wozniak DF, Diamond MI, Holtzman DM. Anti-Tau antibodies that block Tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron. 2013;6:402–414. doi: 10.1016/j.neuron.2013.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Vizarra P, Lopez-Franco O, Mallavia B, Higuera-Matas A, Lopez-Parra V, Ortiz-Munoz G, Ambrosio E, Egido J, Almeida OF, Gomez-Guerrero C. Immunoglobulin G Fc receptor deficiency prevents Alzheimer-like pathology and cognitive impairment in mice. Brain. 2012;6:2826–2837. doi: 10.1093/brain/aws195. [DOI] [PubMed] [Google Scholar]
- Deng J, Hou H, Giunta B, Mori T, Wang YJ, Fernandez F, Weggen S, Araki W, Obregon D, Tan J. Autoreactive-Abeta antibodies promote APP beta-secretase processing. J Neurochem. 2012;6:732–740. doi: 10.1111/j.1471-4159.2011.07629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Congdon EE, Sigurdsson EM. Two novel tau antibodies targeting the 396/404 region are primarily taken up by neurons and reduce tau pathology. J Biol Chem. 2013. [epub ahead of print] [DOI] [PMC free article] [PubMed]
- McEwan WA, Mallery DL, Rhodes DA, Trowsdale J, James LC. Intracellular antibody-mediated immunity and the role of TRIM21. Bioessays. 2011;6:803–809. doi: 10.1002/bies.201100093. [DOI] [PubMed] [Google Scholar]
- Golde TE, Schneider LS, Koo EH. Anti-abeta therapeutics in Alzheimer’s disease: the need for a paradigm shift. Neuron. 2011;6:203–213. doi: 10.1016/j.neuron.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinne JO, Brooks DJ, Rossor MN, Fox NC, Bullock R, Klunk WE, Mathis CA, Blennow K, Barakos J, Okello AA, Rodriguez Martinez de Liano S, Liu E, Koller M, Gregg KM, Schenk D, Black R, Grundman M. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010;6:363–372. doi: 10.1016/S1474-4422(10)70043-0. [DOI] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;6:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]