Nitrosation-dependent caveolin 1 phosphorylation, ubiquitination, and degradation and its association with idiopathic pulmonary arterial hypertension (original) (raw)
Abstract Abstract
In the present study, we tested the hypothesis that chronic inflammation and oxidative/nitrosative stress induce caveolin 1 (Cav-1) degradation, providing an underlying mechanism of endothelial cell activation/dysfunction and pulmonary vascular remodeling in patients with idiopathic pulmonary arterial hypertension (IPAH). We observed reduced Cav-1 protein despite increased Cav-1 messenger RNA expression and also endothelial nitric oxide synthase (eNOS) hyperphosphorylation in human pulmonary artery endothelial cells (PAECs) from patients with IPAH. In control human lung endothelial cell cultures, tumor necrosis factor α–induced nitric oxide (NO) production and S-nitrosation (SNO) of Cav-1 Cys-156 were associated with Src displacement and activation, Cav-1 Tyr-14 phosphorylation, and destabilization of Cav-1 oligomers within 5 minutes that could be blocked by eNOS or Src inhibition. Prolonged stimulation (72 hours) with NO donor DETANONOate reduced oligomerized and total Cav-1 levels by 40%–80%, similar to that observed in IPAH patient–derived PAECs. NO donor stimulation of endothelial cells for >72 hours, which was associated with sustained Src activation and Cav-1 phosphorylation, ubiquitination, and degradation, was blocked by NOS inhibitor L-NAME, Src inhibitor PP2, and proteosomal inhibitor MG132. Thus, chronic inflammation, sustained eNOS and Src signaling, and Cav-1 degradation may be important causal factors in the development of IPAH by promoting PAEC dysfunction/activation via sustained oxidative/nitrosative stress.
Keywords: Src, endothelial nitric oxide synthase (eNOS), endothelial dysfunction, oxidative stress, pulmonary arterial hypertension (PAH)
Introduction
Caveolin 1 (Cav-1) is a key regulator of endothelial cell function and vascular homeostasis.1-3 Through its association with membrane cholesterol, Cav-1 is essential for the formation of caveolae,4 the nonclathrin endocytic vesicles that mediate transcellular transport (transcytosis) of macromolecules.5 Cav-1 has also been shown to regulate numerous signaling pathways (e.g., endothelial nitric oxide synthase [eNOS], Ca2+, Rho)6 that control the integrity of junctional adhesion complexes,7 neoangionesis,8 and cellular proliferation.9 Evidence pointing to a loss of Cav-1 protein expression in the underlying mechanism of endothelial cell activation/dysfunction and the resultant cardiovascular disease phenotype has been steadily accumulating.10,11 For instance, we showed that Cav-1 knockout (Cav-1−/−) mice have poorly perfused and disorganized microvessels in the lungs and that this contributes to a moderate increase in pulmonary vascular resistance in aged mice.12 In addition, Cav-1 expression was shown to be significantly reduced in plexiform lesions in lung sections from patients with idiopathic pulmonary hypertension,13 although it remains unclear at present whether the loss of Cav-1 in endothelial cells induces hypertensive arteriopathy and formation of focal plexigenic vascular lesions.13 Interestingly, Austin et al.14 recently identified a genetic Cav-1 mutation in skin-punch biopsy fibroblast cultures from patients with pulmonary arterial hypertension (PAH) that was associated with reduced Cav-1 expression. However, the mechanisms responsible for changes in total Cav-1 protein levels in familial or idiopathic disease—that is, whether changes in expression are due to altered transcription, translation, trafficking, or protein turnover—have not been investigated.
In the plasma membrane of endothelial cells, Cav-1 regulates and is regulated by eNOS and Src signaling.15-17 Interactions between Cav-1, eNOS, and Src not only permit the endothelium to transiently produce NO (via eNOS activation) in response to vasodilator agonists but also prevent unfettered activation of eNOS and resultant oxidative stress via peroxynitrite production that can damage the vasculature.18 Maintenance of this delicate and somewhat fragile balance between NO output and Src activity by phospho-Cav-1-dependent feedback inhibition of eNOS and Src15-17 appears to be a critical mechanism involved in endothelial cell homeostasis. NO-mediated Src activation and phosphorylation of Cav-1 increases the affinity of Cav-1 for eNOS17 as well as the Src-inactivating kinase Csk,15 thereby promoting the sequestration and inactivation of eNOS and Src at the plasma membrane. This feedback regulatory mechanism, which appears to be critical for maintaining endothelial vasodilatory and homeostatic functions, led us to hypothesize that under conditions of oxidative stress as during chronic inflammation, chemical modification and targeted degradation of Cav-1 may underlie persistent vascular dysfunction and remodeling as observed in patients with idiopathic PAH (IPAH) and scleroderma.10,11,19-22
The findings of the present study indicate that prolonged exposure of endothelial cells to the inflammatory cytokine tumor necrosis factor α (TNF-α) or NO donor induces Cav-1 S-nitrosation (SNO) at Cys-156 within the C-terminus. Following nitrosation, Src, which normally binds to this residue,23 becomes displaced and activated, resulting in Src-dependent phosphorylation of Cav-1 N-terminal Tyr-14 and leading to destabilization of membrane-associated Cav-1 oligomers. We propose a model in which sustained Cav-1 nitrosation and phosphorylation increases its ubiquitination and degradation by the proteosome. Since both eNOS and Src inhibitors blocked oxidative stress–induced Cav-1 degradation, these findings suggest that aberrant Cav-1-regulated eNOS and Src signaling may play a significant role in initiating and sustaining endothelial cell activation/dysfunction associated with inflammatory vascular disease, in particular IPAH.
Material and methods
Cell culture and reagents
Human lung microvascular endothelial cells (HLMVECs) and mouse lung microvascular endothelial cells (MLMVECs)
HLMVECs were purchased from Lonza (Walkersville, MD), cultured on 0.2% gelatin–coated dishes, grown using the VascuLife VEGF-Mv Medium Complete kit (VascuLife basal medium [475 mL], VascuLife VEGF-Mv LifeFactors kit; LifeLine, Frederick, MD), and incubated at 37°C in a humidified atmosphere of 5% CO2/95% air. MLMVECs from wild-type (WT; B6/129SJ2) and Cav-1−/− mice (Jackson Laboratory, Bar Harbor, ME) were isolated as described elsewhere.24
Reagents
Reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless stated otherwise, including carbobenzoxy-L-leucyl-L-leucinal (MG132), 3-(4-chlorophenyl)-1-(1,1-dimethethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (PP2), and Nω-nitro-L-arginine methyl ester hydrochloride (L-NAME). The ubiquitin antibody used (P4D1) and the green fluorescent protein (GFP) antibody used (B-2) were from Santa Cruz Biotechnology (Dallas, TX). β-Actin, Cav-1, and p-Cav-1 Tyr-14 antibodies were purchased from BD Biosciences (San Jose, CA); p-Src Tyr418 and p-Src Tyr530 antibodies were purchased from Cell Signaling Technology (Danvers, MA); and antibody for Src (monoclonal) was purchased from Santa Cruz Biotechnology. DETANONOate ((Z)-1-[N-(2-aminoethyl)-N-(2ammonioethyl)amino]diazen-1-ium-1,2-diolate) was purchased from A.G. Scientific (San Diego, CA). _n_-Octyl-β-d-glucopyranoside (ODG) buffer was purchased from RPI (Mt. Prospect, IL); S-nitroso-N-acetyl-D,L-penicillamine (SNAP) was purchased from Caymen Chemicals (Ann Arbor, MI).
Human embryonic kidney (HEK) 293 cells
HEK cells were purchased from the American Type Culture Collection (Rockville, MD). They were cultured in Dulbecco’s modified eagle medium from Invitrogen (Grand Island, NY) supplemented with 10% fetal bovine serum from Serum Source International (Charlotte, NC) and 1% penicillin/streptomycin from Invitrogen.
Human pulmonary arterial endothelial cells (HPAECs; Cleveland Clinic)
HPAECs were obtained from human lung tissues from either unused explanted donor lungs or explanted lungs from confirmed subjects with PAH undergoing lung transplantation at the Cleveland Clinic, as described elsewhere.25 The study was approved by the Cleveland Clinic Institutional Review Board (IRB).
Human pulmonary microvascular endothelial cells (HPMVECs; Amsterdam)
HPMVECs were isolated from PAH patients (autopsies and lung transplantations) and controls (material from lung tumor surgeries) by mechanical homogenization and treatment with 0.3% collagenase type 2 (Sigma-Aldrich), as approved by the IRB and with consent. The tissue suspension was filtered through 70-μm nylon mesh and purified by CD31-labeled magnetic-activated cell sorting (Miltenyi Biotec, Bergisch Gladbach, Germany). Pure HPMVEC cultures were seeded on 1% gelatin–coated culture flasks and maintained in EGM-2 growth medium plus Bulletkit (Lonza). Passage numbers between four and six were used for experiments. HPMVECs were cultured in growth medium until confluence was reached and kept confluent for 72 hours to allow maturation of cell adhesions. Then the cells were put on ice, washed with ice-cold phosphate-buffered saline (PBS), and lysed with lysis buffer (Roche, San Francisco, CA) containing Triton X-100 and a protease/phosphatase-inhibitor cocktail. The lysates were centrifuged at 17,075 g, and supernatants were stored at −80°C.
Western blotting and immunoprecipitation
HPAECs, HLMVECs, and HEK cells were lysed on ice in lysis buffer containing 20 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 60 mM _n_-octylglucoside, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride, 1 mM sodium orthovanadate, and protease inhibitor cocktail. Insoluble materials were removed by centrifugation for 5 minutes at 16,100 g at 4°C, and lysates were boiled in lysis buffer with 6× Laemmli sample buffer (Boston BioProducts, Ashland, MA) and 30 mM dithiothreitol. Protein concentration was determined using the BCA Protein Assay kit (Pierce, Rockford, IL). Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) separation was performed, and proteins were blotted onto nitrocellulose membranes and blocked with 5% blotting-grade nonfat dry milk (Bio-Rad, Hercules, CA) in Tris-buffered saline (TBS) with 0.05% Tween 20 for 1 hour. Membranes were probed with primary antibodies overnight at 4°C, washed three times in TBS with Tween, and incubated with secondary species-specific horseradish peroxidase–conjugated antibodies for 1 hour at room temperature in blocking buffer. Membranes were washed three times, and proteins were visualized with enhanced chemiluminescence substrate (Pierce). For immunoprecipitation studies, cells were lysed in ODG buffer containing 50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 20 mmol/L NaF, 2% octyl-d-glucoside, 5% glycerol, 1 mM phenylmethanesulfonylfluoride, 1 mM sodium orthovanadate, and protease inhibitor. Resulting lysates were added to magnetic sheep anti–mouse immunoglobulin (Ig) G–coated Dynabeads (Invitrogen) that were preincubated for 30 min at 4°C with nonspecific mouse IgG or anti-Cav-1 monoclonal antibodies. Samples were rotated for 1 hour at 4°C, placed on a magnetic particle concentrator (Invitrogen), and washed with cold PBS without calcium/magnesium supplemented with protease-inhibitor cocktail and sodium orthovanadate. Samples were washed three times, 6× Laemmli sample buffer was added, and the samples were then either boiled for 5 minutes or loaded without boiling onto SDS-PAGE gels for Western blot analysis.
Immunostaining and confocal microscopy
Formalin-fixed, paraffin-embedded (FFPE) lung tissue sections were processed for fluorescence microscopy as follows. Lung tissues sectioned at 20-μm thickness were rehydrated and subjected to antigen retrieval in citrate buffer. After cooling, sections were postfixed in methanol, endogenous peroxidase activity was blocked, and antibody staining was performed by first blocking nonspecific binding with 5% donkey serum for 2 hours and then sections were incubated with primary antibodies diluted in blocking solution overnight at 4°C. After being washed in PBS with 0.05% Tween 20 PBS, sections were incubated with fluorophore-conjugated secondary antibodies raised in donkey (Jackson ImmunoResearch Laboratories, West Grove, PA). Primary antibodies were diluted as follows: mouse monoclonal vascular endothelial (VE)-cadherin, 1∶1,000 (BD Transduction Laboratories, San Diego, CA); platelet endothelial cell adhesion molecule 1 (PECAM-1) monoclonal antibody hec7, 1∶1,000 (gift of W. A. Muller, Northwestern University, Chicago, IL). Negative controls and appropriate isotype-matched control antibodies were included in all immunostaining experiments. Confluent HLMVECs grown on collagen-coated glass coverslips or MLMVECs purified from WT or Cav-1−/− mice grown on Matrigel-coated coverslips at 37°C. Cells were washed, fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and stained with the nuclear marker 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; 1 μg/mL). Nonconfocal DAPI images were acquired using Hg lamp excitation and a UV filter set. Confocal microscopy was performed using a 63× 1.2 N.A. objective on a Zeiss LSM 510 META microscope (Carl Zeiss MicroImaging, Oberkochen, Germany) with a 488-excitation laser line and pinhole set to achieve 1 Airy unit.
Generation of Cav-1 cysteine mutants and transfections
Cells were tansfected using Lipofectamine 2000 on the basis of manufacturer’s protocols.
Methods for pEGFP-N1-CAV1-WT plasmid subcloning
Full-length Homo sapiens Cav-1 complementary DNA (cDNA) was subcloned into pEGFP-N1 (C-terminal-tagged GFP) vector (Clontech, Mountain View, CA) via restriction sites 5′-_Nhe_I and 3′-_Kpn_I. Polymerase chain reaction (PCR) was performed with Phusion (Finnzymes, Espoo, Finland) to generate the full-length Cav-1 PCR fragment without the STOP codon using the primer pairs Cav1-NheI-F (5′-ACTA**GCTAGCGACCGCCATGTCTGGGGGCAAATAC-3′) and Cav1-KpnI-R (5′-ACTGGGTACC**GTTATTTCTTTCTGCAAGTTGATGCG-3′). The bold and italicized base pairs shown in the primers indicate the restriction sites. The resulting PCR fragment was digested with _Nhe_I and _Kpn_I (Invitrogen or New England Biolabs, Ipswich, MA) and ligated to pEGFP-N1, also digested with the same restriction enzymes, using T4 DNA Ligase (New England Biolabs). The ligated reaction was transformed into DH5α chemically competent cells (Invitrogen), and the transformed cells were plated onto Luria-Bertani (LB) plates supplemented with 25 mg/mL kanamycin. After overnight incubation at 37°C, single colonies were cultured in LB with kanamycin overnight. Plasmid DNA was obtained using the PureLink Quick Plasmid Mini Prep kit (Invitrogen) and analyzed by restriction digest to confirm the presence of Cav-1 cDNA. Further amplification of plasmid DNA was done using the NucleoBond Xtra Midi kit (Clontech). The resulting plasmid was verified for DNA purity and accuracy by DNA quantification, gel analysis, and sequencing analysis.
Double or multiple amino acid mutation Cav-1 plasmid subcloning
Several steps of PCR were used to create the multiple mutations using the single amino acid–mutated plasmid as the starting cDNA template. Several fragments of PCR were generated using primer sets that targeted other amino acid mutations. Subsequent steps of digestion and ligation, transformation, colony screening, and plasmid DNA isolation and amplification were performed as described. The resulting plasmid was verified for DNA purity and accuracy by DNA quantitation, gel analysis, and sequencing analysis. The PCR thermocycling conditions were as follows: initial denaturation at 98°C for 30 seconds; 30 cycles of denaturation, annealing, and extension at 98°C for 10 seconds, 67°C or melting temperature plus 3° of the lowest primer in a primer pair for 30 seconds, and 72°C for 15 seconds; and final extension at 72°C for 10 minutes followed by holding at 4°C.
H. sapiens Cav-1 primer sequences
Bold and italicized base pairs indicate the sequence of restriction enzymes, while the underlined base pairs indicate the amino acid being mutated. Primers and sequences were as follows: for CAV1-WT, Cav1-NheI-F (5′-ACTA**GCTAGCGACCGCCATGTCTGGGGGCAAATAC-3′) and Cav1-KpnI-R (5′-ACTGGGTACC**GTTATTTCTTTCTGCAAGTTGATGCG-3′); for CAV1-C133S, Cav1-C133S-F (5′-GTTGTACCATCCATTAAGAGC-3′) and Cav1-C133S-R (5′-GCTCTTAATGGATGGTACAAC-3′); for CAV1-C143S, Cav1-C143S-F (5′-GAGATTCAGTCCACCAGCCGTGTC-3′) and Cav1-C143S-R (5′-GACACGGCTGGTGGACTGAATCTC-3′); for CAV1-C156S, Cav1-C156S-F (5′-TACGTCCACACCGTCTCTGACCCACTC-3′) and Cav1-C156S-R (5′-TTCAAAGAGTGGGTCAGAGACGGTGTG-3′); and for CAV1-C133/143/156S, pEGFP-N1-CAV1-C143S was used as template, and C133S then C156S primers were used in PCR.
Reverse-transcription (RT) PCR
Total RNA from endothelial cells were isolated using TRIzol reagent. RT was performed using oligo(dT) primers and superscript RT (Invitrogen) following the manufacturer’s instructions. Human p38 isoforms and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were amplified using the following primer sets: p38 (sense, 5′-GATCAGTTGAAGCTCATTTTAA-3′; antisense, 5′-CACTTGAATAATATTTGGAGAGT-3′); p38 (sense, 5′-AGCCATATCTGGCAAGAAGCTGGA-3′; antisense, 5′-AAGTGTCCGAGTCCAAGTCCACAT-3′); p38 (sense, 5′-TTGAATTGGATGCGCTACACGCAG-3′; antisense, 5′-AGGGCTTGCATTGGTCAGGATAGA-3′); p38 (sense, 5′-TGTGCAGAAGCTGAACGACAAAGC-3′; antisense, 5′-TGCCATGCAAGATGAGTCCCTACA-3′); and GAPDH (sense, 5′-TATCGTGGAAGGACTCATGACC-3′; antisense, 5′-TACATGGCAACTGTGAGGGG-3′). RT product (2 L) was amplified in a 20-L volume containing 100 pmol of primers and 2.5 U of TaqDNA polymerase. To quantify fold change in Cav-1 expression TaqMan probes, GAPDH (Hs02758991_g1) and Cav-1 (Hs00971716_m1) from Invitrogen were used, and the analysis was run on Applied Biosystems 7500 FAST (Invitrogen) according to the manufacturer’s instructions. We looked for fold change in hypertension/control. Cav-1 expression levels were normalized to GAPDH expression in each sample. The graph represents fold change in expression in hypertension compared with control samples.
Biotin switch assay
S-nitrosylated protein was detected by the biotin switch method as described elsewhere26 using the Caymen Chemical S-Nitrosylated Protein Detection kit. Briefly, endothelial cells were treated with DETANONOate for the indicated time. Samples were immunoprecipitated using streptavidin-conjugated antibodies (Dynabeads M-280 streptavidin from Invitrogen). Biotinylated Cav-1 was resolved by nonreducing SDS-PAGE, transferred, and detected by immunoblotting with mouse anti-Cav-1 (1∶1,000). Analysis of SNO-Cav-1 by biotin switch assay was performed as described elsewhere.26 Samples were immunoprecipitated using streptavidin-conjugated antibodies (Dynabeads M-280 streptavidin from Invitrogen). Biotinylated Cav-1 was resolved by nonreducing SDS-PAGE, transferred, and detected by immunoblotting with mouse anti-Cav-1 (1∶1,000).
Mass spectrometry
Mass spectrometry analysis was performed by the Chicago Biomedical Consortium/University of Illinois at Chicago Research Resources Center Proteomics and Informatics Services Facility. The in-gel tryptic digestion was carried out by following the protocol described by Kinter and Sherman.27 Briefly, Coomassie-stained gel bands were cut into 1-mm3 pieces, rinsed, and dehydrated with acetonitrile, and the protein was reduced with dithiothreitol (DTT) and alkylated with iodoacetamide in the dark prior to overnight digestion with Promega Gold trypsin at 37°C in 100 mM ammonium bicarbonate. The peptides were extracted with ammonium bicarbonate and acetonitrile, concentrated, and analyzed using a Thermo LTQ-FT Ultra mass spectrometer equipped with a Dionex 3000 nanoflow high-performance liquid chromatography system controlling a reverse-phase column (Agilent Zorbax 300SB-C18, 3.5 μm, 75 μm × 150 mm) at a flow rate of 250 nL/min. The peptides were separated and eluted with a linear gradient of 10%–60% solution B (95% acetonitrile, 0.1% formic acid) for 60 minutes.
Transmission electron microscopy
For ultrastructural analysis, cells grown on Matrigel-coated plastic dishes were fixed with Karnovsky fixative, stained with OsO4, dehydrated, and embedded in Epon. Ultrathin sections were stained with uranyl acetate and lead citrate and examined under a transmission electron microscope.
Densitometry and statistics
Densitometry of protein bands was performed with ImageJ software (http://rsbweb.nih.gov/ij/). Comparison of two groups was conducted using Student’s t test, and three or more groups were compared with analysis of variance with Bonferroni post hoc testing to correct for multiple testing and to keep the family-wise error rate below 0.05. Experimental data are presented as mean ± standard error of the mean (SEM). All tests were performed two-sided and nonblinded and were executed with GraphPad Prism software for Mac (ver. 6; GraphPad Software, La Jolla, CA). A P value of <0.05 was considered statistically significant.
Results
Reduced Cav-1 protein but not mRNA expression in IPAH endothelial cells
Consistent with previous studies and various animal models of PAH,14,28,29 PAECs isolated from excised human lung tissue from patients with IPAH (n = 4)25,30 exhibited reduced oligomerized and total Cav-1 protein levels (Fig. 1_A_, 1_B_) but increased Cav-1 mRNA expression (Fig. 1_C_), suggesting that Cav-1 degradation is faster than its synthesis in the PAH endothelia. Consistent with the known role of Cav-1 in the mechanism of eNOS inactivation,31 reduction in expression and oligomerization of Cav-1 paralleled an increase in eNOS phosphorylation at active site Ser 1177 despite a slight reduction in the total level of eNOS protein (Fig. 1_A_). That Cav-1 mRNA expression was upregulated twofold in IPAH PAECs suggests that there is increased turnover of Cav-1 protein in IPAH patient–derived PAECs. Furthermore, we observed reduced Cav-1 expression in tissue sections (deidentified FFPE sections of cadaver lung tissue diagnosed with PAH; n = 6) compared with that in healthy donor lung tissue (n = 6). Immunostaining and confocal microscopy revealed reduced Cav-1 expression in PECAM-1-positive endothelial cells in pulmonary arterioles from patients with IPAH (Fig. 1_D_). While the observed decrease in total Cav-1 protein expression in IPAH PAECs was not found in isolated and cultured microvascular endothelial cells from excised lungs diagnosed with PAH, there was a trend toward reduced oligomerization (Fig. S1).
Figure 1.

Reduced caveolin 1 (Cav-1) oligomer and monomer protein level in pulmonary artery endothelial cells (PAECs) from patients with idiopathic pulmonary arterial hypertension (IPAH). A, Representative Western blot of PAEC lysates obtained from a healthy donor (left lane) and a patient with IPAH (right lane). The lower panel reveals an increase in phospho-Ser-1177 endothelial nitric oxide synthase (p-eNOS) relative to total eNOS expression in IPAH endothelial cells, indicative of eNOS hyperactivation/dysfunction. B, Summarized data. Asterisk indicates P < 0.05 versus control; n = 5. C, Quantitative polymerase chain reaction indicates increased Cav-1 messenger RNA (mRNA) in IPAH PAECs compared with control. Asterisk indicates P < 0.05; n = 3. D, Immunohistochemical staining and confocal microscopy of Cav-1 (green) and platelet endothelial cell adhesion molecule 1 (PECAM-1; red) in formalin-fixed, paraffin-embedded control and PAH lung sections. Note the significant colocalization of Cav-1 and PECAM-1 in control lung pulmonary arteriole and reduced Cav-1 staining in the endothelial cells of the remodeled pulmonary arteriole of the PAH lung section. Staining is representative of 6 PAH and control lung samples. Mr: relative molecular mass; DAPI: 4′,6-diamidino-2-phenylindole dihydrochloride.
Inflammatory mediator TNF-α induces eNOS-dependent Cav-1 SNO
In view of findings shown in Figure 1, we hypothesized that inflammation and oxidative stress that accompanies PAH19 may promote chemical modification and degradation of Cav-1, thus altering its ability to negatively regulate eNOS signaling in the endothelium. Consistent with this assumption, we observed that brief challenge with TNF-α, a powerful proinflammatory cytokine known to activate eNOS,32 significantly increased SNO of Cav-1 (Fig. 2_A_). The dependence of Cav-1 SNO on NOS activity was shown by its abrogation in cells pretreated with the NOS inhibitor L-NAME (Fig. 2_A_). To pinpoint the SNO to a particular thiol residue, each of the critical thiol residues implicated in the regulation of Cav-1 targeting to the plasma membrane (C-133, C-143, and C-156) was mutated to serine (Fig. 2_B_). The mutants were then transfected into HEK cells overexpressing eNOS (HEK-eNOS cells).14 In addition, we prepared a triple mutant–encoding construct. Analysis of each of the transfected lines revealed that the C156S Cav-1 mutation was sufficient to abolish Cav-1 SNO, whereas the C133S and C143S mutants showed only a modest reduction in SNO once normalized to total Cav-1 expression level (Fig. 2_C_).
Figure 2.

Tumor necrosis factor α (TNF-α) induces caveolin 1 (Cav-1) S-nitrosation on Cys-156. A, Biotin switch assay indicates TNF-α-dependent, L-NAME-inhibitable nitrosation of Cav-1. TNF-α (2–20 ng/mL) was added to human lung microvascular endothelial cells (HLMVECs) 5 minutes before lysis. L-NAME was added 30 minutes prior to TNF-α. B, Biotin switch assay performed on human embryonic kidney cells overexpressing endothelial nitric oxide synthase that were expressing wild type (WT)–Cav-1, C133S-Cav-1, C143S-Cav-1, and C156S-Cav-1 mutants in pEGFP vector. C133S-, C143S-, C156S, and triple (C133S/C143S/C156S)–Cav-1 mutants in pEGFP vectors were made. C, Nitrosation was promoted by adding S-nitroso-N-acetyl-D,L-penicillamine (SNAP; 20 μM) at room temperature for 5 minutes. After lysis and the biotin switch, samples were immunoprecipitated with Cav-1 and blotted for streptavidin. Right panels show quantified densitometry of Western blots using ImageJ software. Bars show mean ± standard deviation. Asterisks indicate P < 0.05 versus control (A) or WT (C); n = 3. GFP: green fluorescent protein.
SNO of Cav-1 promotes Src dissociation and activation
We next interrogated whether the nitrosation of Cav-1 on Cys-156 regulates the interaction/binding and activity Src, which is known to phosphorylate and affect Cav-1-regulated signaling.15-17 As shown in Figure 3_A_, treatment of HEK cells expressing WT-Cav-1-GFP with SNAP significantly reduced Src/Cav-1 coimmunoprecipitation. Furthermore, TNF-α-induced Src activation was shown to be NOS dependent (Fig. 3_B_). The ratio of Src active-site phospho-Tyr416 to inactive-site phospho-Tyr527, which predicts Src activity in human lung endothelial cells,12,33 increased in cells challenged with TNF-α alone and was blocked in cells pretreated with the NOS inhibitor L-NAME (Fig. 3_B_). Indeed, we found, concurrently with the demonstration that Cys-156 is required for Cav-1/Src interaction, that mutation of Cys-156 to serine (C156S) was sufficient to abolish Cav-1/Src interaction (Fig. 3_C_). A diminishment in the association of Cav-1 and Src produced by nitrosation—or as shown in Figure 3_D_ by expression of the Cav-1 C156S mutant—led to increased Src activity (Src-pTyr416) and Cav-1 phosphorylation at Tyr-14.
Figure 3.

Nitric oxide (NO) donor and caveolin 1 (Cav-1)–C156S mutant decrease Cav-1/Src interaction and increase Src activity. A, Human embryonic kidney (HEK) cells were transfected with wild type (WT)–Cav-1–green fluorescent protein (GFP) and stimulated with either NO donor or S-nitroso-N-acetyl-D,L-penicillamine (SNAP; 20 μM, 5 minutes) or left untreated. Cells were lysed, Src was immunoprecipitated, and bound Cav-1 was quantified by Western blot. Asterisks indicate P < 0.05 versus control; n = 3. B, Analysis of active/inactive Src as indicated. pTyr419/pTyr530 Western blots from tumor necrosis factor α (TNF-α)–stimulated human lung microvascular endothelial cells with or without NO synthase (NOS) inhibitor pretreatment indicates that L-NAME blocks TNF-α-induced Src activation. Asterisks indicate P < 0.05 versus TNF-α alone; n = 3. C, HEK cells overexpressing endothelial NOS (eNOS; HEK-eNOS cells) were transfected with empty vector (EV), WT-Cav-1, and C156S-Cav-1 mutant in pEGFP vector. Samples were blotted for total Cav-1 (upper row) or immunoprecipitated with anti-Src monoclonal antibody and blotted for Cav-1 and Src (lower rows). D, HEK-eNOS cells were transfected with WT-Cav-1 and C156S-Cav-1 mutant and blotted for Src-pTyr418, total Src, Cav-1-pTyr14, and total Cav-1. Asterisks indicate P < 0.05 versus WT-Cav-1; n = 3.
Cav-1 SNO is accompanied by destabilization of Cav-1 oligomers
To form caveolae and regulate signaling via specific scaffolding interactions with a multitude of target proteins known to interact with Cav-1,1 Cav-1 organization as a multimeric homotypic structure is thought to be essential.34 As shown in Figure 4, TNF-α, possibly via Cav-1 SNO, destabilized and disrupted Cav-1 oligomers in an L-NAME-sensitive manner, indicating that eNOS activation and subsequent Cav-1 SNO may promote Cav-1 oligomer destabilization and accumulation of Cav-1 monomers. Furthermore, in addition to NO signaling, Src activity also appears to participate in the destabilization of Cav-1 oligomers since PP2, a specific inhibitor of Src, blocked the effects of TNF-α to an extent comparable to that observed in the presence of L-NAME (Fig. 4_A_). Again, modification of the Cys-156 residue appears to mediate this effect since the C156S mutation (but not C133S or C143S) mimicked the effect of SNO (i.e., decreased oligomerization) as shown in Figure 4_B_. Taken together, these observations indicate that Cav-1 SNO followed by Src-dependent phosphorylation of Cav-1 Tyr-14 induces the destabilization of Cav-1 oligomers, resulting in the accumulation of Cav-1 in its monomeric form. It also implies that modifications to Cys-156 may disrupt the oligomeric structure and function of Cav-1 polymers in the plasma membrane.
Figure 4.

Caveolin 1 (Cav-1) oligomerization is reduced by tumor necrosis factor α (TNF-α) in a nitric oxide (NO)– and Src-dependent manner. A, Human lung microvascular endothelial cells were pretreated with either nitric oxide synthase inhibitor L-NAME or Src inhibitor PP2 30 minutes prior to being challenged with TNF-α for 5 minutes. Cav-1 oligomer/monomer distribution was analyzed by Western blot of nonboiled samples. Quantification of densitometry using ImageJ (mean ± SD; asterisk indicates P < 0.05 versus untreated; n = 3). B, Western blot analysis of Cav-1 expressed in human embryonic kidney cells (EV = empty green fluorescent protein [GFP] vector, wild type [WT]–, C133S-, C143S-, C156S-, and triple [C133S/C143S/C156S]–Cav-1-GFP mutants) showed that Cys-156 is required to maintain oligomer stability. Bar graphs show quantified densitometry of Cav-1 oligomer/monomer. Asterisks indicate P < 0.05 versus WT-Cav-1; pound signs indicate P < 0.05 versus C133S- and C143S-Cav-1; n = 3.
Differential effects of short- and long-term exposure to NO on Cav-1 oligomer stability
As shown in Figure 5_A_, brief (minutes) and prolonged (days) exposure to the NO donor SNAP dramatically reduced the structural integrity of Cav-1 oligomers and the overall stability of the protein, respectively. Short-term NO exposure rapidly (within 5–10 minutes) reduced Cav-1 oligomerization, which was followed, after 72 hours of continuous NO challenge, by an overall reduction in total Cav-1 protein level (monomers and oligomers). This observation suggests that Cav-1 and its ability to interact with Src and eNOS may be regulated by NO. It also indicates that prolonged exposure to NO may alter Cav-1-regulated signaling by provoking its degradation, consistent with reports of derailed Cav-1/eNOS interactions in conditions characterized by chronic inflammatory signaling and oxidative stress.35-37
Figure 5.

Nitric oxide (NO) donors induce Src-dependent caveolin 1 (Cav-1) oligomer destabilization and Cav-1 degradation. A, Human lung microvascular endothelial cells (HLMVECs) treated with S-nitroso-N-acetyl-D,L-penicillamine (SNAP; 10 μM) showed reduced oligomerization at 5 minutes (0.08 hours) and a decrease in total Cav-1 expression at 72 hours. B, HLMVECs either untreated or pretreated with the Src inhibitor PP2 (10 μM) or the proteosome inhibitor MG132 (20 μm) were challenged every 24 hours with NO donor DETANONOate (250 μM) for 24, 48, or 72 hours. Nonboiled samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and blotted for Cav-1 and actin. C, D, Quantified densitometry of Western blots reveal a significant decrease in the Cav-1 oligomer/monomer ratio (C) and a significant decrease in total Cav-1 (monomer + oligomer; D) after 72 hours of DETANONOate, which is blocked by PP2 and MG132. Asterisks indicate P < 0.05 versus untreated by analysis of variance; n = 3. E, Ubiquitination of immunoprecipitated Cav-1 from HLMVEC lysates after challenge with NO donor DETANONOate (250 μM) in the presence or absence of PP2 (10 μM; left panels) and reprobe of membrane with anti-Cav-1 antibody (right panels). Immunoblots show that ubiquitination (left panel) and degradation (right panel) of Cav-1 (oligomers and monomers) following treatment with DETANONOate is blocked by PP2 (representative of 3 experiments).
NO-dependent loss of Cav-1 protein was mitigated by MG132 (Fig. 5_B_), a proteasome inhibitor,38 suggesting that the decrease in Cav-1 expression as a consequence of persistent NO challenge occurs via proteolytic degradation. Importantly, PP2, a specific Src inhibitor, also inhibited NO-stimulated Cav-1 proteolytic degradation (Fig. 5_B_, middle panel), thus implicating sustained Cav-1 Tyr-14 phosphorylation by active Src tyrosine kinase in the mechanism of Cav-1 degradation. This evidence suggests that Src displacement from Cav-1 on nitrosation of Cys-156 may be upstream of Cav-1 phosphorylation by Src and that, in tandem, SNO and phosphorylation decrease Cav-1 oligomerization (Fig. 5_C_) and promote Cav-1 degradation (Fig. 5_D_). In agreement with the possibility that Cav-1 SNO followed by Src-dependent phosphorylation targets Cav-1 for proteasomal degradation, we found that NO, particularly after 48- and 72-hour exposures, markedly increased Cav-1 ubiquitination (Fig. 5_E_), the characteristic signature of proteasomal targeting. PP2 strongly inhibited Cav-1 ubiquitination, providing evidence for the involvement of Src family tyrosine kinase activity in NO-mediated degradation of Cav-1 (Fig. 5_E_). Further evidence for the ubiquitination of Cav-1 is provided in Figure S2. Using mass spectrometry–assisted peptide mapping, we identified the residue that is ubiquitinated following NO exposure as Lys-86. Taken together, experimental evidence demonstrates that short- and long-term exposure to NO may have markedly distinct effects on Cav-1 stability and regulated signaling. Sustained Cav-1 SNO was associated with Src-dependent phosphorylation of Tyr-14, ubiquitination of Lys-86, and proteosomal degradation.
Functional implications of Cav-1 degradation
Confocal imaging experiments showed that WT-Cav-1-GFP transfection into Cav-1−/− mouse lung endothelial cells was associated with formation of punctate vesicle-like structures that decorated the membrane and cytoplasmic compartments (Fig. 6_A_, left panel). However, expressed C156S-Cav-1-GFP accumulated as large aggregates in the perinuclear region (Fig. 6_A_, right panel) with reduced localization in the plasma membrane where Cav-1 is known to assemble into functional oligomers and promote the formation of caveolae. In parallel, electron microscopy analysis of Cav-1−/− endothelial cells confirmed the requirement of Cav-1 for formation of caveolae (Fig. 6_B_, 6_C_). Restoration of Cav-1 expression by transfection of WT-Cav-1 cDNA rescued caveolae formation, as demonstrated in Figure 6_B_ (left panel). In clear contrast, transfection of C156S-Cav-1 mutant cDNA was unable to rescue caveolae formation in Cav-1−/− endothelial cells (Fig. 6_B_, 6_C_). Thus, Cav-1 Cys-156 is a requirement for stability as an oligomer and caveolae formation. Consistent with these findings, WT-Cav-1-GFP expressed in HEK cells was detectable primarily in its high-molecular-weight oligomeric form in nonboiled SDS-PAGE, whereas C156S Cav-1-GFP was detected as a monomeric protein (Fig. 6_D_).
Figure 6.

Confocal and transmission electron microscopy reveals that the C156S caveolin 1 (Cav-1) mutant fails to rescue caveolae formation in Cav-1−/− mouse lung endothelial cells (MLECs). A, Cav-1−/− MLECs transfected with wild type (WT)–Cav-1–green fluorescent protein (GFP) complementary DNA (cDNA) restored membrane-associated Cav-1 localization, whereas the C156S-Cav-1-GFP mutant accumulated in perinuclear intracellular compartments. B, Representative transmission electron microscopy micrographs show rescue of caveolae in Cav-1−/− endothelial cells following transduction of WT-Cav-1, whereas no caveolae were observed in Cav1−/− MLECs transduced with C156S-Cav-1 cDNA. C, Quantitative analysis of electron micrographs indicate near-complete rescue of caveolae formation by WT-Cav-1 compared with WT control and Cav-1−/− mouse lung endothelial cells and lack of caveolae formation in cells transduced with C156S-Cav-1. Asterisks indicate P < 0.05 versus WT control MLECs by analysis of variance; n = 10 sections per group. D, Western blot analysis of WT-Cav-1-GFP, C156S-Cav-1-GFP, and empty GFP vector (EV) expression in human embryonic kidney cells. Note the absence of stable high-molecular-weight oligomers in C156S-Cav-1-transfected cells.
Discussion
Cav-1 expression is essential for caveolae formation5,39 and the endocytosis and transcytosis of macromolecular cargo (e.g., albumin,40-43 insulin,44 myeloperoxidase,40 and cholesterol45) into and through the vascular endothelium.46 In addition, studies during the last 15 years have unveiled critical functions of Cav-1 as a membrane-associated scaffolding protein involved in the regulation of receptors, ion channels, and signaling enzymes.15-17,47-55 In the current investigation, we describe a novel posttranslational modification, SNO of Cav-1 Cys-156. When sustained by chronic inflammatory cytokine signaling (e.g., TNF-α) or direct NO stimulation, Cav-1 SNO leads to Src-dependent Tyr-14 phosphorylation, Lys-86 ubiquitination, and Cav-1 degradation via the proteosomal pathway. We propose, on the basis of these findings, that chronic vascular inflammation induces Cav-1 degradation via ubiquitination and proteolysis through the proteosomal pathway. These findings are consistent with reduced Cav-1 expression and endothelial cell activation/dysfunction in group 1 idiopathic pulmonary artery hypertension (IPAH) patients characterized by hypertensive arteriopathy and plexiform lesions.13,56 The mechanism by which Cav-1 degradation may promote the plexigenic endothelial cell phenotype is currently under investigation.
First, as noted in PAECs harvested from the excised lungs of IPAH patients undergoing transplant surgery,57 Cav-1 levels were significantly reduced and there was a concurrent increase in eNOS phosphorylation at active site Ser-1177, indicative of its activation. We noted a twofold increase in Cav-1 mRNA, suggesting that the decrease in Cav-1 expression in IPAH PAECs occurs at the protein rather than the transcriptional level. Interestingly, we did not observe a statistically significant reduction in Cav-1 protein level or oligomerization in human PAH MVECs. However, lung sections from patients with PAH showed a significant reduction in Cav-1 immunostaining of PECAM-1-positive cells associated with vessels <100 μm in diameter, suggesting that at least endothelial cells of the smaller muscularized microvessels may be similarly affected by oxidative stress. Further study of patient-derived endothelial cells from different vascular beds in association with tissue immunohistochemisry is required to better understand whether inflammation and/or gradated oxidative stress differentially affects macro- versus microvascular endothelial cell Cav-1 expression.
In normal human lung endothelial cell cultures, we demonstrated that the proinflammatory cytokine TNF-α induces Cav-1 nitrosation within minutes, an event that we surmised links eNOS hyperactivation to the sustained proteolytic loss of Cav-1 in chronic inflammatory states. We observed in experiments that employed the NOS inhibitor L-NAME that TNF-α-induced nitrosation of Cav-1 was eNOS dependent. Additionally, by generating single-residue mutations to C-terminal Cys residues, experiments pinpointed Cys-156 as the primary residue in Cav-1 modified by NO. The significance of this finding with regard to Cav-1 stability and signaling was then investigated. We showed that Cav-1 nitrosation or mutation of Cys-156 to Ser (C156S) decreased Cav-1 association with Src tyrosine kinase, indicating that Cav-1 nitrosation is sufficient to activate Src by releasing it from its inhibitory association with the Cav-1 C-terminus. Src displacement from Cav-1 was also shown to promote activation (as demonstrated by Western blot studies shown in Fig. 3_B_) and resultant phosphorylation of Cav-1 Tyr-14 (Fig. 3_D_).
Interestingly, we observed that Cav-1 nitrosation and phosphorylation was associated with its accumulation in monomeric form (i.e., 22 kDa) in nonreduced SDS gels. As a structural protein, Cav-1 is thought to function as a polymeric chain of oligomers.58 In conjunction with the cavin family of proteins, Cav-1 is stabilized in cholesterol-enriched membrane microdomains and therein is thought to account for the flask-shaped curvature of plasmalemmal vesicles, or caveolae.59,60 Destabilization, disassembly, and/or dissociation of Cav-1 polymers was suggested to regulate membrane curvature and invagination61 and—if sustained, as suggested by the current study—disruption/dissolution of caveolae due to Cav-1 degradation. The mechanism of Cav-1 nitrosation and phosphorylation-dependent destabilization of oligomers is unclear. Phosphorylation of Cav-1 has been suggested to introduce significant charge repulsion within the polymer due to negative charges in closely apposed N-terminal phospho-Tyr-14 residues within the oligomeric clusters of Cav-1.61 As shown in the current study (Figs. 4, 5), both L-NAME (NOS inhibitor) and PP2 (Src inhibitor) blocked TNF-α-induced Cav-1 monomerization and degradation. Therefore, NO and downstream Src signaling–induced destabilization and degradation of Cav-1 may be an important determinant of the structural and functional integrity of caveolae as well as Cav-1 regulation of endothelial cell signaling.
Our studies demonstrate that NO donors are sufficient to promote Cav-1 monomerization. Inhibition of Src activity downstream of NO prevented Cav-1 oligomer destabilization and pretreatment of cells with the proteasome inhibitor MG132 blocked NO-dependent Cav-1 degradation. Taken together, these observations establish a sequence of events that connect nitrosative stress to Cav-1 degradation. According to our findings, Cav-1 Cys-156 nitrosation is associated with Src displacement and activation. Src phosphorylation of Cav-1, if sustained, is associated with degradation of Cav-1. Consistently, we detected Cav-1 ubiquitination in samples exposed to the NO donor DETANONOate and observed that PP2 could block Cav-1 ubiquitination and degradation. Importantly, mass spectrometry indicated Lys-86 within the caveolin scaffolding domain (which is known to be important for eNOS binding)62 as a site of Cav-1 ubiquitination; other Lys residues in Cav-1 have also been shown to be ubiquitinated.63,64 In the absence of Cav-1-mediated feedback regulation of eNOS activity, eNOS has been shown to be hyperphosphorylated on Ser-1179 (see Fig. 1_B_). Hyperacivation of eNOS leads to its uncoupling,21 thereby promoting peroxynitrite production in lieu of NO,18 which we hypothesize promotes persistent activation and dysfunction of the vascular endothelium.
Cav-1-null endothelial cells harvested from the lungs of Cav-1−/− mice lack caveolae.24,65 Rescue experiments in which the Cav-1 C156S mutant was reintroduced failed to restore caveolae formation, whereas WT-Cav-1 cDNA was sufficient to induce formation of caveolae (Fig. 6_B_). This result is not surprising in view of our finding that Cav-1 C156S is unable to form stable oligomeric structural units (Figs. 3_B_, 6_D_).
In summary, our studies demonstrate that Cav-1 degradation occurs following sustained NO production, as with chronic inflammatory stimuli. Decreased Cav-1 expression results in uncontrolled activation of at least two key endothelial cell-signaling proteins: eNOS and Src, whose persistent activation may perpetuate Cav-1 proteolytic degradation by promoting Cav-1 oligomer destabilization and ubiquitination. As observed in IPAH lung sections and PAECs, Cav-1 expression is reduced and, in parallel, eNOS is hyperactivated. Further study of the mechanisms regulating Cav-1 expression level and the consequences of Cav-1 degradation in endothelial cells due to chronic vascular inflammation on altered endothelial cell function and vascular arteriopathy/remodeling, as noted in patients with IPAH, type 2 diabetes, COPD, and so on, is warranted.
Acknowledgments
We express our sincere appreciation to Maricela Castellon, Bob Lee, PhD, and Larry Helseth, PhD, in the Research Resources Center (RRC) Mass Spectrometry, Metabolomics, and Proteomic Facility; Linda Juarez, PhD, in the RRC Electron Microscopy Core; and Vasily Shinin, MD/PhD, for excellent technical assistance and histological analysis. This article is based on a thesis submitted in partial fulfillment of the requirements for a doctoral degree (Bakhshi FR. Mechanism of caveolin-1 degradation. 2013. PhD thesis, Department of Pharmacology, University of Illinois, Chicago).
Supplementary figures.
Figure S1.
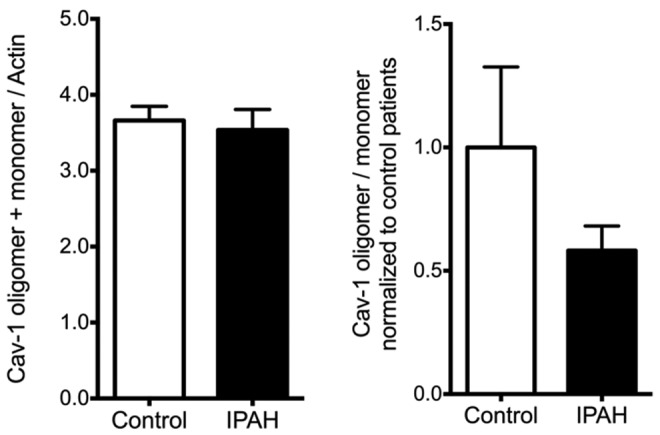
Human pulmonary microvascular endothelial cell (HPMVEC) lysates showed no change in total caveolin 1 (Cav-1) protein expression, but there was a trend toward reduced oligomerization in pulmonary arterial hypertension (PAH) samples. IPAH: idiopathic PAH.
Figure S2.
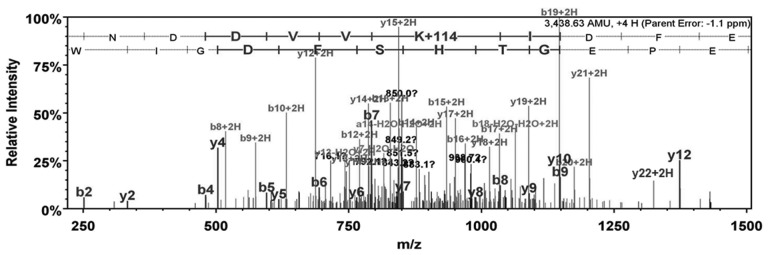
Three unique peptides from caveolin 1 (Cav-1) were identified by liquid chromatography tandem mass spectrometry (MS/MS) analysis of the 50-kDa Cav-1–green fluorescent protein (GFP) agarose gel band. The MS/MS spectrum of an ion at mass 860.67 (4+) was matched by Mascot software to Cav-1 tryptic peptide 66–86 IDFEDVIAEPEGTHSFDGIWK containing one ubiquitin group. The measured mass for this peptide was 3,438.63 Da, which is −1.8 ppm different from the theoretical value. The site of ubiquitination modification is indicated by the fragment ion at mz 1,324.1, which corresponds to the y(22+2H) ion with a modified lysine. The lysine is residue 86 in the Cav-1 sequence.
Source of Support: Funding was provided by National Heart, Lung, and Blood Institute grants R01HL71626 and P01HL60678 (RDM), 5T32HL007829 and 5T32HL072742 (training grant support of FRB), and R01HL60917 and P01HL103453 (SE, SC); by Dutch Heart Foundation grants CVON2011-T072 (GPvNA) and CVON2012-08; and by Longfonds grant 3.3.12.036 (GPvNA and HJB).
Conflict of Interest: None declared.
References
- 1.Maniatis N, Chernaya O, Shinin V, Minshall R. Caveolins and lung function. Adv Exp Med Biol 2012;729:157–179. [DOI] [PMC free article] [PubMed]
- 2.Stan R-V. Structure and function of endothelial caveolae. Microsc Res Tech 2002;57:350–364. [DOI] [PubMed]
- 3.Minshall RD, Sessa WC, Stan RV, Anderson RGW, Malik AB. Caveolin regulation of endothelial function. Am J Physiol Lung Cell Mol Physiol 2003;285:L1179–L1183. [DOI] [PubMed]
- 4.Stan RV. Structure of caveolae. Biochim Biophys Acta 2005;1746:334–348. [DOI] [PubMed]
- 5.Rothberg KG, Heuser JE, Donzell WC, Ying Y-S, Glenney JR, Anderson RGW. Caveolin, a protein component of caveolae membrane coats. Cell 1992;68:673–682. [DOI] [PubMed]
- 6.Schlegel A, Volonté D, Engelman JA, et al. Crowded little caves: structure and function of caveolae. Cell Sig 1998;10:457–463. [DOI] [PubMed]
- 7.Kronstein R, Seebach J, Großklaus S, et al. Caveolin-1 opens endothelial cell junctions by targeting catenins. Cardiovasc Res 2012;93:130–140. [DOI] [PubMed]
- 8.Schwencke C, Braun-Dullaeus RC, Wunderlich C, Strasser RH. Caveolae and caveolin in transmembrane signaling: implications for human disease. Cardiovasc Res 2006;70:42–49. [DOI] [PubMed]
- 9.Koleske AJ, Baltimore D, Lisanti MP. Reduction of caveolin and caveolae in oncogenically transformed cells. Proc Natl Acad Sci USA 1995;92:1381–1385. [DOI] [PMC free article] [PubMed]
- 10.Machado RF, Londhe Nerkar MV, Dweik RA, et al. Nitric oxide and pulmonary arterial pressures in pulmonary hypertension. Free Radic Biol Medi 2004;37:1010–1017. [DOI] [PubMed]
- 11.Xu W, Kaneko FT, Zheng S, et al. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J 2004;18:1746–1748. [DOI] [PubMed]
- 12.Mathew R. Cell-specific dual role of caveolin-1 in pulmonary hypertension. Pulm Med 2011;2011:573432. [DOI] [PMC free article] [PubMed]
- 13.Achcar ROD, Demura Y, Rai PR, et al. Loss of caveolin and heme oxygenase expression in severe pulmonary hypertension. Chest 2006;129:696–705. [DOI] [PubMed]
- 14.Austin ED, Ma L, LeDuc C, et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ Cardiovasc Genet 2012;5:336–343. [DOI] [PMC free article] [PubMed]
- 15.Place AT, Chen Z, Bakhshi FR, Liu G, O’Bryan JP, Minshall RD. Cooperative role of caveolin-1 and C-terminal Src kinase binding protein in C-terminal Src kinase-mediated negative regulation of c-Src. Mol Pharmacol 2011;80:665–672. [DOI] [PMC free article] [PubMed]
- 16.Bernatchez PN, Bauer PM, Yu J, Prendergast JS, He P, Sessa WC. Dissecting the molecular control of endothelial NO synthase by caveolin-1 using cell-permeable peptides. Proc Natl Acad Sci USA 2005;102:761–766. [DOI] [PMC free article] [PubMed]
- 17.Chen Z, Bakhshi FR, Shajahan AN, et al. Nitric oxide–dependent Src activation and resultant caveolin-1 phosphorylation promote eNOS/caveolin-1 binding and eNOS inhibition. Mol Biol Cell 2012;23:1388–1398. [DOI] [PMC free article] [PubMed]
- 18.Mao M, Sudhahar V, Ansenberger-Fricano K, et al. Nitroglycerin drives endothelial nitric oxide synthase activation via the phosphatidylinositol 3-kinase/protein kinase B pathway. Free Radic Biol Med 2012;52:427–435. [DOI] [PMC free article] [PubMed]
- 19.Stenmark KR, McMurtry IF. Vascular remodeling versus vasoconstriction in chronic hypoxic pulmonary hypertension: a time for reappraisal? Circ Res 2005;97:95–98. [DOI] [PubMed]
- 20.Bowers R, Cool C, Murphy RC, et al. Oxidative stress in severe pulmonary hypertension. Am J Respir Crit Care Med 2004;169:764–769. [DOI] [PubMed]
- 21.Konduri GG, Bakhutashvili I, Eis A, Pritchard K. Oxidant stress from uncoupled nitric oxide synthase impairs vasodilation in fetal lambs with persistent pulmonary hypertension. Am J Physiol Heart Circ Physiol 2007;292:H1812–H1820. [DOI] [PubMed]
- 22.Oishi PE, Wiseman DA, Sharma S, et al. Progressive dysfunction of nitric oxide synthase in a lamb model of chronically increased pulmonary blood flow: a role for oxidative stress. Am J Physiol Lung Cell Mol Physiol 2008;295:L756–L766. [DOI] [PMC free article] [PubMed]
- 23.Lee H, Woodman SE, Engelman JA, et al. Palmitoylation of caveolin-1 at a single site (Cys-156) controls its coupling to the c-Src tyrosine kinase: targeting of dually acylated molecules (GPI-linked, transmembrane, or cytoplasmic) to caveolae effectively uncouples c-Src and caveolin-1 (TYR-14). J Biol Chem 2001;276:35150–35158. [DOI] [PubMed]
- 24.Sverdlov M, Shinin V, Place AT, Castellon M, Minshall RD. Filamin A regulates caveolae internalization and trafficking in endothelial cells. Mol Biol Cell 2009;20:4531–4540. [DOI] [PMC free article] [PubMed]
- 25.Comhair SAA, Xu W, Mavrakis L, Aldred MA, Asosingh K, Erzurum SC. Human primary lung endothelial cells in culture. Am J Respir Cell Mol Biol 2012;46:723–730. [DOI] [PMC free article] [PubMed]
- 26.Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE 2001;2001:pl1. [DOI] [PubMed]
- 27.Kinter M, Sherman NE. Protein sequencing and identification using tandem mass spectrometry. Hoboken, NJ: Wiley, 2000.
- 28.Mathew R, Huang J, Gewitz MH. Pulmonary artery hypertension: caveolin-1 and eNOS interrelationship: a new perspective. Cardiol Rev 2007;15:143–149. [DOI] [PubMed]
- 29.Mathew R, Huang J, Shah M, Patel K, Gewitz M, Sehgal PB. Disruption of endothelial-cell caveolin-1α/raft scaffolding during development of monocrotaline-induced pulmonary hypertension. Circulation 2004;110:1499–1506. [DOI] [PubMed]
- 30.Masri FA, Xu W, Comhair SAA, et al. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 2007;293:L548–L554. [DOI] [PubMed]
- 31.Dudzinski DM, Michel T. Life history of eNOS: partners and pathways. Cardiovasc Res 2007;75:247–260. [DOI] [PMC free article] [PubMed]
- 32.Barsacchi R, Perrotta C, Bulotta S, Moncada S, Borgese N, Clementi E. Activation of endothelial nitric-oxide synthase by tumor necrosis factor–α: a novel pathway involving sequential activation of neutral sphingomyelinase, phosphatidylinositol-3′ kinase, and Akt. Mol Pharmacol 2003;63:886–895. [DOI] [PubMed]
- 33.Martin GS. The hunting of the Src. Nat Rev Mol Cell Biol 2001;2:467–475. [DOI] [PubMed]
- 34.Fernandez I, Ying Y, Albanesi J, Anderson RGW. Mechanism of caveolin filament assembly. Proc Natl Acad Sci USA 2002;99:11193–11198. [DOI] [PMC free article] [PubMed]
- 35.Arciniegas E, Frid MG, Douglas IS, Stenmark KR. Perspectives on endothelial-to-mesenchymal transition: potential contribution to vascular remodeling in chronic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2007;293:L1–L8. [DOI] [PubMed]
- 36.Cruz JA, Bauer EM, Rodriguez AI, et al. Chronic hypoxia induces right heart failure in caveolin-1−/− mice. Am J Physiol Heart Circ Physiol 2012;302:H2518–H2527. [DOI] [PMC free article] [PubMed]
- 37.Savai R, Pullamsetti SS, Kolbe J, et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2012;186:897–908. [DOI] [PubMed]
- 38.Rougier J-S, Gavillet B, Abriel H. Proteasome inhibitor (MG132) rescues Nav1.5 protein content and the cardiac sodium current in dystrophin-deficient mdx5cv mice. Front Physiol 2013;4:51. [DOI] [PMC free article] [PubMed]
- 39.Glenney JR, Soppet D. Sequence and expression of caveolin, a protein component of caveolae plasma membrane domains phosphorylated on tyrosine in Rous sarcoma virus–transformed fibroblasts. Proc Natl Acad Sci USA 1992;89:10517–10521. [DOI] [PMC free article] [PubMed]
- 40.Tiruppathi C, Naqvi T, Wu Y, Vogel SM, Minshall RD, Malik AB. Albumin mediates the transcytosis of myeloperoxidase by means of caveolae in endothelial cells. Proc Natl Acad Sci USA 2004;101:7699–7704. [DOI] [PMC free article] [PubMed]
- 41.Ghitescu L, Fixman A, Simionescu M, Simionescu N. Specific binding sites for albumin restricted to plasmalemmal vesicles of continuous capillary endothelium: receptor-mediated transcytosis. J Cell Biol 1986;102:1304–1311. [DOI] [PMC free article] [PubMed]
- 42.Minshall RD, Tiruppathi C, Vogel SM, et al. Endothelial cell-surface gp60 activates vesicle formation and trafficking via Gi-coupled Src kinase signaling pathway. J Cell Biol 2000;150:1057–1070. [DOI] [PMC free article] [PubMed]
- 43.Schubert W, Frank PG, Razani B, Park DS, Chow C-W, Lisanti MP. Caveolae-deficient endothelial cells show defects in the uptake and transport of albumin in vivo. J Biol Chem 2001;276:48619–48622. [DOI] [PubMed]
- 44.Bendayan M, Rasio EA. Transport of insulin and albumin by the microvascular endothelium of the rete mirabile. J Cell Sci 1996;109:1857–1864. [DOI] [PubMed]
- 45.Vasile E, Simionescu M, Simionescu N. Visualization of the binding, endocytosis, and transcytosis of low-density lipoprotein in the arterial endothelium in situ. J Cell Biol 1983;96:1677–1689. [DOI] [PMC free article] [PubMed]
- 46.Hu G, Place AT, Minshall RD. Regulation of endothelial permeability by Src kinase signaling: vascular leakage versus transcellular transport of drugs and macromolecules. Chem Biol Interact 2008;171:177–189. [DOI] [PMC free article] [PubMed]
- 47.Schnitzer JE, Liu J, Oh P. Endothelial caveolae have the molecular transport machinery for vesicle budding, docking, and fusion including VAMP, NSF, SNAP, annexins, and GTPases. J Biol Chem 1995;270:14399–14404. [DOI] [PubMed]
- 48.Schnitzer JE, Oh P, Jacobson BS, Dvorak AM. Caveolae from luminal plasmalemma of rat lung endothelium: microdomains enriched in caveolin, Ca2+-ATPase, and inositol trisphosphate receptor. Proc Natl Acad Sci USA 1995;92:1759–1763. [DOI] [PMC free article] [PubMed]
- 49.Ju H, Venema VJ, Liang H, Harris MB, Zou R, Venema RC. Bradykinin activates the Janus-activated kinase/signal transducers and activators of transcription (JAK/STAT) pathway in vascular endothelial cells: localization of JAK/STAT signalling proteins in plasmalemmal caveolae. Biochem J 2000;351:257–264. [DOI] [PMC free article] [PubMed]
- 50.García-Cardeña G, Oh P, Liu J, Schnitzer JE, Sessa WC. Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: implications for nitric oxide signaling. Proc Natl Acad Sci USA 1996;93:6448–6453. [DOI] [PMC free article] [PubMed]
- 51.Lisanti MP, Scherer PE, Vidugiriene J, et al. Characterization of caveolin-rich membrane domains isolated from an endothelial-rich source: implications for human disease. J Cell Biol 1994;126:111–126. [DOI] [PMC free article] [PubMed]
- 52.Liu J, Oh P, Horner T, Rogers RA, Schnitzer JE. Organized endothelial cell surface signal transduction in caveolae distinct from glycosylphosphatidylinositol-anchored protein microdomains. J Biol Chem 1997;272:7211–7222. [DOI] [PubMed]
- 53.Annabi B, Lachambre M, Bousquet-Gagnon N, Pagé M, Gingras D, Béliveau R. Localization of membrane-type 1 matrix metalloproteinase in caveolae membrane domains. Biochem J 2001;353:547–553. [DOI] [PMC free article] [PubMed]
- 54.Chun M, Liyanage UK, Lisanti MP, Lodish HF. Signal transduction of a G protein–coupled receptor in caveolae: colocalization of endothelin and its receptor with caveolin. Proc Natl Acad Sci USA 1994;91:11728–11732. [DOI] [PMC free article] [PubMed]
- 55.Oh P, Schnitzer JE. Segregation of heterotrimeric G proteins in cell surface microdomains: Gq binds caveolin to concentrate in caveolae, whereas Gi and Gs target lipid rafts by default. Mol Biol Cell 2001;12:685–698. [DOI] [PMC free article] [PubMed]
- 56.Stenmark KR, Meyrick B, Galiè N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol 2009;297:L1013–L1032. [DOI] [PubMed]
- 57.Fijalkowska I, Xu W, Comhair SAA, et al. Hypoxia inducible-factor 1α regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am J Path 2010;176:1130–1138. [DOI] [PMC free article] [PubMed]
- 58.Mercier I, Jasmin J-F, Pavlides S, et al. Clinical and translational implications of the caveolin gene family: lessons from mouse models and human genetic disorders. Lab Invest 2009;89:614–623. [DOI] [PMC free article] [PubMed]
- 59.Hill MM, Bastiani M, Luetterforst R, et al. PTRF-cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell 2008;132:113–124. [DOI] [PMC free article] [PubMed]
- 60.Liu L, Brown D, McKee M, et al. Deletion of cavin/PTRF causes global loss of caveolae, dyslipidemia, and glucose intolerance. Cell Metab 2008;8:310–317. [DOI] [PMC free article] [PubMed]
- 61.Parkar NS, Akpa BS, Nitsche LC, et al. Vesicle formation and endocytosis: function, machinery, mechanisms, and modeling. Antioxid Redox Signal 2009;11:1301–1312. [DOI] [PMC free article] [PubMed]
- 62.Bernatchez P, Sharma A, Bauer PM, Marin E, Sessa WC. A noninhibitory mutant of the caveolin-1 scaffolding domain enhances eNOS-derived NO synthesis and vasodilation in mice. J Clin Invest 2011;121:3747–3755. [DOI] [PMC free article] [PubMed]
- 63.Kirchner P, Bug M, Meyer H. Ubiquitination of the N-terminal region of caveolin-1 regulates endosomal sorting by the VCP/p97 AAA-ATPase. J Biol Chem 2013;288:7363–7372. [DOI] [PMC free article] [PubMed]
- 64.Hayer A, Stoeber M, Ritz D, Engel S, Meyer HH, Helenius A. Caveolin-1 is ubiquitinated and targeted to intralumenal vesicles in endolysosomes for degradation. J Cell Biol 2010;191:615–629. [DOI] [PMC free article] [PubMed]
- 65.Razani B, Engelman JA, Wang XB, et al. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem 2001;276:38121–38138. [DOI] [PubMed]