The Salmonella Effector SteA Contributes to the Control of Membrane Dynamics of Salmonella-Containing Vacuoles (original) (raw)
Abstract
Salmonella enterica serovar Typhimurium is a bacterial pathogen causing gastroenteritis in humans and a typhoid-like systemic disease in mice. S. Typhimurium virulence is related to its capacity to multiply intracellularly within a membrane-bound compartment, the Salmonella_-containing vacuole (SCV), and depends on type III secretion systems that deliver bacterial effector proteins into host cells. Here, we analyzed the cellular function of the Salmonella effector SteA. We show that, compared to cells infected by wild-type S. Typhimurium, cells infected by Δ_steA mutant bacteria displayed fewer Salmonella_-induced filaments (SIFs), an increased clustering of SCVs, and morphologically abnormal vacuoles containing more than one bacterium. The increased clustering of SCVs and the appearance of vacuoles containing more than one bacterium were suppressed by inhibition of the activity of the microtubule motor dynein or kinesin-1. Clustering and positioning of SCVs are controlled by the effectors SseF and SseG, possibly by helping to maintain a balanced activity of microtubule motors on the bacterial vacuoles. Deletion of steA in S. Typhimurium Δ_sseF or Δ_sseG_ mutants revealed that SteA contributes to the characteristic scattered distribution of Δ_sseF_ or Δ_sseG_ mutant SCVs in infected cells. Overall, this shows that SteA participates in the control of SCV membrane dynamics. Moreover, it indicates that SteA is functionally linked to SseF and SseG and suggests that it might contribute directly or indirectly to the regulation of microtubule motors on the bacterial vacuoles.
INTRODUCTION
Salmonella enterica serovar Typhimurium is a facultative intracellular bacterial pathogen that causes gastroenteritis in humans and a typhoid-like systemic disease in susceptible mouse strains. The virulence of S. Typhimurium is related to its ability to invade nonphagocytic cells and to replicate inside host cells within a membrane-bound compartment, the _Salmonella_-containing vacuole (SCV) (1). These abilities are associated with two distinct type III secretion systems (T3SSs), encoded by Salmonella pathogenicity island 1 (SPI-1) and SPI-2, which deliver >40 effector proteins into host cells (1–3). In general, the SPI-1 T3SS is required for invasion of nonphagocytic cells (4) and enables S. Typhimurium to cross the gut barrier and promote intestinal inflammation (5, 6), while the SPI-2 T3SS mediates intravacuolar bacterial replication (7–9) and is necessary for systemic infection of mice (8, 9). However, the boundaries between SPI-1 and SPI-2 T3SSs are not that sharply defined (10–13), and several effectors are secreted by both T3SSs (1–3, 14).
Maturation of the SCV within host cells involves sequential interactions with the endocytic pathway, resulting in a compartment showing features of late endosomes, including an acidified lumen and an accumulation of lysosomal glycoproteins, such as LAMP1, in its membrane. However, the SCV is not enriched in lysosomal hydrolases (15). The acidified lumen of the mature SCV contributes to the induction of expression of the genes encoding the SPI-2 T3SS (8, 16). The phenotypic consequences of the ensuing translocation of SPI-2 effectors across the SCV membrane have been characterized in different cell types, especially in infected human epithelial HeLa cells. Notably, a subset of SPI-2 effectors (PipB2, SifA, SopD2, SseF, SseG, and SseJ) has been shown to control membrane dynamics and the intracellular positioning of the SCV (3, 17). S. Typhimurium Δ_pipB2_ (18), Δ_sifA_ (19), Δ_sopD2_ (20), Δ_sseF_ (9), Δ_sseG_ (9), and Δ_sseJ_ (21) single mutants are all attenuated for virulence in the mouse model of systemic infection, and with the exception of S. Typhimurium Δ_pipB2_, each of these single mutants is deficient for intracellular replication in mouse macrophages (22). Therefore, the action of these effectors should promote bacterial virulence by mediating the intracellular replication of Salmonella through subversion of host cell intracellular trafficking pathways.
Among the SPI-2 effectors that control SCV membrane dynamics and positioning, SifA is required for the stability of the SCV membrane (19), is essential for the appearance in infected epithelial cells of LAMP1-enriched _Salmonella_-induced filaments (SIFs) (23), and attenuates lysosome function by inhibiting trafficking of mannose-6-phosphate receptors (24). SifA regulates the activity of the microtubule plus-end-directed motor kinesin-1 on the SCV (25, 26), and its function is linked to at least PipB2 (27), SopD2 (28), and SseJ (21, 29). SseF and SseG play a central role in maintaining the position of SCVs within the Golgi region of infected epithelial cells (30, 31) and are also involved in the formation of SIFs (32). An SseF-SseG protein complex might tether SCVs to the Golgi region or contribute to a balanced activity of microtubule motors on the bacterial vacuoles, possibly by mediating the recruitment of the microtubule minus-end-directed motor dynein (17, 30, 31, 33). Most of the studies on the mode of action of these effectors have been done using HeLa cells, which are immortalized cells of epithelial origin but which have an abnormal karyotype. However, regardless of the exact physiological relevance of HeLa cells as an infection model, the _Salmonella_-induced tubules or the bacterial microcolonies in the Golgi region observed in HeLa cells infected by S. Typhimurium have provided a means to study the effectors involved in their appearance (28–32, 34, 35).
SteA is a Salmonella effector translocated by both the SPI-1 and the SPI-2 T3SSs (36, 37). It localizes to the _trans_-Golgi network (TGN) when the effector is ectopically expressed in uninfected mammalian cells or delivered by S. Typhimurium into infected host cells (36). Moreover, bacterially translocated SteA localizes on Salmonella_-induced tubules enriched in the TGN marker 1,4-galactosyltransferase (GalT) but is mostly excluded from LAMP1-enriched tubules (38). SteA does not contribute to the intracellular replication of S. Typhimurium in mouse bone marrow macrophages (22), but an intraperitoneally injected Δ_steA mutant S. Typhimurium strain shows a virulence defect in a mouse model of systemic infection (36). When the same inoculation route and infection model are used, mutants that cannot assemble the SPI-1 T3SS display no obvious defect (5). Therefore, a virulence function of SteA could be related to its delivery into host cells by the SPI-2 T3SS. However, the cellular role(s) of SteA has not been characterized. In this work, we show that SteA contributes to the control of SCV membrane dynamics, possibly by participating in the regulation of microtubule motors on bacterial vacuoles. Our results also indicate that SteA is functionally linked to SseF and SseG.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and genetic procedures.
This work was done using S. Typhimurium NCTC 12023 (which is identical to ATCC 14208s) and its isogenic mutant derivatives. Additional information on these bacterial strains is provided in Table S1 in the supplemental material. Escherichia coli Top10 (Invitrogen) was used for the construction and amplification of plasmids. Bacterial cells were grown in Luria-Bertani (LB) medium (NZYtech) supplemented when appropriate with ampicillin (100 μg/ml) and/or kanamycin (50 μg/ml). Chromosomal deletion of single genes in S. Typhimurium was performed by using the one-step gene disruption method described by Datsenko and Wanner (39), using plasmids pKD4, pKD46, and pCP20 (see Table S2 in the supplemental material). S. Typhimurium double or triple mutants were constructed by using P22 HT105 int phage lysates of kanamycin-resistant single mutants to transduce antibiotic-sensitive single or double mutant strains. All deletions were confirmed by PCR. For transformation, DNA was introduced into E. coli or S. Typhimurium by electroporation.
Plasmids, DNA manipulations, and oligonucleotides.
The plasmids and DNA oligonucleotides used in this work are detailed in Tables S2 and S3 in the supplemental material, respectively. To construct a plasmid expressing SteA C-terminally tagged with a double hemagglutinin (HA) tag (pSteA-2HA), a DNA fragment including steA and its promoter region was amplified by PCR from the chromosomal DNA of S. Typhimurium NCTC 12023. The purified PCR product was digested with BamHI and EcoRI and inserted into those sites of pWSK129 (40). To construct pBAD-SteA-2HA, _steA_-2HA was amplified by PCR using pSteA-2HA DNA as the template. The purified PCR product was digested with NcoI and HindIII and inserted into those sites of pBAD/_myc_-HisA (Invitrogen). The accuracy of the nucleotide sequence in the inserts in the constructed plasmids was checked by DNA sequencing. These plasmids were constructed using proofreading Phusion DNA polymerase (Finnzymes), restriction enzymes (MBI Fermentas), T4 DNA ligase (Invitrogen), a DNA clean & Concentrator-5 kit, and a Zymoclean gel DNA recovery kit (Zymo Research) and purified with a GeneElute plasmid miniprep kit (Sigma), according to the instructions of the manufacturers. Additional PCRs were performed using DreamTaq DNA polymerase (MBI Fermentas), according to the instructions of the manufacturer.
Cell culture and bacterial infections.
Human epithelial HeLa cells (clone HtTA-1), human intestinal Caco-2 cells, and RAW 264.7 murine macrophage-like cells were obtained from the European Collection of Cell Cultures (ECACC). HeLa or RAW 264.7 cells were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% (vol/vol) fetal bovine serum, without antibiotics, at 37°C in a humidified atmosphere with 5% (vol/vol) CO2. Caco-2 cells were maintained under the same conditions as HeLa or RAW 264.7 cells, except that the medium also contained nonessential amino acids (Life Technologies). Infection of HeLa, Caco-2, or RAW 264.7 cells with S. Typhimurium was performed essentially as previously described (19), except that for infection of macrophages, the cells were seeded at a density of 2 × 105 cells per well in 24-well tissue culture plates and the multiplicity of infection was 2:1. When needed, l-arabinose was added to the cell culture medium at 0.2% (wt/vol) to induce the expression of SteA or of DsRed (see below).
Transient transfection of mammalian cells.
HeLa cells were transfected with plasmid DNA by using the jetPEI reagent (Polyplus), as detailed by the manufacturer but using 250 ng of DNA per well of a 24-well tissue culture plate. Cells were transfected 2 h after bacterial inoculation with pLAMP1-GFP (41), obtained from Patrice Boquet; pLAMP1-mGFP (42); pHA-TPR (25), obtained from Stéphane Méresse; pHA-p50 (43), obtained from Serge Benichou; pEGFP-C1 (Clontech); pEGFP-p50 (43), obtained from Serge Benichou; or pEGFP-TPR-KLC2 (44), obtained from Michael Way (see Table S2 in the supplemental material). Cells were subsequently incubated at 37°C in 5% CO2 for the indicated time periods prior to fixation and antibody staining.
Antibodies.
For immunofluorescence (IF) microscopy, goat polyclonal anti-Salmonella antibody CSA-1 (Kirkegaard & Perry Laboratories) was used at 1:200; mouse monoclonal antibody anti-LAMP1 H4A3, developed by J. T. August and J. E. K. Hildreth, and rat monoclonal antibody anti-LAMP1 1D4B, developed by J. T. August, were both obtained from the Developmental Studies Hybridoma Bank (DSHB), developed under the auspices of NICHD and maintained by the University of Iowa, Department of Biology, Iowa City, IA, and were used at 1:400 and 1:200, respectively; the rat anti-HA 3F10 antibody (Roche) was used at 1:200; the rabbit anti-giantin antibody (Berkeley Antibody Company) was used at 1:600. Secondary antibodies were obtained from Jackson ImmunoResearch Laboratories (donkey anti-goat antibody conjugated to cyanine 5, anti-rat antibody conjugated to rhodamine RedX, and anti-rabbit antibody conjugated to rhodamine RedX, all of which were used at 1:200) and from Invitrogen (goat anti-mouse antibody AF568 used at 1:200).
Immunofluorescence microscopy.
Cell monolayers seeded on glass coverslips were fixed with 4% (wt/vol) paraformaldehyde in phosphate-buffered saline (PBS) at room temperature for 15 min and then washed three times in PBS. Antibodies were diluted in PBS containing 0.1% (wt/vol) saponin and 10% (vol/vol) horse serum. The cells in the coverslips were washed twice in PBS with 0.1% (wt/vol) saponin and incubated for 1 h with primary antibodies. The cells in the coverslips were then again washed twice in PBS containing 0.1% (wt/vol) saponin and incubated with appropriate secondary antibodies for 30 min. The coverslips were mounted onto glass slides using Aqua-poly/Mount mounting medium (Polysciences). Samples were analyzed using a fluorescence microscope (Leica DMRA2) or a confocal laser scanning microscope (LSM 510 META or LSM 710 META; Zeiss) at the Instituto Gulbenkian da Ciência (IGC) or the Centro de Doenças Crónicas (CEDOC), respectively. All images were obtained by confocal microscopy and processed using Zeiss LSM Image Browser and Adobe Photoshop software.
Live-cell imaging.
HeLa cells were seeded at 2.0 × 105 cells per dish in 35-mm glass-bottom dishes (MakTeK Corporation) and transfected with pLAMP1-GFP (41) as described above. At 24 h after transfection, the cells were infected with wild-type (wt) or steA mutant S. Typhimurium harboring a plasmid expressing the fluorescent DsRed protein (45). Prior to live-cell imaging at the time points indicated below, the medium was changed to Opti-MEM (Invitrogen) with 5% (vol/vol) fetal bovine serum. We did not image cells displaying high levels of LAMP1-green fluorescent protein (GFP), to avoid artifacts due to dimerization of LAMP1-GFP (42). The cells were imaged on a LSM 710 META microscope (Carl Zeiss). The images were recorded and processed using Volocity software (Improvision).
Scoring of phenotypes by microscopy.
The quantification of SCV positioning in the Golgi region was done and the appearance of SIFs was determined as previously described (46). Also as described before, a bacterial microcolony was defined as a cluster of at least 5 SCVs showing overlapping fluorescence signals (46). To quantify the appearance of compact microcolonies, we defined an infected cell to contain such a bacterial microcolony if all SCVs within that cell were clustered; i.e., the fluorescence signal of the bacteria did not reveal a single nonoverlapping vacuole. The appearance of an excessive accumulation of LAMP1 within the microcolony was estimated visually. To quantify the appearance of abnormal SCVs, we examined images of individual infected cells to identify vacuoles containing more than one bacterium. In the experiments where we used pLAMP1-GFP, to avoid artifacts due to dimerization of LAMP1-GFP (42), we analyzed only cells displaying low to moderate levels of LAMP1-GFP. In wt- and Δ_steA_ mutant-infected cells, we considered only cells containing 15 to 25 bacteria. To quantify SIFs or the accumulation of LAMP1 within the microcolony or abnormal SCVs in HeLa cells infected by wt or Δ_steA_ mutant S. Typhimurium, we also considered only centrally located microcolonies. The intracellular growth of S. Typhimurium in RAW 264.7 cells was quantified by microscopy by estimating the number of bacteria inside infected cells. To quantify bacterial invasion by microscopy, extracellular bacteria were immunolabeled with green and red fluorophores and intracellular bacteria were immunolabeled only with a red fluorophore. In all cases, at least 50 infected cells were scored blind in each experiment, and all experiments were repeated at least three times. Results are reported as means ± standard errors of the means (SEMs). Differences between data sets were considered significant if P was <0.05 in a two-tailed unpaired Student's t test.
RESULTS
SteA contributes to the formation of SIFs.
Initial experiments indicated that an S. Typhimurium Δ_steA_ mutant strain was not defective for invasion of epithelial cells and confirmed that, as recently reported (22), a lack of SteA did not significantly impair the ability of Salmonella to grow within mouse macrophages (see Fig. S1 in the supplemental material). While this does not exclude a role of SteA during the initial stages of host cell infection by Salmonella, given that the S. Typhimurium Δ_steA_ mutant showed a defect in a mouse model of systemic infection (36), we analyzed if SteA was involved in phenotypes that in Salmonella cell culture infection models have been associated with the SPI-2 T3SS. In particular, we analyzed if SteA was required for SCV positioning within the Golgi region (30) or for the appearance of SIFs in infected epithelial cells (47). For this, we infected HeLa cells with wt or Δ_steA_ mutant S. Typhimurium constitutively expressing GFP (48) (see Table S2 in the supplemental material). The cells were fixed at 8 or 14 h following infection and immunolabeled for the Golgi region with an anti-giantin antibody or for SIFs with an anti-LAMP1 antibody and were then analyzed by indirect immunofluorescence (IF) microscopy. At either 8 or 14 h postinvasion (p.i.), S. Typhimurium Δ_steA_ mutant SCVs formed microcolonies in the vicinity of the Golgi region at a frequency (83% ± 4% and 82% ± 4% of infected cells at 8 and 14 h p.i., respectively) that was not significantly different (P = 0.0558) from that for wild-type vacuoles (96% ± 1% and 92% ± 3% of infected cells at 8 and 14 h p.i., respectively). However, there was a significant decrease in the frequency of appearance of SIFs in cells infected by Δ_steA_ mutant bacteria (with 13% ± 7% and 20% ± 2% of cells showing SIFs at 8 and 14 h p.i., respectively), in comparison to that in cells infected by wt S. Typhimurium (with 34% ± 5% and 38% ± 3% of infected cells showing SIFs at 8 and 14 h p.i., respectively) (Fig. 1). The SteA-dependent deficiency in the appearance of SIFs was complemented when a plasmid expressing SteA-2HA was introduced in the Δ_steA_ mutant strain (with 40% ± 1% of infected cells showing SIFs at 14 h p.i.) (Fig. 1). Because the appearance of SIFs has been shown to depend on several SPI-2 effectors (47), we wanted to confirm that a defect in their formation is not a general feature of mutants with mutations in SPI-2 effector genes. For this, we also infected HeLa cells with Δ_sifB_ mutant S. Typhimurium expressing GFP. The sifB gene encodes an SPI-2 effector displaying ∼30% amino acid identity to SifA (49), which is essential for the formation of SIFs (23, 28). HeLa cells infected by Δ_sifB_ mutant S. Typhimurium for 14 h showed a frequency in the appearance of SIFs (40% ± 3%) similar to that of cells infected by wt bacteria (38% ± 3%) (Fig. 1A). Therefore, not all SPI-2 effectors are required for the formation of SIFs. These results showed that SteA, together with the SPI-2 effectors SifA (23), SseF (32), SseG (32), PipB2 (35), and SopD2 (20), contributes to the formation of SIFs in HeLa cells infected by S. Typhimurium.
FIG 1.
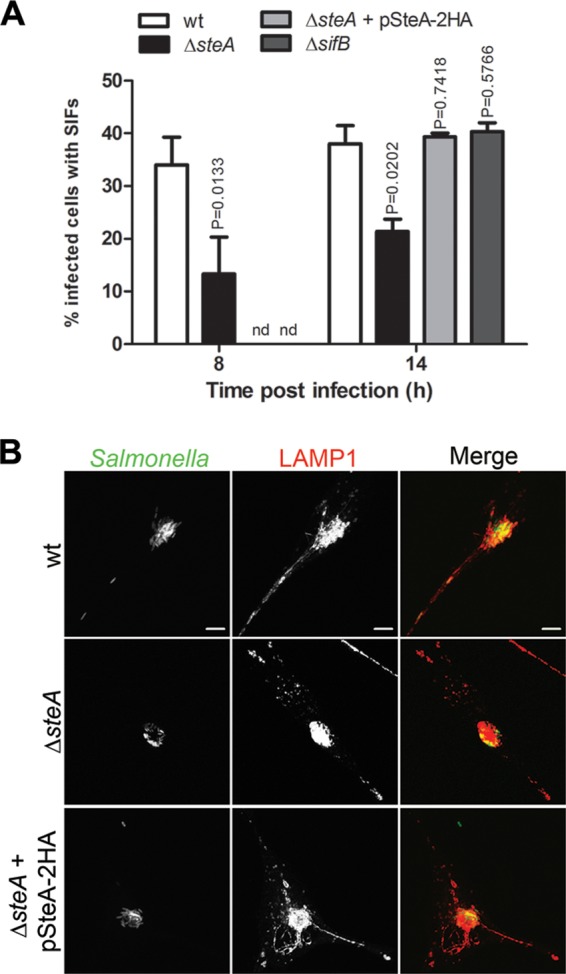
SteA contributes to the formation of SIFs. HeLa cells were infected for 8 or 14 h with the indicated GFP-expressing S. Typhimurium strains, fixed, and immunostained for LAMP1. (A) Infected cells showing SIFs were counted by immunofluorescence microscopy. Values are the means ± standards errors of the means (n = 3). At least 50 infected cells were analyzed in each experiment. P values were calculated by a two-tailed unpaired Student's t test relative to wt-infected cells for each time postinvasion. (B) Infected HeLa cells were imaged by confocal microscopy for Salmonella expressing GFP (green) and LAMP1 (red). nd, not determined. Bars, 5 μm.
Δ_steA_ mutant vacuoles form microcolonies that show an excessive accumulation of LAMP1 and that are frequently more compact than those formed by wt vacuoles.
While comparing the appearance of Golgi region-associated microcolonies and of SIFs in HeLa cells infected by wt bacteria and in those infected by Δ_steA_ mutant S. Typhimurium, it seemed that the microcolonies formed by Δ_steA_ mutant SCVs were often more compact than those formed by wt vacuoles (Fig. 2A) and showed an apparent excessive accumulation of LAMP1 (Fig. 2B). To confirm these observations, we first examined by IF microscopy the morphology of the bacterial microcolonies in HeLa cells that had been infected by wt or Δ_steA_ mutant S. Typhimurium constitutively expressing GFP for 8 or 14 h p.i. For this analysis, we defined an infected cell to contain a compact microcolony if all SCVs within that cell were clustered, i.e., if the fluorescence signal of the bacteria within that cell did not show a single nonoverlapping vacuole. This allowed us to confirm that, at either 8 or 14 h p.i., Δ_steA_ mutant SCVs were clustered in compact microcolonies more frequently than wt bacterial vacuoles (Fig. 2A and C). To analyze the accumulation of LAMP1 within the microcolonies, we examined by IF microscopy HeLa cells that had been infected by GFP-expressing wt or Δ_steA_ mutant S. Typhimurium, fixed at 8 or 14 h p.i., and immunolabeled for LAMP1. A visual analysis of the intensity of the LAMP1 IF signal within the bacterial microcolonies indicated that, at either 8 or 14 h p.i., ∼40% of cells infected by Δ_steA_ mutant S. Typhimurium showed microcolonies displaying an excessive accumulation of LAMP1, while only 5 to 10% of cells infected by wt bacteria showed a similar excessive accumulation of LAMP1 within the bacterial microcolonies (Fig. 2B and D).
FIG 2.
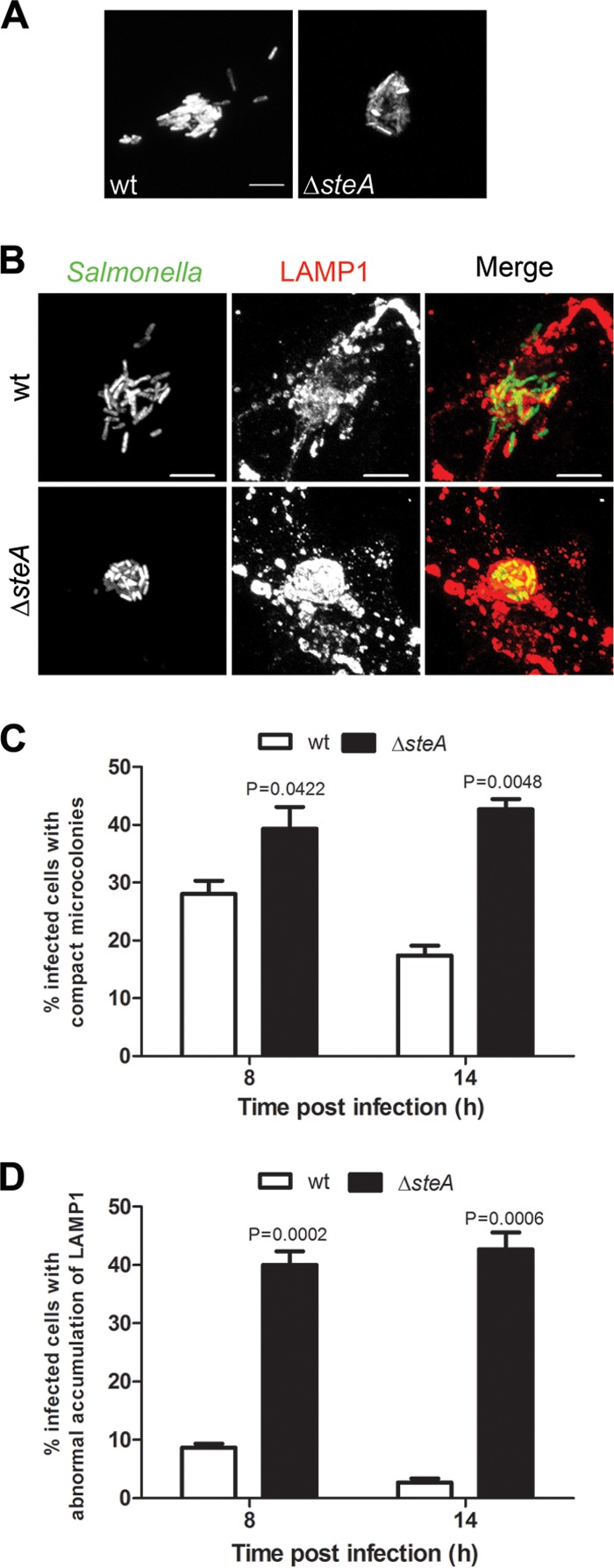
SteA participates in the control of SCV membrane dynamics. HeLa cells were infected for 8 or 14 h with the indicated GFP-expressing strains and fixed. (A and B) Infected HeLa cells were imaged by confocal microscopy for Salmonella expressing GFP (A) or were immunostained for LAMP1 and imaged by confocal microscopy for Salmonella expressing GFP (green) and LAMP1 (red) (B). (C) Infected cells with compact microcolonies were counted by IF microscopy. (D) Infected cells showing apparent abnormal LAMP1 accumulation within the bacterial microcolony were counted by IF microscopy. All values are the means ± standard errors of the means (n = 3). At least 50 infected cells were analyzed in each experiment. P values were calculated by a two-tailed unpaired Student's t test relative to wt-infected cells at each time postinvasion. Bars, 5 μm.
Δ_steA_ mutant S. Typhimurium is enclosed in abnormal vacuoles.
As LAMP1 is enriched on the SCV membrane, the relatively frequent appearance of microcolonies showing an excessive accumulation of LAMP1 in HeLa cells infected by Δ_steA_ mutant S. Typhimurium suggested that the vacuoles containing these mutant bacteria could be abnormal. In particular, from the IF microscopy analyses of LAMP1 accumulation on the bacterial microcolonies, it appeared that Δ_steA_ mutant SCVs often contained more than a single bacterium. In contrast, in HeLa cells or macrophages infected for several hours by wt S. Typhimurium, each bacterium is normally enclosed in a single vacuole (50, 51). To analyze this, HeLa cells were infected with wt or Δ_steA_ mutant S. Typhimurium expressing the red fluorescent protein DsRed. After 2 h, the cells were transfected with a plasmid encoding a fusion of LAMP1 to GFP (pLAMP1-GFP) and then fixed at 14 h p.i. We used this approach because direct fluorescence microscopy of ectopically expressed LAMP1-GFP in Salmonella_-infected HeLa cells provided a sharper delineation of the SCV membrane than indirect IF microscopy with a LAMP1 antibody. In the subsequent analysis by fluorescence microscopy, we considered an abnormal SCV to be a bacterial vacuole that showed defined and continuous LAMP1-GFP fluorescence encircling more than one bacterium. These experiments indicated that cells infected by Δ_steA mutant S. Typhimurium often displayed abnormal SCVs (51% ± 7%), while they were seldom seen in cells infected by wt bacteria (1% ± 1%) (Fig. 3A and B). Similar results were obtained when the experiment was performed using a transfection vector encoding LAMP-mGFP, a nondimerizable form of LAMP1-GFP (42) (see Fig. S2 in the supplemental material). Therefore, the appearance of a high number of abnormal SCVs in cells infected by Δ_steA_ mutant S. Typhimurium is not a consequence of the potential formation of LAMP1-GFP dimers. To further examine this, we used time-lapse fluorescence microscopy to image HeLa cells transfected with pLAMP1-GFP and infected with wt or Δ_steA_ mutant S. Typhimurium expressing DsRed. The images were recorded during 4 h of infection, between 8 and 12 h p.i. (Fig. 3C; see Movies S1 and S2 in the supplemental material). These analyses confirmed that in infected HeLa cells, Δ_steA_ mutant S. Typhimurium, but not wt Salmonella, can frequently be found in a vacuole containing more than one bacterium (Fig. 3C; see Movies S1 and S2 in the supplemental material). To assess the properties of Δ_steA_ mutant vacuoles in macrophages, the main cell type colonized by S. Typhimurium during systemic infection of mice (52), RAW 264.7 murine macrophage-like cells were infected for 16 h with wt or Δ_steA_ mutant S. Typhimurium expressing GFP. After fixation, the cells were immunolabeled using an antibody against LAMP1 and analyzed by indirect IF microscopy. This revealed that about 21% ± 4% of RAW 264.7 macrophages infected by Δ_steA_ mutant S. Typhimurium showed vacuoles that contained more than one bacterium, while only 3% ± 2% of macrophages infected by wt S. Typhimurium displayed abnormal vacuoles (Fig. 3B and D).
FIG 3.

SteA contributes to the normal partitioning of the bacterial vacuole in infected cells. (A) HeLa cells were infected for 14 h with the indicated DsRed-expressing strains, transfected with a plasmid encoding LAMP1-GFP, and fixed. Infected HeLa cells were imaged by confocal microscopy for LAMP1-GFP (green) and Salmonella expressing DsRed (red). (B) HeLa or RAW 264.7 infected cells with abnormal vacuoles, which apparently contained more than a single bacterium, were enumerated by immunofluorescence microscopy. All values are the means ± standard errors of the means (n = 3). At least 50 infected cells were analyzed in each experiment. P values were calculated by a two-tailed unpaired Student's t test relative to wt-infected HeLa or RAW 264.7 cells, except where indicated (comparison of Δ_steA_ mutant- and Δ_steA_ Δ_invG_ mutant-infected cells). (C) Still images from Movie S1 (wt) and Movie S2 (Δ_steA_ mutant) in the supplemental material. HeLa cells were transfected with pLAMP1-GFP and infected for 8 to 12 h with wt or Δ_steA_ mutant S. Typhimurium. The images show an infected cell with SCVs containing a single bacterium (wt) and an infected cell showing a large vacuole containing several bacteria (Δ_steA_ mutant). (D) RAW 264.7 macrophages were infected for 16 h with the indicated GFP-expressing strains, fixed, and immunostained for LAMP1. Infected RAW 264.7 macrophages were imaged by confocal microscopy for Salmonella expressing GFP (green) and LAMP1 (red). In the merged images, the arrows indicate normal vacuoles containing a single bacterium and the arrowheads indicate abnormal vacuoles containing more than one bacterium. Bars, 5 μm.
The results of these experiments were in agreement with previous observations indicating that HeLa cells or macrophages infected by wt S. Typhimurium normally display individual vacuoles (50, 51) and showed that these cells infected with the Δ_steA_ mutant often display abnormal vacuoles containing several bacteria. This suggests that the activity of SteA could contribute to fission of the vacuolar membrane as the bacterium divides. All together, the fluorescence microscopy analyses of cells infected with wt or Δ_steA_ mutant S. Typhimurium showed that SteA participates in the control of SCV membrane dynamics.
Phenotypes associated with infection of HeLa cells by Δ_steA_ mutant S. Typhimurium are not due to a lack of translocation of SteA during invasion.
As SteA has been shown to be translocated by both the SPI-1 T3SS and the SPI-2 T3SS and at early times of host cell infection (from 15 min to 2 h p.i.) (36, 37), it was conceivable that the phenotypes in infected HeLa cells described above were due to a lack of translocation of SteA during invasion. This could result in an early defect in SCV maturation that influenced the ensuing course of infection. To analyze this, we infected HeLa cells with wt or Δ_steA_ mutant S. Typhimurium harboring a plasmid expressing SteA-2HA under the control of the arabinose-inducible E. coli P_BAD_ promoter (pBAD-SteA-2HA). The experiment was conducted in the absence of l-arabinose, in the continuous presence of l-arabinose, or upon addition of l-arabinose to the infection medium at 1 or 4 h p.i. (Fig. 4) The cells were fixed at 14 h, immunolabeled for Salmonella and LAMP1, and analyzed by IF microscopy for the appearance of SIFs and of compact microcolonies. When the experiment was done in the continuous presence of l-arabinose (Fig. 4, +Ara) or in its absence (Fig. 4, −Ara), the results were similar to those observed after infection with wt or Δ_steA_ mutant S. Typhimurium, respectively (compare Fig. 4 with Fig. 1 and 2). When SteA expression was induced by addition of l-arabinose to the medium at 1 or 4 h p.i., this rescued the defects on the appearance of SIFs and on the morphology of the microcolonies normally displayed by the Δ_steA_ mutant (Fig. 3). If the described Δ_steA_ mutant-associated phenotypes were due to a lack of early translocation of SteA (i.e., during invasion or the first hours of SCV maturation), it would have not been possible to complement them when inducing expression of SteA-2HA by adding l-arabinose at 1 h or, especially, at 4 h after infection. These results show that the Δ_steA_ mutant-associated phenotypes observed in HeLa cells (Fig. 2 and 3) are not due to an early defect in SCV maturation caused by a lack of translocation of SteA during invasion or before 4 h p.i. Instead, they suggest that the observed phenotypes are due to a lack of translocation of SteA across the SCV membrane from 4 h after invasion.
FIG 4.
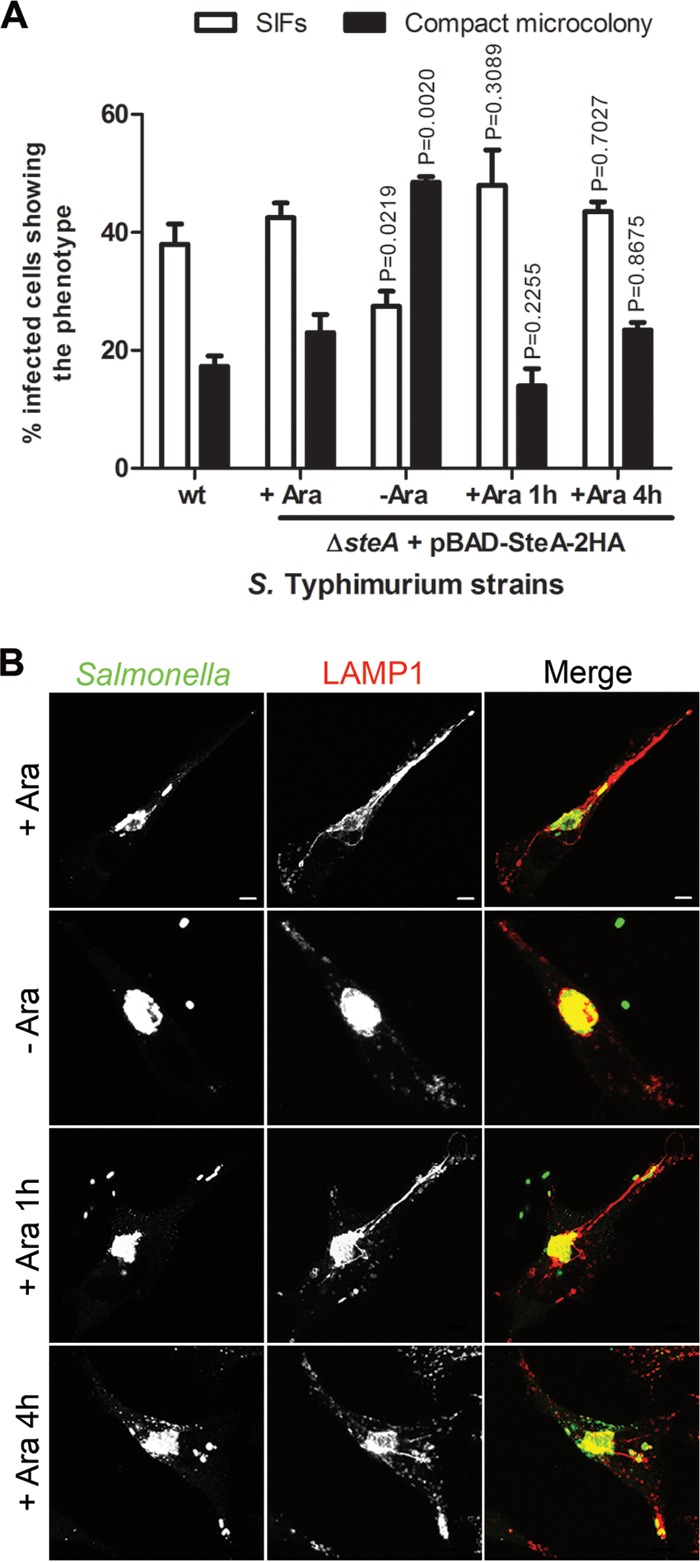
Early translocation of SteA is not required for the control of SCV membrane dynamics in HeLa cells. HeLa cells were infected for 14 h with S. Typhimurium wt or a Δ_steA_ mutant strain expressing SteA in trans under the control of the Escherichia coli arabinose-inducible P_BAD_ promoter (pBAD-SteA-2HA), fixed, and immunostained for Salmonella and LAMP1. The experiment was done in the continuous presence of 0.2% (vol/vol) l-arabinose (+Ara), in its absence (−Ara), or when the sugar was added to the medium at the indicated time points (1 or 4 h p.i.). (A) Infected cells showing SIFs and compact microcolonies were counted by immunofluorescence microscopy. Values are the means ± standard errors of the means (n = 3). At least 50 infected cells were analyzed in each experiment. P values were calculated by a two-tailed unpaired Student's t test relative to the condition with l-arabinose (+ Ara). (B) HeLa cells were imaged by confocal microscopy for Salmonella (green) and LAMP1 (red). Bars, 5 μm.
The abnormal SCVs found in RAW 264.7 macrophages infected by Δ_steA_ mutant S. Typhimurium are due to a lack of translocation of SteA by the SPI-2 T3SS.
InvG is an essential component of the SPI-1 T3SS (53), and therefore, a Δ_invG_ mutant S. Typhimurium strain cannot assemble the SPI-1 T3SS and is severely impaired in its ability to invade nonphagocytic cells (see Fig. S1 in the supplemental material). To analyze which secretion system (the SPI-1 T3SS or the SPI-2 T3SS) is required for the control of SCV membrane dynamics by SteA, we compared the properties of the SCVs in RAW 264.7 macrophages infected for 16 h by Δ_invG_ or Δ_steA_ Δ_invG_ mutant S. Typhimurium expressing GFP. After fixation, RAW 264.7 cells infected by Δ_invG_ or Δ_steA_ Δ_invG_ mutant S. Typhimurium were immunolabeled using an antibody against LAMP1 and analyzed by indirect IF microscopy. Approximately 19% ± 2% of RAW 264.7 macrophages infected by Δ_steA_ Δ_invG_ mutant S. Typhimurium showed vacuoles that contained more than one bacterium, while only 6% ± 1% of macrophages infected by Δ_invG_ mutant S. Typhimurium displayed such abnormal vacuoles (Fig. 3B; see Fig. S3 in the supplemental material). Importantly, the numbers of RAW 264.7 cells infected by Δ_invG_ or Δ_steA_ Δ_invG_ mutant S. Typhimurium displaying abnormal vacuoles were not significantly different (P > 0.05) from the numbers of RAW 264.7 cells infected in parallel by wt or Δ_steA_ mutant S. Typhimurium displaying abnormal vacuoles, respectively (Fig. 3B). These results indicate that the abnormal SCVs more frequently detected in macrophages infected by Δ_steA_ mutant S. Typhimurium than in macrophages infected by wt bacteria are due to a lack of translocation of SteA by the SPI-2 T3SS.
Inhibition of dynein or kinesin-1 activity suppresses defects displayed by Δ_steA_ mutant S. Typhimurium in infected HeLa cells.
Intravacuolar multiplication of S. Typhimurium, the formation of SIFs, the stability of the SCV membrane, and SCV positioning in HeLa cells have been shown to be controlled by the activity of the microtubule motors dynein and kinesin-1 (25, 27, 31, 50, 54–56). Based on this, we hypothesized that the distinct compact morphology of many Δ_steA_ mutant microcolonies (Fig. 2A and C) could be related to a diminished activity of kinesin-1 or of another microtubule plus-end-directed kinesin or to an increased activity of the microtubule minus-end-directed dynein. The defect in the appearance of SIFs and the abnormal vacuoles frequently seen in cells infected by Δ_steA_ mutant S. Typhimurium (Fig. 1 and Fig. 3) could also be related to the defective activity of these microtubule motors on the SCV (50, 57, 58). To analyze this, we examined the activity of dynein and kinesin-1 on SteA-dependent phenotypes (compact microcolonies and abnormal vacuoles). To interfere with the activity of these microtubule motors, we ectopically expressed p50-dynamitin, which inhibits dynein function (59), or the tetratricopeptide repeat (TPR) of the cargo binding domain of kinesin light chain 2 (KLC2), which inhibits kinesin-1 (44). HeLa cells were cotransfected with vectors expressing HA-p50-dynamitin (pHA-p50) and LAMP1-GFP or HA-TPR-KLC2 (pHA-TPR) and LAMP1-GFP 2 h after they had been inoculated with wt or Δ_steA_ mutant S. Typhimurium. Control infected cells were transfected only with LAMP1-GFP. The infected cells were fixed at 14 h p.i., immunolabeled with anti-Salmonella and anti-HA antibodies, and analyzed for the presence of compact microcolonies and abnormal vacuoles. Cells infected with wt S. Typhimurium and transfected with either pHA-p50 or pHA-TPR did not show a frequency of appearance of compact microcolonies and abnormal vacuoles significantly different from that in control infected cells (Fig. 5A). In contrast, in cells infected with Δ_steA_ mutant S. Typhimurium, expression of either HA-p50 or HA-TPR-KLC2 caused a significant reduction in the frequency of cells showing compact microcolonies (47% ± 3% in control cells, 6% ± 3% in cells expressing HA-p50, and 8% ± 4% in cells expressing HA-TPR-KLC2) and abnormal vacuoles (46% ± 3% in control cells, 22% ± 1% in cells expressing HA-p50, and 24% ± 3% in cells expressing HA-TPR-KLC2) (Fig. 5). Regarding the morphology of the microcolony, we obtained identical results when dynein or kinesin-1 activities were inhibited with GFP-tagged p50 or TPR-KLC2, respectively (see Fig. S4 in the supplemental material). These results indicate that kinesin-1 and dynein are directly or indirectly involved in the formation of the compact microcolonies and abnormal SCVs that characterize HeLa cells infected by Δ_steA_ mutant S. Typhimurium. Therefore, SteA could contribute to the control of the activity of these motors on the SCV.
FIG 5.
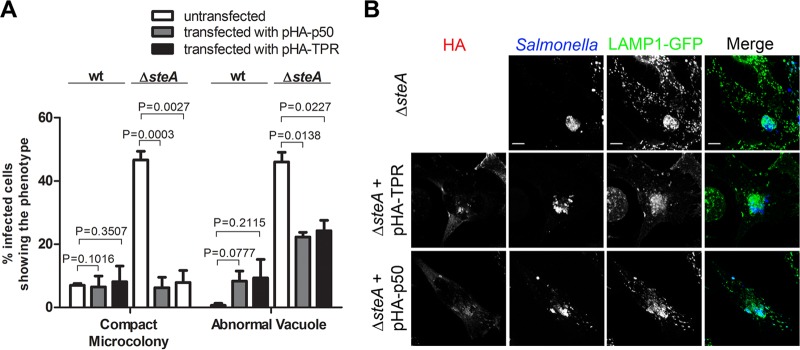
Inhibition of dynein or kinesin-1 activity suppresses defects in Δ_steA_ mutant SCVs in HeLa cells. HeLa cells were infected for 14 h with wt and Δ_steA_ mutant S. Typhimurium; transfected with pLAMP1-GFP plasmid alone, pLAMP1-GFP and pHA-p50 plasmids, or pLAMP1-GFP and pHA-TPR plasmids; fixed; and immunostained for Salmonella and HA. (A) Infected cells with compact microcolonies and abnormal vacuoles were counted by immunofluorescence microscopy. Values are the means ± standard errors of the means (n = 3). At least 50 infected cells were analyzed in each experiment. P values were calculated by a two-tailed unpaired Student's t test. (B) HeLa cells were imaged by confocal microscopy for HA (red), Salmonella (blue), and LAMP1-GFP (green). Bars, 5 μm.
SteA is functionally linked to the SPI-2 effectors SseF and SseG.
Kinesin-1 and dynein have been shown or suggested to be targets of SPI-2 effectors (25–27, 31, 50, 54, 55). To gain insights into the mode of action of SteA, we examined the consequences of deleting steA from the chromosome of relevant SPI-2 effector mutants (Δ_sseF_ and Δ_sseG_ mutants; see Discussion) for phenotypes associated with Salmonella infection of HeLa cells. In these cells, from 6 h p.i., bacterial vacuoles containing S. Typhimurium Δ_sseF_ or Δ_sseG_ mutants do not usually form a microcolony at the Golgi region and are scattered throughout the cell (30, 31, 34). A mechanism by which SseF and SseG might control SCV positioning is by ensuring a balanced activity of microtubule motors on the SCV (17, 31). We thought that if SteA contributes to the control of the activity of a microtubule motor, then the vacuoles of Δ_steA_ Δ_sseF_ or Δ_steA_ Δ_sseG_ double mutants would have a distinct positioning phenotype relative to that of the SCVs of the corresponding single mutants. To analyze this, we infected HeLa cells for 14 h with wt, Δ_steA_, Δ_sseF_, Δ_sseG_, Δ_steA_ Δ_sseF_, or Δ_steA_ Δ_sseG_ mutant S. Typhimurium constitutively expressing GFP. After fixation, the infected cells were immunolabeled for the Golgi region with an anti-giantin antibody and examined by indirect IF microscopy for the presence of a bacterial microcolony in the Golgi region and for the appearance of compact microcolonies (Fig. 6). As expected, in cells infected by Δ_sseF_ or Δ_sseG_ mutants, there were significantly fewer SCVs that clustered in the Golgi region and fewer compact microcolonies than in wt-infected cells (Fig. 6A to C). The SCVs of the Δ_steA_ Δ_sseF_ or Δ_steA_ Δ_sseG_ double mutant displayed a Golgi region-positioning defect similar to that of Δ_sseF_ or Δ_sseG_ mutant SCVs (Fig. 6B and C) and formed less compact microcolonies than Δ_steA_ mutant vacuoles (Fig. 6A). However, importantly, they formed more compact microcolonies than the SCVs of wt S. Typhimurium or of the Δ_sseF_ or Δ_sseG_ mutant (Fig. 6A and C). When we infected HeLa cells with an Δ_sseFG_ double mutant, the results were identical to those obtained with the single Δ_sseF_ or Δ_sseG_ mutant, and when we infected HeLa cells with a Δ_steA_ Δ_sseFG_ triple mutant, the results were the same as those obtained with the Δ_steA_ Δ_sseF_ or Δ_steA_ Δ_sseG_ double mutant (Fig. 6A and B). To understand if the increase in the appearance of compact microcolonies observed in Δ_sseF_ or Δ_sseG_ mutant SCVs by deletion of steA was due to the formation of vacuoles containing more than one bacterium, we infected HeLa cells with wt, Δ_steA_, or Δ_steA_ Δ_sseF_ mutant S. Typhimurium expressing DsRed. At 2 h p.i., the cells were transfected with pLAMP1-GFP and then fixed at 14 h p.i. IF microscopy analysis of the infected cells revealed that Δ_steA_ Δ_sseF_ mutant S. Typhimurium appeared to reside mostly in individual vacuoles (Fig. 6D and E).
FIG 6.

SteA is functionally linked to SPI-2 effectors SseF and SseG. (A to C) HeLa cells were infected for 14 h with the indicated GFP-expressing strains, fixed, and immunostained for the Golgi region with giantin. The infected cells showing microcolonies associated with the Golgi region (A) or compact microcolonies (B) were counted by IF microscopy. (C) HeLa cells were imaged by confocal microscopy for Salmonella expressing GFP (green) and giantin (red). (D and E) HeLa cells were infected with the indicated DsRed-expressing strains for 14 h, transfected with pLAMP1-GFP, and fixed. (D) Infected cells with abnormal vacuoles were counted by IF microscopy. (E) HeLa cells were imaged by confocal microscopy for LAMP1-GFP (green) and Salmonella expressing DsRed (red). All values are the means ± standard errors of the means (n = 3). At least 50 infected cells were analyzed in each experiment. P values were calculated by the Student t test relative to wild-type-infected cells. Bars, 5 μm.
Overall, these data indicate that SteA is functionally linked to SseF and SseG, as its activity is required for scattering of Δ_sseF_ or Δ_sseG_ mutant vacuoles. Assuming that the scattered distribution of Δ_sseF_ or Δ_sseG_ mutant vacuoles is at least partially caused by an imbalance in the activity of microtubule motors, the results also suggested that SteA could contribute to activation of a microtubule plus-end-directed kinesin or to inhibition of microtubule minus-end-directed dynein on the SCV. Each of these possible functions of SteA would be expected to lead to the increased clustering of Δ_steA_ mutant SCVs. As the SPI-2 effector PipB2 recruits kinesin-1 to the SCV (27), we thought that if SteA contributes to the activation of kinesin-1 on the SCV, then a Δ_pipB2_ Δ_sseF_ mutant could lead to phenotypes in infected cells similar to those of Δ_steA_ Δ_sseF_ or Δ_steA_ Δ_sseG_ mutants. To analyze this, we compared the SCV positioning and the morphology of the microcolonies in HeLa cells infected by a Δ_pipB2_ Δ_sseF_ double mutant with those of the microcolonies in HeLa cells infected by the corresponding single mutants or by wt S. Typhimurium (Fig. 6A to C). Like Δ_steA_ mutant vacuoles, Δ_pipB2_ mutant SCVs appeared more frequently in compact microcolonies than wt SCVs (Fig. 6A). However, the appearance of compact microcolonies was less frequent in cells infected by Δ_pipB2_ mutant S. Typhimurium (∼20%) than in cells infected by Δ_steA_ mutant S. Typhimurium (∼50%) (Fig. 6A). Furthermore, unlike Δ_steA_ Δ_sseF_ or Δ_steA_ Δ_sseG_ double mutant vacuoles, the SCVs of the Δ_pipB2_ Δ_sseF_ double mutant showed a positioning at the Golgi region and a frequency of appearance of compact microcolonies that were similar to those of Δ_sseF_ mutant SCVs (Fig. 6A to C). These results suggest that if SteA contributes to the activity of a kinesin on the SCV, this kinesin should be different from kinesin-1.
DISCUSSION
In this work, we show that SteA contributes to the control of the dynamics of the vacuolar membrane enclosing S. Typhimurium during bacterial intracellular replication in HeLa cells or in RAW 264.7 macrophages. Taking into account what is known about other Salmonella effectors that control SCV membrane dynamics (1, 3, 25–28, 31), the phenotypes displayed in infected HeLa cells by vacuoles of Δ_steA_ mutant S. Typhimurium (fewer SIFs, compact microcolonies, and vacuoles containing more than one bacterium) suggest that the activity of microtubule motors on the SCV is defective.
To analyze if SteA controls the activity of microtubule motors on the SCV, we examined how phenotypes displayed in HeLa cells infected by Δ_steA_ mutant S. Typhimurium were affected by inhibition of dynein (by expression of p50-dynamitin) or kinesin-1 (by expression of TPR-KLC2) and how deletion of either steA or pipB2 from the chromosome of Δ_sseF_ and Δ_sseG_ mutants affected the characteristic scattered distribution of their vacuoles in infected HeLa cells (31, 32, 34). These experiments suggested that SteA might inhibit dynein or contribute to the activity of a kinesin other than kinesin-1. First, inhibition of dynein reduced the frequency at which Δ_steA_ mutant SCVs formed compact microcolonies and contained more than one bacterium. Second, Δ_steA_ Δ_sseF_ or Δ_steA_ Δ_sseG_ mutant SCVs were not as scattered as Δ_sseF_ or Δ_sseG_ mutant vacuoles. Therefore, assuming that SseF and SseG promote the recruitment of dynein to the SCV (31), if SteA inhibits the activity of this motor, the reduced amount of dynein that might exert its activity on Δ_steA_ Δ_sseF_ or Δ_steA_ Δ_sseG_ mutant vacuoles would be compensated for by the lack of SteA; if SteA contributes to the activity of a microtubule plus-end-directed kinesin, this would prevent scattering of Δ_steA_ Δ_sseF_ or Δ_steA_ Δ_sseG_ mutant vacuoles. Third, as PipB2 recruits kinesin-1 to the SCV (27) and Δ_pipB2_ Δ_sseF_ mutant SCVs were as scattered as Δ_sseF_ mutant vacuoles, this suggested that if SteA contributes to the activity of a kinesin, then it should be different from kinesin-1. However, some of the data gathered in these experiments were apparently contradictory. In particular, inhibition of kinesin-1 had the same consequences as inhibition of dynein on the phenotypes displayed by Δ_steA_ mutant SCVs. It is counterintuitive that inhibition of a microtubule plus-end-directed motor would result in the appearance of less compact microcolonies by Δ_steA_ mutant S. Typhimurium. It is possible that inhibition of kinesin-1 has pleiotropic effects on intracellular trafficking in such a way that indirectly disturbs recruitment of dynein or of other kinesins to the SCV and/or the control of their activity. Furthermore, the consequences of the expression of p50-dynamitin or TPR-KLC2 on the morphology and positioning of wt SCVs that we observed were less evident than what has been previously reported (31, 50). This may be related to the fact that to ensure proper SCV maturation and migration, we transfected the cells only with the plasmids encoding p50-dynamitin or TPR-KLC2 after bacterial invasion and accumulation of bacterial vacuoles in the Golgi region had occurred (2 h after bacterial inoculation [34]). Finally, although Δ_steA_ Δ_sseF_ or Δ_steA_ Δ_sseG_ mutant SCVs formed compact microcolonies more frequently than wt vacuoles, they were not clustered at the Golgi region. A possible explanation for this is that SCV positioning is not solely controlled through a balanced activity of microtubule motors, in agreement with the hypothesis that SseF and SseG may function by directly tethering SCVs to Golgi region components (17, 34).
A particular characteristic of Δ_steA_ mutant vacuoles is that in infected HeLa or RAW 264.7 cells, they appeared to contain more than a single bacterium. This is in contrast to wt vacuoles, which even after several rounds of bacterial replication normally contain a single bacterium (50, 51). This further supports the notion that SteA controls SCV membrane dynamics and suggests that SteA could be directly involved in the partitioning of the vacuolar membrane as a bacterium divides. Confirmation of this will require correlative light electron microscopy to obtain a detailed ultrastructural characterization of Δ_steA_ mutant vacuoles in different types of cells and at distinct times of infection. In previous studies, inhibition of dynein with p50-dynamitin followed by infection with wt S. Typhimurium led to the appearance of SCVs also apparently containing more than one bacterium (50). To some extent, we obtained similar results by inhibiting dynein (or kinesin), but the differences relative to the results for untreated cells were not statistically significant. Again, this may be explained by the different timings of expression of p50-dynamitin.
In spite of the role of SteA in the control of SCV membrane dynamics, Δ_steA_ mutant S. Typhimurium did not show an intracellular replication defect in mouse macrophages (see Fig. S1 in the supplemental material) (22). We thought that a possible role of SteA in the intracellular multiplication of Salmonella may be detectable only in the context of polyeffector mutants. However, SteA was also not required for the intracellular growth of S. Typhimurium when considering relevant effector double mutant strains (Δ_steA_ Δ_pipB2_, Δ_steA_ Δ_sifA_, Δ_steA_ Δ_sseF_, Δ_steA_ Δ_sseG_, Δ_steA_ Δ_sseJ_, or Δ_steA_ Δ_sopD2_ mutant strains; data not shown). With the exception of the Δ_pipB2_ and Δ_steA_ mutations, null mutations in each of the other effector genes expressing proteins involved in the control of SCV membrane dynamics (SifA, SopD2, SseF, SseG, and SseJ) result in significant intracellular replication defects in mouse macrophages (22). This is in contrast to the findings for the majority of S. Typhimurium mutant strains lacking individual SPI-2 effectors, which display no intramacrophage replication defect (22). As in the case of SteA, the absence of PipB2 also results in detectable phenotypes in infected HeLa cells related to SCV membrane dynamics, such as shorter SIFs (35) or a lack of accumulation of kinesin-1 in the vacuoles of Δ_sifA_ mutant S. Typhimurium (27). S. Typhimurium translocates at least 28 effectors across the SCV membrane (3), and the function of only about half of these effectors is known. It is therefore possible that a role of SteA or PipB2 in the intracellular replication of Salmonella in macrophages can be detected only using specific polyeffector mutants, which, because of a lack of current knowledge, are not yet possible to rationally design. Another possibility is that the levels of translocation of the different effectors might vary among cell types (60), resulting in detectable phenotypes in some cell types but not in others. Regardless of the exact explanation(s), additional studies are needed to understand how the function of SteA relates to the virulence defects displayed by Δ_steA_ mutant S. Typhimurium (36, 61).
SteA can be translocated by both the SPI-1 and SPI-2 T3SSs (36). Specifically, 1 h after Salmonella invasion of macrophages, the translocation of SteA was shown to depend on the SPI-1 T3SS, while 8 h after bacterial uptake by macrophages, the translocation of SteA was shown to depend on the SPI-2 T3SS (37). In a more recent study, it was shown that SteA could also be translocated into HeLa cells by the SPI-2 T3SS at early times of infection and by the SPI-1 T3SS at later times of infection (37). These apparent discrepancies could be explained by whether prior to infection the bacteria were grown or not under conditions that promote expression of the SPI-1 T3SS or the use of different cell types (62). Regardless of the exact T3SS that translocates SteA at specific times of infection and the particular experimental conditions, we showed that the SteA-dependent defects in SCV membrane dynamics in HeLa cells were not caused by a lack of early translocation of the effector (before 4 h p.i.; Fig. 4). Moreover, we showed that the abnormal SCVs seen more frequently in RAW 264.7 macrophages infected by Δ_steA_ mutant S. Typhimurium than in macrophages infected by wt bacteria were due to a lack of translocation of SteA by the SPI-2 T3SS (Fig. 3C; see Fig. S3 in the supplemental material). This indicates that SteA participates in the control of SCV membrane dynamics after its translocation across the bacterial vacuolar membrane several hours after host cell invasion, when the bacteria are replicating intracellularly within the boundaries of the vacuole. However, in agreement with previous studies (37) using a chromosomally encoded SteA-3× FLAG fusion, we could detect the expression and translocation of SteA in infected HeLa cells at 2 h p.i. (data not shown). Overall, this suggests that while SteA has a specific role during the intravacuolar replication of Salmonella, it should have an additional role(s), which remains to be elucidated, at earlier times of host cell infection.
Among Salmonella effectors translocated by the SPI-2 T3SS, PipB2, SifA, SopD2, SseF, SseG, SseJ, and, as we now show, SteA control SCV membrane dynamics, i.e., maintain the integrity of the vacuolar membrane and/or mediate the formation of Salmonella_-induced tubules, such as SIFs. The role of all these effector proteins has been directly or indirectly related to manipulation of dynein and kinesin-1. As mentioned before, dynein could be recruited to the SCV by SseF and SseG (31) and kinesin-1 is recruited to the SCV by PipB2 (27). In turn, SifA interacts with SKIP/PLEKHM2 (25), and the SifA-SKIP/PLEKHM2 complex binds and possibly activates kinesin-1 on the SCV, leading to vesicle fission and tubulation (25, 26). In addition, the activities of SseJ, which is a glycerophospholipid:cholesterol acyltransferase (63, 64), and of SopD2, which has an unknown host cell target and biochemical activity, destabilize the vacuolar membrane enclosing Δ_sifA mutant S. Typhimurium (28). As rupture of the vacuolar membrane of a Δ_sifA_ mutant can be suppressed by inhibiting kinesin-1 or dynein (25, 50), it is possible that the activity of SseJ and SopD2 is also directly related to the control of these microtubule motors. However, the SCV membrane of the Δ_sifA_ Δ_pipB2_, Δ_sifA_ Δ_sseF_, or Δ_sifA_ Δ_sseG_ double mutant is as unstable as the one enclosing Δ_sifA_ mutant bacteria (27, 28). It has been speculated that residual kinesin activity or dynein activity on the SCV would be responsible for the loss of the vacuolar membrane in the Δ_sifA_ Δ_pipB2_ mutant (27). Given that our results suggest that SteA could inhibit dynein or activate a kinesin, we analyzed the stability of the SCV membrane of Δ_sifA_ Δ_steA_ and of Δ_sifA_ Δ_pipB2_ Δ_steA_ mutant strains. However, the vacuolar membrane enclosing Δ_sifA_ Δ_steA_ or Δ_sifA_ Δ_pipB2_ Δ_steA S_. Typhimurium was as unstable as that of Δ_sifA_ mutant vacuoles (data not shown). Instead, we found that the activity of SteA is necessary for scattering of Δ_sseF_ and Δ_sseG_ mutant SCVs, thereby suggesting a functional link between SteA and the SseF-SseG complex (33). In contrast, we showed that the activity of the kinesin-1 linker PipB2 effector (27) is not involved in the scattering of Δ_sseF_ and Δ_sseG_ mutant SCVs. Overall, this suggests that control of SCV membrane dynamics might be defined by two groups of functionally linked effectors, PipB2-SifA-SopD2-SseJ and SseF-SseG-SteA.
Supplementary Material
Supplemental material
ACKNOWLEDGMENTS
This work was supported by the Fundação para a Ciência e a Tecnologia (FCT) through grants PTDC/BIA-MIC/116780/2010 and PEst-OE/BIA/UI0457/2011. L.D. holds a Ph.D. fellowship (SFRH/BD/62587/2009) from FCT.
We are grateful to Serge Benichou, Stéphane Méresse, Patrice Boquet, and Michael Way for gifts of plasmids, to António J. Santos for help and expertise in time-lapse microscopy, and to Mei Liu for help in the construction of plasmids. We thank Irina Franco for a critical review of the manuscript.
Footnotes
Published ahead of print 28 April 2014
REFERENCES
- 1.Haraga A, Ohlson MB, Miller SI. 2008. Salmonellae interplay with host cells. Nat. Rev. Microbiol. 6:53–66. 10.1038/nrmicro1788 [DOI] [PubMed] [Google Scholar]
- 2.Agbor TA, McCormick BA. 2011. Salmonella effectors: important players modulating host cell function during infection. Cell. Microbiol. 13:1858–1869. 10.1111/j.1462-5822.2011.01701.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Figueira R, Holden DW. 2012. Functions of the Salmonella pathogenicity island 2 (SPI-2) type III secretion system effectors. Microbiology 158:1147–1161. 10.1099/mic.0.058115-0 [DOI] [PubMed] [Google Scholar]
- 4.Patel JC, Galan JE. 2005. Manipulation of the host actin cytoskeleton by Salmonella—all in the name of entry. Curr. Opin. Microbiol. 8:10–15. 10.1016/j.mib.2004.09.001 [DOI] [PubMed] [Google Scholar]
- 5.Galan JE, Curtiss R., III 1989. Cloning and molecular characterization of genes whose products allow Salmonella typhimurium to penetrate tissue culture cells. Proc. Natl. Acad. Sci. U. S. A. 86:6383–6387. 10.1073/pnas.86.16.6383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Layton AN, Galyov EE. 2007. Salmonella-induced enteritis: molecular pathogenesis and therapeutic implications. Expert Rev. Mol. Med. 9:1–17. 10.1017/S1462399407000373 [DOI] [PubMed] [Google Scholar]
- 7.Ochman H, Soncini FC, Solomon F, Groisman EA. 1996. Identification of a pathogenicity island required for Salmonella survival in host cells. Proc. Natl. Acad. Sci. U. S. A. 93:7800–7804. 10.1073/pnas.93.15.7800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cirillo DM, Valdivia RH, Monack DM, Falkow S. 1998. Macrophage-dependent induction of the Salmonella pathogenicity island 2 type III secretion system and its role in intracellular survival. Mol. Microbiol. 30:175–188. 10.1046/j.1365-2958.1998.01048.x [DOI] [PubMed] [Google Scholar]
- 9.Hensel M, Shea JE, Waterman SR, Mundy R, Nikolaus T, Banks G, Vazquez-Torres A, Gleeson C, Fang FC, Holden DW. 1998. Genes encoding putative effector proteins of the type III secretion system of Salmonella pathogenicity island 2 are required for bacterial virulence and proliferation in macrophages. Mol. Microbiol. 30:163–174. 10.1046/j.1365-2958.1998.01047.x [DOI] [PubMed] [Google Scholar]
- 10.Steele-Mortimer O, Brumell JH, Knodler LA, Meresse S, Lopez A, Finlay BB. 2002. The invasion-associated type III secretion system of Salmonella enterica serovar Typhimurium is necessary for intracellular proliferation and vacuole biogenesis in epithelial cells. Cell. Microbiol. 4:43–54. 10.1046/j.1462-5822.2002.00170.x [DOI] [PubMed] [Google Scholar]
- 11.Brawn LC, Hayward RD, Koronakis V. 2007. Salmonella SPI-1 effector SipA persists after entry and cooperates with a SPI2 effector to regulate phagosome maturation and intracellular replication. Cell Host Microbe 1:63–75. 10.1016/j.chom.2007.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hernandez LD, Hueffer K, Wenk MR, Galan JE. 2004. Salmonella modulates vesicular traffic by altering phosphoinositide metabolism. Science 304:1805–1807. 10.1126/science.1098188 [DOI] [PubMed] [Google Scholar]
- 13.Hapfelmeier S, Hardt WD. 2005. A mouse model for S. typhimurium-induced enterocolitis. Trends Microbiol. 13:497–503. 10.1016/j.tim.2005.08.008 [DOI] [PubMed] [Google Scholar]
- 14.Ramos-Morales F. 2012. Impact of Salmonella enterica type III secretion system effectors on the eukaryotic host cell. ISRN Cell Biol. 2012:787934. 10.5402/2012/787934 [DOI] [Google Scholar]
- 15.Steele-Mortimer O. 2008. The Salmonella-containing vacuole: moving with the times. Curr. Opin. Microbiol. 11:38–45. 10.1016/j.mib.2008.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lober S, Jackel D, Kaiser N, Hensel M. 2006. Regulation of Salmonella pathogenicity island 2 genes by independent environmental signals. Int. J. Med. Microbiol. 296:435–447. 10.1016/j.ijmm.2006.05.001 [DOI] [PubMed] [Google Scholar]
- 17.Ramsden AE, Holden DW, Mota LJ. 2007. Membrane dynamics and spatial distribution of Salmonella-containing vacuoles. Trends Microbiol. 15:516–524. 10.1016/j.tim.2007.10.002 [DOI] [PubMed] [Google Scholar]
- 18.Knodler LA, Vallance BA, Hensel M, Jackel D, Finlay BB, Steele-Mortimer O. 2003. Salmonella type III effectors PipB and PipB2 are targeted to detergent-resistant microdomains on internal host cell membranes. Mol. Microbiol. 49:685–704. 10.1046/j.1365-2958.2003.03598.x [DOI] [PubMed] [Google Scholar]
- 19.Beuzón CR, Meresse S, Unsworth KE, Ruiz-Albert J, Garvis S, Waterman SR, Ryder TA, Boucrot E, Holden DW. 2000. Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J. 19:3235–3249. 10.1093/emboj/19.13.3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brumell JH, Kujat-Choy S, Brown NF, Vallance BA, Knodler LA, Finlay BB. 2003. SopD2 is a novel type III secreted effector of Salmonella typhimurium that targets late endocytic compartments upon delivery into host cells. Traffic 4:36–48. 10.1034/j.1600-0854.2003.40106.x [DOI] [PubMed] [Google Scholar]
- 21.Ruiz-Albert J, Yu XJ, Beuzon CR, Blakey AN, Galyov EE, Holden DW. 2002. Complementary activities of SseJ and SifA regulate dynamics of the Salmonella typhimurium vacuolar membrane. Mol. Microbiol. 44:645–661. 10.1046/j.1365-2958.2002.02912.x [DOI] [PubMed] [Google Scholar]
- 22.Figueira R, Watson KG, Holden DW, Helaine S. 2013. Identification of Salmonella pathogenicity island-2 type III secretion system effectors involved in intramacrophage replication of S. enterica serovar Typhimurium: implications for rational vaccine design. mBio 4(2):e00065. 10.1128/mBio.00065-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stein MA, Leung KY, Zwick M, Garcia-del Portillo F, Finlay BB. 1996. Identification of a Salmonella virulence gene required for formation of filamentous structures containing lysosomal membrane glycoproteins within epithelial cells. Mol. Microbiol. 20:151–164. 10.1111/j.1365-2958.1996.tb02497.x [DOI] [PubMed] [Google Scholar]
- 24.McGourty K, Thurston TL, Matthews SA, Pinaud L, Mota LJ, Holden DW. 2012. Salmonella inhibits retrograde trafficking of mannose-6-phosphate receptors and lysosome function. Science 338:963–967. 10.1126/science.1227037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boucrot E, Henry T, Borg JP, Gorvel JP, Meresse S. 2005. The intracellular fate of Salmonella depends on the recruitment of kinesin. Science 308:1174–1178. 10.1126/science.1110225 [DOI] [PubMed] [Google Scholar]
- 26.Dumont A, Boucrot E, Drevensek S, Daire V, Gorvel JP, Pous C, Holden DW, Meresse S. 2010. SKIP, the host target of the Salmonella virulence factor SifA, promotes kinesin-1-dependent vacuolar membrane exchanges. Traffic 11:899–911. 10.1111/j.1600-0854.2010.01069.x [DOI] [PubMed] [Google Scholar]
- 27.Henry T, Couillault C, Rockenfeller P, Boucrot E, Dumont A, Schroeder N, Hermant A, Knodler LA, Lecine P, Steele-Mortimer O, Borg JP, Gorvel JP, Meresse S. 2006. The Salmonella effector protein PipB2 is a linker for kinesin-1. Proc. Natl. Acad. Sci. U. S. A. 103:13497–13502. 10.1073/pnas.0605443103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schroeder N, Henry T, de Chastellier C, Zhao W, Guilhon AA, Gorvel JP, Meresse S. 2010. The virulence protein SopD2 regulates membrane dynamics of Salmonella-containing vacuoles. PLoS Pathog. 6:e1001002. 10.1371/journal.ppat.1001002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohlson MB, Huang Z, Alto NM, Blanc MP, Dixon JE, Chai J, Miller SI. 2008. Structure and function of Salmonella SifA indicate that its interactions with SKIP, SseJ, and RhoA family GTPases induce endosomal tubulation. Cell Host Microbe 4:434–446. 10.1016/j.chom.2008.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salcedo SP, Holden DW. 2003. SseG, a virulence protein that targets Salmonella to the Golgi network. EMBO J. 22:5003–5014. 10.1093/emboj/cdg517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abrahams GL, Muller P, Hensel M. 2006. Functional dissection of SseF, a type III effector protein involved in positioning the Salmonella-containing vacuole. Traffic 7:950–965. 10.1111/j.1600-0854.2006.00454.x [DOI] [PubMed] [Google Scholar]
- 32.Kuhle V, Hensel M. 2002. SseF and SseG are translocated effectors of the type III secretion system of Salmonella pathogenicity island 2 that modulate aggregation of endosomal compartments. Cell. Microbiol. 4:813–824. 10.1046/j.1462-5822.2002.00234.x [DOI] [PubMed] [Google Scholar]
- 33.Deiwick J, Salcedo SP, Boucrot E, Gilliland SM, Henry T, Petermann N, Waterman SR, Gorvel JP, Holden DW, Meresse S. 2006. The translocated Salmonella effector proteins SseF and SseG interact and are required to establish an intracellular replication niche. Infect. Immun. 74:6965–6972. 10.1128/IAI.00648-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramsden AE, Mota LJ, Munter S, Shorte SL, Holden DW. 2007. The SPI-2 type III secretion system restricts motility of Salmonella-containing vacuoles. Cell. Microbiol. 9:2517–2519. 10.1111/j.1462-5822.2007.00977.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knodler LA, Steele-Mortimer O. 2005. The Salmonella effector PipB2 affects late endosome/lysosome distribution to mediate Sif extension. Mol. Biol. Cell 16:4108–4123. 10.1091/mbc.E05-04-0367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geddes K, Worley M, Niemann G, Heffron F. 2005. Identification of new secreted effectors in Salmonella enterica serovar Typhimurium. Infect. Immun. 73:6260–6271. 10.1128/IAI.73.10.6260-6271.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cardenal-Munoz E, Ramos-Morales F. 2011. Analysis of the expression, secretion and translocation of the Salmonella enterica type III secretion system effector SteA. PLoS One 6:e26930. 10.1371/journal.pone.0026930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Engelenburg SB, Palmer AE. 2010. Imaging type-III secretion reveals dynamics and spatial segregation of Salmonella effectors. Nat. Methods 7:325–330. 10.1038/nmeth.1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645. 10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang RF, Kushner SR. 1991. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100:195–199. 10.1016/0378-1119(91)90366-J [DOI] [PubMed] [Google Scholar]
- 41.Rajashekar R, Liebl D, Seitz A, Hensel M. 2008. Dynamic remodeling of the endosomal system during formation of Salmonella-induced filaments by intracellular Salmonella enterica. Traffic 9:2100–2116. 10.1111/j.1600-0854.2008.00821.x [DOI] [PubMed] [Google Scholar]
- 42.Falcon-Perez JM, Nazarian R, Sabatti C, Dell'Angelica EC. 2005. Distribution and dynamics of Lamp1-containing endocytic organelles in fibroblasts deficient in BLOC-3. J. Cell Sci. 118:5243–5255. 10.1242/jcs.02633 [DOI] [PubMed] [Google Scholar]
- 43.Jacquot G, Maidou-Peindara P, Benichou S. 2010. Molecular and functional basis for the scaffolding role of the p50/dynamitin subunit of the microtubule-associated dynactin complex. J. Biol. Chem. 285:23019–23031. 10.1074/jbc.M110.100602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rietdorf J, Ploubidou A, Reckmann I, Holmstrom A, Frischknecht F, Zettl M, Zimmermann T, Way M. 2001. Kinesin-dependent movement on microtubules precedes actin-based motility of vaccinia virus. Nat. Cell Biol. 3:992–1000. 10.1038/ncb1101-992 [DOI] [PubMed] [Google Scholar]
- 45.Sorensen M, Lippuner C, Kaiser T, Misslitz A, Aebischer T, Bumann D. 2003. Rapidly maturing red fluorescent protein variants with strongly enhanced brightness in bacteria. FEBS Lett. 552:110–114. 10.1016/S0014-5793(03)00856-1 [DOI] [PubMed] [Google Scholar]
- 46.Mota LJ, Ramsden AE, Liu M, Castle JD, Holden DW. 2009. SCAMP3 is a component of the Salmonella-induced tubular network and reveals an interaction between bacterial effectors and post-Golgi trafficking. Cell. Microbiol. 11:1236–1253. 10.1111/j.1462-5822.2009.01329.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schroeder N, Mota LJ, Meresse S. 2011. Salmonella-induced tubular networks. Trends Microbiol. 19:268–277. 10.1016/j.tim.2011.01.006 [DOI] [PubMed] [Google Scholar]
- 48.Valdivia RH, Falkow S. 1997. Fluorescence-based isolation of bacterial genes expressed within host cells. Science 277:2007–2011. 10.1126/science.277.5334.2007 [DOI] [PubMed] [Google Scholar]
- 49.Freeman JA, Ohl ME, Miller SI. 2003. The Salmonella enterica serovar Typhimurium translocated effectors SseJ and SifB are targeted to the Salmonella-containing vacuole. Infect. Immun. 71:418–427. 10.1128/IAI.71.1.418-427.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guignot J, Caron E, Beuzon C, Bucci C, Kagan J, Roy C, Holden DW. 2004. Microtubule motors control membrane dynamics of Salmonella-containing vacuoles. J. Cell Sci. 117:1033–1045. 10.1242/jcs.00949 [DOI] [PubMed] [Google Scholar]
- 51.Eswarappa SM, Negi VD, Chakraborty S, Chandrasekhar Sagar BK, Chakravortty D. 2010. Division of the Salmonella-containing vacuole and depletion of acidic lysosomes in Salmonella-infected host cells are novel strategies of Salmonella enterica to avoid lysosomes. Infect. Immun. 78:68–79. 10.1128/IAI.00668-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salcedo SP, Noursadeghi M, Cohen J, Holden DW. 2001. Intracellular replication of Salmonella typhimurium strains in specific subsets of splenic macrophages in vivo. Cell. Microbiol. 3:587–597. 10.1046/j.1462-5822.2001.00137.x [DOI] [PubMed] [Google Scholar]
- 53.Kaniga K, Bossio JC, Galan JE. 1994. The Salmonella typhimurium invasion genes invF and invG encode homologues of the AraC and PulD family of proteins. Mol. Microbiol. 13:555–568. 10.1111/j.1365-2958.1994.tb00450.x [DOI] [PubMed] [Google Scholar]
- 54.Harrison RE, Brumell JH, Khandani A, Bucci C, Scott CC, Jiang X, Finlay BB, Grinstein S. 2004. Salmonella impairs RILP recruitment to Rab7 during maturation of invasion vacuoles. Mol. Biol. Cell 15:3146–3154. 10.1091/mbc.E04-02-0092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marsman M, Jordens I, Kuijl C, Janssen L, Neefjes J. 2004. Dynein-mediated vesicle transport controls intracellular Salmonella replication. Mol. Biol. Cell 15:2954–2964. 10.1091/mbc.E03-08-0614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kaniuk NA, Canadien V, Bagshaw RD, Bakowski M, Braun V, Landekic M, Mitra S, Huang J, Heo WD, Meyer T, Pelletier L, Andrews-Polymenis H, McClelland M, Pawson T, Grinstein S, Brumell JH. 2011. Salmonella exploits Arl8B-directed kinesin activity to promote endosome tubulation and cell-to-cell transfer. Cell. Microbiol. 13:1812–1823. 10.1111/j.1462-5822.2011.01663.x [DOI] [PubMed] [Google Scholar]
- 57.McGhie EJ, Brawn LC, Hume PJ, Humphreys D, Koronakis V. 2009. Salmonella takes control: effector-driven manipulation of the host. Curr. Opin. Microbiol. 12:117–124. 10.1016/j.mib.2008.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Drecktrah D, Levine-Wilkinson S, Dam T, Winfree S, Knodler LA, Schroer TA, Steele-Mortimer O. 2008. Dynamic behavior of Salmonella-induced membrane tubules in epithelial cells. Traffic 9:2117–2129. 10.1111/j.1600-0854.2008.00830.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Echeverri CJ, Paschal BM, Vaughan KT, Vallee RB. 1996. Molecular characterization of the 50-kD subunit of dynactin reveals function for the complex in chromosome alignment and spindle organization during mitosis. J. Cell Biol. 132:617–633. 10.1083/jcb.132.4.617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nunez-Hernandez C, Alonso A, Pucciarelli MG, Casadesus J, Garcia-del Portillo F. 2014. Dormant intracellular Salmonella enterica serovar Typhimurium discriminates among Salmonella pathogenicity island 2 effectors to persist inside fibroblasts. Infect. Immun. 82:221–232. 10.1128/IAI.01304-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lawley TD, Chan K, Thompson LJ, Kim CC, Govoni GR, Monack DM. 2006. Genome-wide screen for Salmonella genes required for long-term systemic infection of the mouse. PLoS Pathog. 2:e11. 10.1371/journal.ppat.0020011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hautefort I, Thompson A, Eriksson-Ygberg S, Parker ML, Lucchini S, Danino V, Bongaerts RJ, Ahmad N, Rhen M, Hinton JC. 2008. During infection of epithelial cells Salmonella enterica serovar Typhimurium undergoes a time-dependent transcriptional adaptation that results in simultaneous expression of three type 3 secretion systems. Cell. Microbiol. 10:958–984. 10.1111/j.1462-5822.2007.01099.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lossi NS, Rolhion N, Magee AI, Boyle C, Holden DW. 2008. The Salmonella SPI-2 effector SseJ exhibits eukaryotic activator-dependent phospholipase A and glycerophospholipid: cholesterol acyltransferase activity. Microbiology 154:2680–2688. 10.1099/mic.0.2008/019075-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nawabi P, Catron DM, Haldar K. 2008. Esterification of cholesterol by a type III secretion effector during intracellular Salmonella infection. Mol. Microbiol. 68:173–185. 10.1111/j.1365-2958.2008.06142.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material