Glutaredoxin 2 Reduces Both Thioredoxin 2 and Thioredoxin 1 and Protects Cells from Apoptosis Induced by Auranofin and 4-Hydroxynonenal (original) (raw)
Abstract
Aims: Mitochondrial thioredoxin (Trx) is critical for defense against oxidative stress-induced cell apoptosis. To date, mitochondrial thioredoxin reductase (TrxR) is the only known enzyme catalyzing Trx2 reduction in mitochondria. However, TrxR is sensitive to inactivation by exo/endogenous electrophiles, for example, 4-hydroxynonenal (HNE). In this study, we characterized the mitochondrial glutaredoxin 2 (Grx2) system as a backup for the mitochondrial TrxR. Meanwhile, as Grx2 is also present in the cytosol/nucleus of certain cancer cell lines, the reducing activity of Grx2 on Trx1 was also tested. Results: Glutathione alone could reduce oxidized Trx2, and the presence of physiological concentrations of Grx2 markedly increased the reaction rate. HeLa cells with Grx2 overexpression (particularly in the mitochondria) exhibited higher viabilities than the wild-type cells after treatment with TrxR inhibitors (Auranofin or HNE), whereas knockdown of Grx2 sensitized the cells to TrxR inhibitors. Accordingly, Grx2 overexpression in the mitochondria had protected Trx2 from oxidation by HNE treatment, whereas Grx2 knockdown had sensitized Trx2 to oxidation. On the other hand, Grx2 reduced Trx1 with similar activities as that of Trx2. Overexpression of Grx2 in the cytosol had protected Trx1 from oxidation, indicating a supportive role of Grx2 in the cytosolic redox balance of cancer cells. Innovation: This work explores the reductase activity of Grx2 on Trx2/1, and demonstrates the physiological importance of the activity by using in vivo redox western blot assays. Conclusion: Grx2 system could help to keep Trx2/1 reduced during an oxidative stress, thereby contributing to the anti-apoptotic signaling. Antioxid. Redox Signal. 21, 669–681.
Introduction
The thioredoxins (Trxs) and glutaredoxins (Grxs) belong to the Trx superfamily of thiol–disulfide oxidoreductases (25). Each redoxin is the electron receptor of their redox system, and the effector of the downstream redox regulations. In mammalian cells, the cytosolic Trx system composed of nicotinamide adenine dinucleotide phosphate (NADPH), thioredoxin reductase 1 (TrxR1), and Trx1, whereas the Grx system is formed by NADPH, glutathione reductase (GR), glutathione (GSH), and Grx1. Relatively independent of the cytosolic milieu, the mitochondria have their own Trx and Grx systems, consisting of NADPH, TrxR2 (22), and Trx2 (38) or NADPH, GR, GSH, and Grx2 (28), respectively.
Innovation.
In mammalian cells, the thioredoxins (Trxs) need to be in the reduced state to have antioxidant and anti-apoptotic effects. This study suggests that glutaredoxin 2 (Grx2) could work as a backup for thioredoxin reductase (TrxR) in keeping Trx reduced when cells are encountering exo/endogenous electrophiles, which are potent inhibitors of TrxR. Particularly in mitochondria, which are the main source of cellular reactive oxygen species, Grx2 here is the first and only hitherto found enzyme that can work as a backup for the reduction of Trx2. This finding may further explain the critical role of Grx2 in the redox signaling of cell survival/apoptosis.
The Trx and Grx systems play critical roles in providing reducing equivalent for DNA synthesis, maintaining cellular thiol-redox homeostasis, defense against oxidative stress, controlling protein folding, and regulation of cell growth/apoptosis (5, 6, 19, 23). Although they share some common functions in the above-mentioned processes, the two systems also differ in their selection of substrate groups and their affinity and activity to the substrates, thereby take effects in different regulatory checkpoints of the physiological pathways. The two systems may support each other's functions but could not substitute for each other, probably due to their respective unique functions.
In general, Trxs are more active in catalyzing the reduction of intra- or interchain disulfides of protein substrates. Trxs are specific and super-fast electron donor for peroxiredoxins (Prxs), which are one of the major players in the elimination of hydrogen peroxide (H2O2), both in the cytosol (Prx I, II) and in the midochondria (Prx III, V) (36). Trxs regulate the activity/activation of many important transcription factors [_e.g.,_ nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) (32), activator protein 1 (AP-1) (18)] or apoptosis signaling factors [_e.g.,_ apoptosis signal-regulating kinase 1 (ASK-1) (37) or phosphatase and tensin homolog (PTEN) (33)] through modulating the redox-regulatory disulfides of the protein. Reduced Trx is usually a factor and indicator of cell survival, whereas oxidized Trx being a factor and indicator of cell death. Knock out of either Trx1 or Trx2 will cause embryonic lethality in mice, and the fibroblasts from these mice are not viable (31, 34), indicating that both Trx1 and Trx2 are required to be functional for mammalian cell growth and survival.
Apart from the common functions with Trxs, Grxs are specifically active in catalyzing the (reversible) deglutathionylation of their protein substrates at the expense of GSH. Protein S-glutathionylation is an important reversible post-translational modification that works as thiol-redox signaling for many cellular events, including apoptosis (2). Grx2 was discovered in 2001 (28), with only 34% sequence identity with Grx1. Compared to Grx1, Grx2 has higher affinity toward the S-glutathionylated protein substrates, but with lower turnover rates (20). Grx2 is not inactivated by oxidation (28), exhibiting better resistance in an oxidative milieu. Grx2 has been found to catalyze the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins (3), responding to both mitochondrial redox signals and oxidative stress. Grx2 overexpression could prevent H2O2-induced apoptosis by protecting complex I activity in the mitochondria (42). Also, Grx2 could detoxify the mutant superoxide dismutase 1 (SOD1) in mitochondria by preventing its aggregation (14). HeLa cells with Grx2a overexpression are more resistant to the oxidative stress-inducing agents doxorubicin (Dox) and phenylarsine oxide due to reduced cytochrome c release (12), whereas siRNA-induced silencing of Grx2 sensitized HeLa cells to Dox treatment (26). These facts imply a critical role of mitochondrial Grx2 in cell survival/apoptosis.
Remarkably, cross talks of the Trx and Grx systems do exist and need to be clarified to have a better view of the redox regulation. Examples of the cross talking include the following: Trx was found to reduce GSSG, thereby having the potential to regenerate GSH in GR-deficient cells (21). Depletion of GSH by buthionine sulfoximine (BSO) results in Trx2 oxidation (45). A previous study in our laboratory revealed that Grx2 could use the reducing equivalent from TrxR (20). Later in our study, we found that Grx system could work as a backup for TrxR1 in the cytosol, keeping Trx1 reduced when TrxR1 were inhibited (11). However, it is still not known if there is any backup enzyme system that helps to keep Trx2 reduced in the mitochondria. In this study, we have analyzed the activities of Grx2 system to reduce Trx2, as well as Trx1, by which Grx2 system may work as a backup for TrxR. We have also applied Grx2 overexpression as well as knockdown in HeLa cells and challenged the cells with TrxR inhibitors and checked the cell viabilities and redox states of Trx2 and Trx1 in vivo, in searching of the physiological significance of this reducing activity of Grx2 on Trx2/1.
Results
Grx2 system as a reductant of Trx2
The active site oxidized hTrx2 (hTrx2-S2) was prepared by incubation with insulin and confirmed by redox urea-polyacrylamide gel electrophoresis (urea-PAGE) (Fig. 1A). The concentration of GSH in the mitochondrial matrix is in the millimolar range, and even up to 14 m_M_ considering the volume of the matrix (29). With Trx2-S2 as substrate, we have tested the reducing rates of a series of GSH concentrations on Trx2. As shown in Figure 1B, the reducing rate had an exponential increase as the concentration of GSH increases, consistent with the stoichiometry of the reaction (two molecules of GSH reduced one molecule of oxidized Trx2). To test the reductase activity of Grx2 on Trx2, we used GSH concentrations of 1 and 10 m_M_, respectively, and different concentrations of Grx2 to simulate the physiological levels of mitochondrial Grx system. Figure 1C shows the reaction curves at the GSH level of 1 m_M_. The addition of 0.05, 0.2, and 0.5 μ_M_ Grx2 resulted in a concentration-dependent increase of the reaction rate, with the activity of 0.2 and 0.5 μ_M_ Grx2 comparable to that of 0.8 and 1.1 n_M_ TrxR, respectively. Figure 1D shows the reaction curves under the GSH concentration of 10 m_M_. Ten micromolars of GSH alone gives a rate similar to that of 2 n_M_ TrxR. The addition of 0.05 and 0.2 μ_M_ Grx2 increased the rates to be comparable to the activities of 3.6 and 6.1 n_M_ TrxR, respectively.
FIG. 1.

The reduction of Trx2 by Grx2 system. (A) Trx2-S2 was prepared as described in the Materials and Methods section and confirmed by redox urea-PAGE. Lane 1: mobility standard. Lane 2: Trx2-S2. (B) Reduction of Trx2-S2 by GSH. For the GSH reduction assay, 6 μg/ml GR, 0.2 m_M_ NADPH, and 25 μ_M_ Trx2-S2 were added to cuvettes in the presence of 1, 3, 6, 10, and 14 m_M_ GSH. The absorbance at 340 nm was followed, and the linear part of the reactions was used to determine the reaction rates. (C) For the Grx2 reduction assay, 6 μg/ml GR, 0.2 m_M_ NADPH, 1 m_M_ GSH, 25 μ_M_ Trx2-S2, and increasing amounts of Grx2 (the solid lines thickening from bottom to top) were added to cuvettes. For the TrxR reduction assay, 0.2 m_M_ NADPH, 25 μ_M_ Trx2-S2, and indicated amounts of rrTrxR (the dotted lines) were added to cuvettes, the absorbance at 340 nm was followed, and the linear part of the reactions was used to determine the reaction rates. (D) Conditions were the same as that of (C), except that 10 m_M_ GSH was used in the Grx2 reduction assay. GR, glutathione reductase; Grx, glutaredoxin; rrTrxR, recombinant rat TrxR1; GSH, glutathione; NADPH, nicotinamide adenine dinucleotide phosphate; Trx, thioredoxin; TrxR, thioredoxin reductase; urea-PAGE, urea-polyacrylamide gel electrophoresis.
Grx2 system as a reductant of Trx1
Specifically within testis and many kinds of cancer cells, Grx2 presents not only in mitochondria but also in the cytosol (27). It is thus interesting to know if Grx2 could work as a reductase of Trx1. Trx1 has five cysteines (Cys), among them three are structural, the other two (Cys32 and Cys35) form the active site (16). The fully reduced Trx1, with all its five cysteines reduced, designated as Trx1-(SH)2, transform to the active site oxidized form (Trx1-S2) by reducing substrate protein disulfides. The Trx1-S2 was prepared as described in the Materials and Methods section and confirmed by redox urea-PAGE (Fig. 2A) and used as substrate for Grx2 system reduction assay. The reaction curves under the GSH concentration of 1 m_M_ are shown in Figure 2B. The addition of increasing concentrations of Grx2 resulted in increasing reaction rates. The activity of 0.2 μ_M_ Grx2 is comparable to that of 1 n_M_ TrxR, whereas the activity of 0.5 μ_M_ Grx2 is comparable to that of 2 n_M_ TrxR. Figure 2C shows the reaction curves under the GSH concentration of 10 m_M_. The activity of 10 m_M_ GSH alone is close to that of 3 n_M_ TrxR. The addition of 0.05 and 0.2 μ_M_ Grx2 increased the rates to be comparable to the activities of 4.7 and 7 n_M_ TrxR, respectively.
FIG. 2.
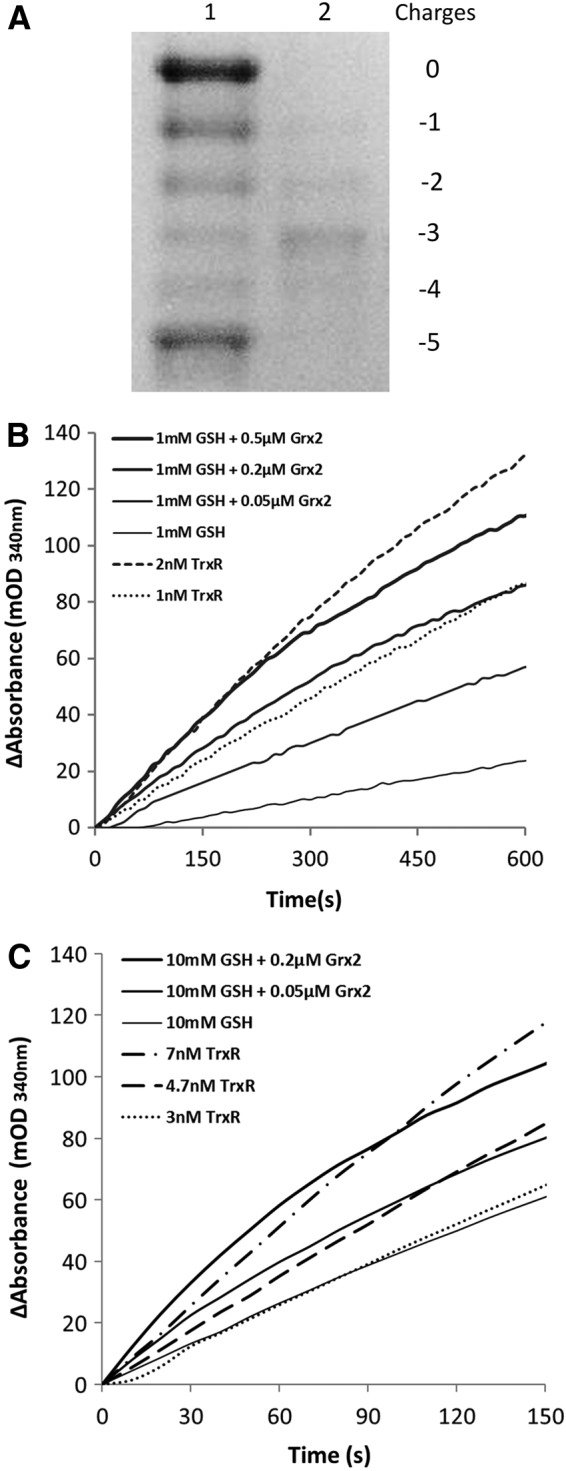
The reduction of Trx1 by Grx2 system. (A) Trx1-S2 was prepared as described in the Materials and Methods section and confirmed by redox urea-PAGE. Lane 1: mobility standard. Lane 2: Trx1-S2. (B) For the Grx2 reduction assay, 6 μg/ml GR, 0.2 m_M_ NADPH, 1 m_M_ GSH, 25 μ_M_ Trx1-S2, and increasing amounts of Grx2 (the solid lines thickening from bottom to top) were added to cuvettes. For the TrxR reduction assay, 0.2 m_M_ NADPH, 25 μ_M_ Trx1-S2, and indicated amounts of rrTrxR (the dotted lines) were added to cuvettes, the absorbance at 340 nm was followed, and the linear part of the reactions was used to determine the reaction rates. (C) Conditions were the same as that of (B), except that 10 m_M_ GSH were used in the Grx2 reduction assay.
Overexpression of Grx2 in HeLa cells provides protection against TrxR inhibitors
TrxR is the native reductant for Trxs, with very high catalytic efficiency. However, both the cytosolic and mitochondrial forms of TrxR possess a super active selenocysteine that is very sensitive to inactivation by electrophilic compounds (35, 44). Upon extensive inactivation of TrxR, Trx will not be reduced effectively, and increased levels of oxidized Trx will result in cell death. As we found that the Grx2 system could reduce Trx2 and Trx1, one would expect that the overexpression of Grx2 will rescue cells from TrxR inhibitors. Two Grx2 stable transfected HeLa cell lines were used, one with Grx2 overexpression in the mitochondria (M-Grx2 HeLa cells) and the other with Grx2 overexpression in the cytosol/nucleus (C-Grx2 HeLa cells). Here, we used auranofin (AF) and 4-hydroxynonenal (HNE) in the treatment of the three HeLa cell lines. AF is a well-known TrxR inhibitor, which is lipophilic and can permeate through biological membranes (10, 39). HNE is an end product of lipid peroxidation and a potent inhibitor of TrxR (13), and the treatment of cells with μ_M_ range of HNE induces both mitochondrial and cytosol oxidative stress (1). Figure 3A shows the viabilities of the cell lines treated with 4–12 μ_M_ AF for 3 h. The viability of wild-type (WT) HeLa cells decreased to 61%–41%, whereas C-Grx2 HeLa cells and especially M-Grx2 HeLa cells are less susceptible. Treatment of the three cell lines with 10–80 μ_M_ HNE (Fig. 3B) resulted in a similar profile as those treated with AF, with even higher resistance in M-Grx2 HeLa cells. Compared to WT cells, M-Grx2 HeLa cells have higher viability in all HNE concentrations tested. C-Grx2 HeLa cells showed significant resistance only in the higher HNE concentrations (40 μ_M_ and 80 μ_M_) and not as resistant as the mGrx2 HeLa cells. These results indicate that Grx2 overexpression either in the mitochondria or in the cytosol may protect cells from TrxR inhibitor. However, the rescuing effect was more profound with the overexpression of Grx2 in the mitochondria.
FIG. 3.

Effects of AF and HNE treatment on the viabilities of WT and Grx2 overexpressing HeLa cells. WT, M-Grx2, or C-Grx2 HeLa cells were treated with the indicated concentrations of AF for 3 h (A) or HNE for 20 h (B). After treatment, the cell viability was measured with the MTT assay. Error bars show mean±S.D. _n_=4. **p<0.01; *p<0.05, Student's _t_-test, treated cells versus control. AF, auranofin; HNE, 4-hydroxynonenal; WT, wild type.
Grx2 overexpression protects Trx2 and Trx1 from oxidation in HeLa cells
To detect the evidence that Grx2 reduces Trx2 and Trx1 in vivo, we analyzed the redox state of Trx2 and Trx1 in the HNE-treated WT, M-Grx2 and C-Grx2 HeLa cells, using a redox-state western blot analysis. Figure 4A shows the redox states of Trx2. The bands with −2 mobility represent fully reduced Trx2. Oxidation of 1 thiol by forming a mixed disulfide with a substrate shifts the band upwards to the −1 mobility, whereas the bands with 0 mobility represent the fully oxidized form of Trx2. Differences in the total amount of Trx were observed among the lanes. This could be resulted from the loss of total protein content during the experimental process. However, as the total protein amount does not affect the relative ratios of different redox forms within the sample, these results can be used to analyze the redox states of Trx. The fractions of different redox forms of Trx2 in each cell lysate are shown in Figure 4B. In all the three cell lines without treatment, about 60% of Trx2 was fully reduced. After treatment with 40 μ_M_ HNE for 20 h, the WT HeLa cells had about 50% of its Trx2 in the fully oxidized form, 23% in the one cysteine oxidized form, leaving only about 28% in the reduced form. On the contrary, the redox profile of Trx2 did not show an obvious change in the M-Grx2 cells, showing that overexpression of Grx2 in the mitochondria had prevented Trx2 from oxidation to a great extent. In the C-Grx2 cells treated with HNE, about half of Trx2 were oxidized, less than in the WT cells, indicating that the overexpression of Grx2 in the cytosol also has some effect in the prevention of Trx2 oxidation, probably by attenuating overall oxidative stress.
FIG. 4.

Redox state of cellular Trx2 before and after HNE treatment. (A) WT, M-Grx2, or C-Grx2 HeLa cells were treated with or without 40 μ_M_ HNE for 20 h, and the redox state of Trx2 was detected using a redox western blot analysis. (B) The intensity of each band in (A) was quantified by using the Quantity One software (Bio-Rad). For each cell lysate, the sum of intensity of bands with all three mobilities was set as 100%, and the fraction (%) of each band (redox form of Trx2) was calculated. Error bars show mean±S.D. based on two independent experiments.
Figure 5A shows the redox states of Trx1. Like Trx2, in all three cell lines of resting state, the fully reduced form represents the largest part of Trx1, whereas their responses to HNE treatment are very diverse: In the WT cells, treatment with HNE caused a dramatic decrease of reduced form of Trx1 and a great increase of the highly oxidized forms. Distinctively, the C-Grx2 cells only had a slight decrease of the reduced form and a small increase of the 2-SH form, indicating that Grx2 overexpression in the cytosol had protected Trx1 from oxidation. In the M-Grx2 cells treated with HNE, there was a small increase of the one cysteine oxidized form and the activate site disulfide form of Trx1, but not obvious increase of the highly oxidized forms.
FIG. 5.

Redox state of cellular Trx1 before and after HNE treatment. (A) WT, M-Grx2, or C-Grx2 HeLa cells were treated with or without 40 μ_M_ HNE for 20 h, and the redox state of Trx1 was detected using a redox western blot analysis. (B) The intensity of each band in (A) was quantified by using the Quantity One software (Bio-Rad). For each cell lysate, the sum of intensity of bands with all six mobilities was set as 100%, and the fraction (%) of each band (redox form of Trx1) was calculated. Error bars show mean±S.D. based on two independent experiments.
Grx2 knockdown causes decreased viability in TrxR inhibitor-treated cells
To further explore the in vivo evidence for the backup role of Grx2 for TrxR, we have used siRNAs specific for the mRNA of human Grx2 to knockdown Grx2 in the HeLa cells. After 2 days of transfection, the cells were assayed for Grx2 levels by ELISA. As shown in Figure 6A, the Grx2 siRNA was effective in decreasing Grx2 protein levels (to ∼10% of WT control). The transfection with a control of unspecific scrambled (scr) siRNA had no effect on Grx2 levels. The WT, scr-siRNA, and Grx2 siRNA-transfected cells were then subjected to the treatment with AF for 1.5 h or HNE for 20 h and assayed for cell viability. As shown in Figure 6B and C, the Grx2 knockdown cells showed a highly increased sensitivity to both AF and HNE.
FIG. 6.

Viability of WT, scr-siRNA-transfected, and Grx2 knockdown HeLa cells. (A) Levels of Grx2 in WT cells, cells treated with an unspecific scr-siRNA (scr), and cells treated with a specific Grx2 siRNA (Grx2 kd). The levels were determined by ELISA as described in the Materials and Methods section. After treatment with AF for 1.5 h (B) or HNE for 20 h (C), the cell viability was determined by the MTT assay. (D) After treatment with AF for 1.5 h, TrxR activity was determined with the fluorescent insulin assay. Error bars show mean±S.D. _n_=4. **p<0.01; *p<0.05, Student's _t_-test, treated cells versus WT control.
To know the extent of TrxR inhibition that is required for Grx2 knockdown to show its effect, we have treated the cells with the same series of concentrations of AF as that in the viabilities assays and assayed the cellular TrxR activities. As shown in Figure 6D, 1.5 h treatment with 4–12 μ_M_ of AF decreased the TrxR activity to about 60%–5% of the original level. These results show that when TrxR activity is diminished to 60% or lower, the knockdown of Grx2 will bring more damage to the cells, probably due to the loss of backup effect of Grx2 in reducing Trxs.
Grx2 knockdown causes increased Trx oxidation in TrxR inhibitor-treated cells
Redox western blot were used to identify the redox state of Trx2 and Trx1 in the WT control, unspecific src-siRNA-transfected, and Grx2 knockdown cells. As shown in Figure 7, compared to the control or src-siRNA-transfected cells, the Grx2 knockdown cells had a slight increase of both oxidized Trx2 and Trx1. Treatment with 10 μ_M_ HNE for 20 h caused a mild increase of oxidized Trx2 and Trx1 in the WT control and src-siRNA-transfected cells, but a dramatic increase of oxidized Trx2 and Trx1 in the Grx2 knockdown cells showing that Grx2 has the effect of protecting Trx from oxidation in vivo.
FIG. 7.
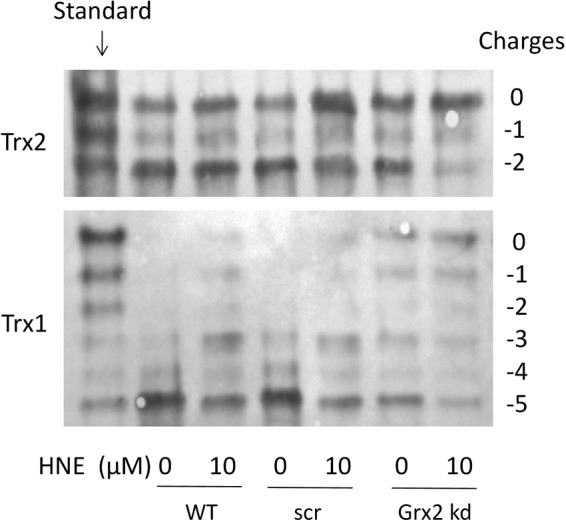
Redox state of Trx1 and Trx2 in control and Grx2 knockdown cells after treatment by HNE. Cells without transfection (WT), cells transfected by an unspecific siRNA (scr), and cells transfected by a specific Grx2 siRNA (Grx2 kd) were treated with or without 10 μ_M_ HNE for 20 h, and then, the redox state of Trx1 and Trx2 was determined using a redox western blot analysis.
Grx2 is resistant to HNE inhibition
To investigate if HNE inhibit Grx2 activity directly, we incubated reduced Grx2 with HNE, took aliquots of the incubation at different time intervals, and assayed the remaining activity of Grx2. As shown in Figure 8, the activity of Grx2 did not decrease during 3 h incubation with HNE. The rest of the incubation was subjected to Mass Spectrum analysis. As shown in Figure 9 A, Grx2 without modification gives a peak at m/z value of 16150. Five additional peaks corresponding to the addition of one to five HNE molecules to Grx2 was found, indicating that Grx2 were modified with one to five HNE molecules. To identify specific amino acid residues that are modified, the HNE-treated Grx2 was digested by trypsin, and the generated peptides were applied to Nano-LC-MS/MS. Four peptides were identified with HNE modification (Fig. 9B–E). The sites of HNE addition were Lys74, His98, His101, and His111, respectively. One more peptide (TSCSZCTMAK, the active site Cys37 and Cys40 containing peptide) was found possible to be modified by HNE (Fig. 9F), with Cys37 as the modification target. However, more spectra of the same peptide were found unmodified (spectra not shown). According to the MS data, the relative amount of modified peptide (TSC*SZCTMAK) based on integrated peptide intensities was 1.8%, meaning most (98%) of the TSCSZCTMAK peptide were not modified with HNE. The data here show that neither Cys37 nor Cys40 is a favorite target for HNE modification. Grx2 active site is relatively resistant to HNE modification, in accordance with that the activity was not lost.
FIG. 8.

Activity assay of Grx2 with/without incubation of HNE. Six micromolars of pre-reduced Grx2 was incubated with 36 μ_M_ HNE. Aliquots of the incubation were taken at indicated time intervals and assayed for the remaining activity of Grx2 with 0.7 m_M_ HED as substrate. HED, 2-hydroxyethyl disulfide.
FIG. 9.

Mass spectrometry analyses of HNE modification on Grx2. (A) MALDI-TOFMS of HNE-modified hGrx2. (B–F) Nano-LC-MS/MS spectra of Grx2 trypsin peptides modified by HNE. The bold characters in each peptide indicate the sites of HNE addition. MALDI-TOFMS, matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry.
Discussion
In this study, we have analyzed the protein–disulfide reductase activity of Grx2 system on cytosolic Trx1 and mitochondrial Trx2. According to our data, under the physiological concentrations of GSH (1–10 m_M_), Grx2 works as a reductase of Trx2, with the activity equivalent to about 1–10 n_M_ TrxR. Grx2 had a similar activity toward Trx1 as that with Trx2. The reducing activity of Grx2 toward Trx gives the possibility for Grx2 to work as a backup for TrxR. As shown in Figure 10, in the resting cells, the reducing equivalent of NADPH goes to Trx mainly through TrxR. In the cells treated with AF/HNE, when the TrxRs are compromised and inactivated, the reducing power of NADPH may flow through a bypass composed by GR, GSH, and Grx2 to Trxs. This hypothesis was further tested by knocking down Grx2 in the cells, where Trxs became remarkably more oxidized than in control cells when treated with same concentration of HNE (Fig. 7).
FIG. 10.
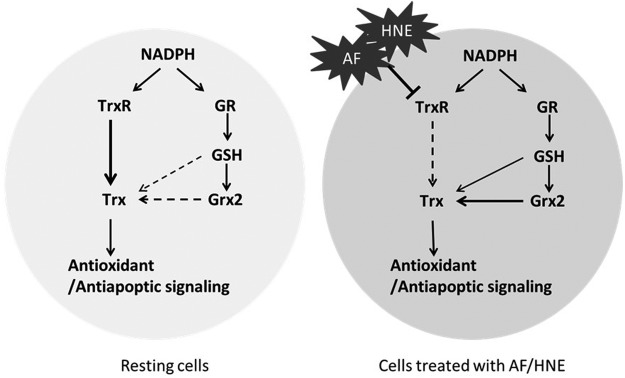
Mechanism for Grx2 working as a backup for TrxR.
Active Trx2 is indispensable for cell survival, as the homozygous Trx2−/− mouse embryonic fibroblasts (MEFs) were not viable (34). Conversely, the homozygous TrxR2−/− MEFs could be propagated (although with lower proliferation rate) (9), which means that there must be a backup system transferring electrons from NADPH to Trx2 to keep it active and functional in these cells. However, the nature of the backup system was unknown and to be identified. Interestingly, the TrxR2−/− cells but not the WT cells died when subjected to GSH depletion (9), implying the importance of GSH for the backup system. But the mechanism was not clear. Based on our finding that Grx2 system has the ability to enzymatically catalyze the reduction of Trx2 (Fig. 1), it is very likely that Grx2 had worked as the electron donor for Trx2 in the TrxR2−/− cells, and the cutoff of electrons from both TrxR2 and Grx2 system resulted in cell death, as has also be seen in our study with the combination of TrxR inhibitor and Grx2 knockdown. These results implied that natural abundance of Grx2 may work as an alternative reductase for Trx2 in vivo. To date, no candidate other than Grx2 was found to play this role.
There are more properties of Grx2 that make it fit for a backup system. First, Grx2 protein could form dimers through a ligating iron–sulfur cluster together with two molecules of noncovalently bound GSH between the active sites of each Grx2 monomer (4). Grx2 in the dimer is catalytically inactive (24), and the iron–sulfur cluster was suggested to work as a redox sensor: During conditions of oxidative stress when free radicals are formed and the GSH pool becomes oxidized, reduced GSH may become the limiting factor for cluster coordination, leading to the dissociation of the holo-Grx2 complex, yielding enzymatically active Grx2. Thus, for Grx2, the oxidative stress plays an activating role, and the activated Grx2 will help in combating the oxidative stress. Second, Grx2 is very resistant to oxidative inactivation compared to other thiol-redox proteins (17). In our study, we have found that Grx2 is also resistant against inactivation by electrophiles like HNE (Figs. 8 and 9). The insensitiveness of Grx2 to HNE increases its chance of protecting Trx from oxidation caused by inactivation of TrxR.
In the cytosol, the Grx1 and GSH system are very effective in protecting Trx1 from oxidation when TrxR1 was inactivated (11). In the mitochondria, GSH cannot be synthesized de novo because of lacking of the enzyme machinery needed for GSH synthesis. Mitochondrial GSH is imported from the cytosol by specific carriers, which exhibit sensitivity to membrane dynamics (30), and mitochondrial GSH has a turnover rate more than 10-fold slower than that in the cytosol (8). Probably as mitochondria is the main generator of cellular reactive oxygen species but have a more fragile redox pool, it seems that Trx2 is more difficult to be kept reduced and functional during an oxidative insult. Overexpression of Grx2 in the mitochondria may help keeping Trx2 reduced to function in cell survival. In our study, overexpression of Grx2 in the cytosol is effective in protecting Trx1 from oxidation (Fig. 5B), but could not stop an obvious oxidation of Trx2 (Fig. 4B), and was not very effective in the prevention of cell death. Differently, overexpression of Grx2 in the mitochondria is better in protection of Trx2 from oxidation (Fig. 4B) and also works better in preventing mitochondrial oxidative stress-mediated apoptosis. Based on these results, overexpression of Grx2 in the mitochondria may have therapeutic potential in neurodegenerative diseases like Alzheimer's disease or Parkinson's disease, where mitochondrial oxidative stress plays a dominant role in the malfunction and apoptosis of the brain cells (43).
The finding that overexpressed Grx2 could reduce Trx1 in the cytosol has its unique importance, as Grx2 specifically exist in the cytosol/nucleus of a number of cancer cell lines, or testis cells, but not in the cytosol/nucleus of most normal tissue cells (27). The potential roles of cytosolic/nuclear Grx2 in the generation and progression of cancer remain unclear. The finding here may indicate a pathway for Grx2 to support cancer cell growth/survival through providing reducing equivalents to Trx1.
Conclusively, the finding in this work provides a new insight into the mechanism for Grx2 to have antioxidative and anti-apoptotic effects, that is, by catalyzing the reduction of oxidized Trx1/2. This activity is of particular importance when TrxR is inactivated by exo/endogenous electrophilic compounds. In mammalian cells, the Trx and Grx systems cannot replace each other, as both are essential for maintaining cellular redox homeostasis and cell survival. In another way, they developed cross talks within certain members from one to another system, providing backups in case if one of the systems gets paralyzed. This mutual backup may strengthen the cellular antioxidant capacity, benefiting cell survival especially under oxidative stress.
Materials and Methods
Materials
Human Trx1 and Trx1 antibody was from IMCO Ltd. Human Trx2 antibody was from Santa Cruz Biotechnology. Recombinant rat TrxR1 (rrTrxR) was a kind gift from Prof. Elias Arnér (Karolinska Institutet, Stockholm, Sweden). HNE was purchased from Calbiochem. AF was form Santa Cruz Biotechnology. Yeast GR, GSH, insulin, NADPH, iodoacetic acid (IAA), and iodoacetamide (IAM) were purchased from Sigma–Aldrich. Dulbecco's modified Eagle medium (DMEM) was obtained from Invitrogen.
Protein expression and purification
Human Grx2 was expressed and purified using the auto-induction method as described in Gustafsson et al. (15) and Lundberg et al. (28). Briefly, Escherichia coli BL21-CodonPlus(DE3)-RIL competentcells were transfected with pET15b hGrx2Δ41–164. The strain was cultured in 10 ml LB medium at 37°C overnight. Two hundred milliliters of the auto-induction medium was inoculated with the 10 ml overnight culture, and cells were then propagated at 18°C for 30 h, harvested, and lysed by sonication. The lysates were cleared by centrifugation and filtration. Grx2 was purified from the clarified lysates by using nickel column on Äkta purification system (GE Healthcare) equilibrated with PBS. Grx2 was eluted with 200 m_M_ imidazole and desalted with NAP-25 columns equilibrated with PBS.
The E. coli BL21 (DE3) strain harboring expressing vector of His6-hTrx2 was a generous gift of Dr. Qing Cheng (Karolinska Institutet, Stockholm, Sweden). The expression and purification of hTrx2 was conducted with the same procedures as stated above for hGrx2, except that the purified hTrx2 were desalted and buffer-exchanged to TE buffer (50 m_M_ Tris, pH7.5, 1 m_M_ EDTA), instead of PBS.
Preparation of the active site oxidized hTrx1 and hTrx2
The human insulin is a protein of two peptide chains, the A-chain and the B-chain, linked together by two disulfide bonds. These disulfide bonds could be reduced by the active site dithiol of Trx1 or Trx2, generating active site oxidized Trxs and reduced (separated A-chains and B-chains of) insulin. As one dithiol can only reduce one disulfide, two molar of Trxs are needed to reduce one molar of insulin. Once reduced, the insulin peptides will become insoluble, which allows the separation of the active site oxidized Trx from the precipitated insulin by centrifugation. Specifically, human Trx1/Trx2 were reduced by incubation with 10 m_M_ DTT at 37°C for 30 min, then excess DTT were removed by desalting on a NAP-5 Sephadex G-25 column. The reduced Trx1/2 were incubated with insulin on a molar ratio of 2:1 (Trx/Insulin) at room temperature for 30 min and centrifuged at 16,000 g for 5 min to remove precipitated insulin. The supernatant containing the Trx-S2 was collected, and the redox state of this product was confirmed by redox urea-PAGE, a method described in the following section.
Urea-PAGE for detection of the redox state of purified Trx1/2
The method used for the detection of Trx redox state was developed in Bersani et al. (7) and Takahashi and Hirose (40) and modified in Du et al. (11) and Ungerstedt et al. (41). To prepare mobility standards, hTrx1/2 proteins were dissolved in TEU buffer (50 m_M_ Tris-HCl, pH 8.2, 1 m_M_ EDTA, 8 M Urea) and incubated with 3.5 m_M_ DTT at 37°C for 30 min to be fully reduced. The incubation was divided into four aliquots and then treated with reagents containing varying molar ratios of IAA/IAM (concentrations in m_M_: 30/0, 30/10, 10/30, 0/10). The aliquots were pooled and mixed, yielding the mobility standards containing Trxs with different patterns of IAA/IAM labeling on their thiols. The Trx samples with unknown redox state were denatured with TEU buffer, and the free thiols were alkylated by IAA by adding IAA to 30 m_M_ and incubated at 37°C for 30 min. The proteins were then precipitated by ice-cold acetone-HCl, washed twice with ice-cold acetone-HCl, and resuspended in TEU buffer containing 3.5 m_M_ DTT. After incubated at 37°C for 30 min, IAM was added to a final concentration of 10 m_M_ and incubated at 37°C for 15 min. The treated sample and the mobility standards were then loaded on Urea-gel (12% acrylamide [stacking gel 2.5%] with 8 M urea) and separated by running at 5 mA for 4–5 h in 25 m_M_ Tris-HCl, 192 m_M_ glycine buffer. The gel was stained by Coomassie Blue, and the bands were photographed by a Bio-Rad ChemDoc XRS scanning image analysis apparatus.
Enzyme activity assay
To determine the reductase activity of hGrx2 on hTrx1/2, a coupled enzyme reaction system was used. The system composed of 0.2 m_M_ NADPH, 6 μg/ml GR, 1/10 m_M_ GSH, various concentrations (0–1 μ_M_) of hGrx2, and 25 μ_M_ active site oxidized Trx1/2 in TE buffer (pH7.5). The activity of the reaction was determined by monitoring the decrease of absorbance at 340 nm caused by NADPH consumption. The same system was used in determination of the reaction rates of GSH (0–14 μ_M_) with active site oxidized hTrx2, only without hGrx2.
The reducing activity of rrTrxR on hTrx1/2 was determined by adding different concentrations (0–10 n_M_) of rrTrxR to a cuvette containing 0.2 m_M_ NADPH, 25 μ_M_ active site oxidized Trx1/2 in TE buffer (pH7.5), and monitoring the change of absorbance at 340 nm.
Cell culture and viability assay
Human cervical carcinoma HeLa cells were cultured in the DMEM medium with 1 mg/ml d-glucose, supplemented with 10% fetal bovine serum and 100 U/ml penicillin and streptomycin at 37°C in an incubator with humidified atmosphere containing 5% CO2. The stably transfected HeLa cell lines with mitochondrial or cytosolic overexpression of Grx2 were from the construction and preparation of Enoksson et al. (12) and cultured in the same conditions as that of the WT cells.
For viability assay, the HeLa cell lines were plated in 96-well plates at a density of 8×103 cells/well and cultured overnight. The medium was then changed to 200 μl of fresh medium containing AF or HNE at the designed concentrations and treated for 1.5, 3, or 20 h as indicated before subjected to MTT assay: 50 μl of MTT solution (2 mg/ml in PBS) was added to each well and incubated for 4 h. The medium containing MTT was removed carefully. One hundred fifty microliters of dimethyl sulfoxide:glycine-NaCl buffer (pH 10.5, 4:1) was added to each well, and the plates were shaken for 1 h at room temperature. The cell viabilities were then determined by measuring the absorbance at 550 nm. The mean value of at least 6 wells of the same treatment was used for statistical analysis.
Redox Western blot of cellular Trx1/2
To prepare mobility standards, HeLa cell lysates were denatured with urea and fully reduced with DTT and then incubated with varying molar ratios of IAA/IAM as described in the Urea-PAGE for Detection of the Redox State of Purified Trx1/2 section. To determine the redox state of Trxs in chemical compound-treated cells, the cells were washed twice with PBS, lysed in TEU buffer containing 30 m_M_ IAA, and incubated at 37°C for 30 min. After removing the cell debris by centrifugation, the cell lysates were precipitated by ice-cold acetone-HCl and washed with the same buffer. The precipitation was resuspended in TEU buffer containing 3.5 m_M_ DTT and incubated at 37°C for 30 min. Then, 10 m_M_ IAM was added to each sample and incubated for 15 min at 37°C. Proteins in the sample were separated by urea-PAGE gel and blotted to a nitrocellulose membrane (Bio-Rad). Membranes were probed with the Trx1/2 primary antibody, biotinylated secondary antibody (Dako), and streptavidin–alkaline phosphatase anti-biotin tertiary antibody (MABTECH AB) and then visualized using 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium substrate (Sigma). The intensity of each band was quantitated with a Bio-Rad ChemDoc XRS scanning image analysis apparatus and Quantity One software (Bio-Rad).
siRNA mediated knockdown of Grx2 in HeLa cells
Negative control siRNA (scr-siRNA) and predesigned Grx2 siRNA were purchased from Qiagen. Shortly before transfection, HeLa cells were seeded in six-well plates and transfection was performed using HiPerFect Transfection Reagent (Qiagen) according to the manufacturer's protocol. Briefly, 50 n_M_ of the siRNAs and 12 μl of Hiperfect Transfection Reagent were added to 100 μl of the Optimem medium (Invitrogen). After 10 min of incubation, the siRNA mixtures were added to the cells. The cells were harvested after 48 h, and the level of Grx2 was analyzed by ELISA.
ELISA of Grx2
Quantification of Grx2 was performed by using specific sandwich ELISA as described in Lillig et al. (26). Briefly, 96-well plates were coated with affinity-purified anti-Grx2 antibodies (2 μg/ml) overnight at 4°C. After blocking with bovine serum albumin (BSA), cell lysates and Grx2 standards (0.05–50 ng/ml) were added to the plate and incubated overnight at 4°C. Biotinylated anti-Grx2 antibody (1 μg/ml) was used as secondary antibody, and the results were obtained by using alkaline phosphatase-conjugated streptavidin (Dako) followed by staining with p-nitrophenyl phosphate (Sigma). Quantification was carried out at 405 nm in a SpectraMax 340PC384 Microplate Reader (Molecular Devices).
Determination of TrxR activity in cell lysates
HeLa cells were plated at a density of 0.5×106 cells in six-well plates in the DMEM medium. After overnight incubation at 37°C, the cells were washed with PBS and treated with different concentrations of AF. After treatment, the cells were carefully washed with PBS twice and lysed in TE buffer (50 m_M_ Tris-HCl, pH 7.5/1 m_M_ EDTA/protease inhibitor cocktails [Roche Diagnostics]) by sonication. The protein concentration was determined by the DC protein assay kit from Bio-Rad. TrxR activity in cell lysates was determined in 96-well plates with a fluorescent insulin assay kit (IMCO Corporation Ltd AB). Approximately 10 μg of cell lysates was added to the reaction solution containing 0.25 m_M_ NADPH and 2.5 μ_M_ human Trx1. The reaction solutions without human Trx1 were used as background. After the reaction solutions were incubated at 37°C for 30 min, fluorescent insulin was added to each well. The emission at 525 nm was recorded after 485 nm excitation using VICTOR3 Multilabel Plate Reader (PerkinElmer). TrxR activity was determined by measuring the initial velocity from a linear fluorescence increase due to the reduction of fluorescent insulin.
hGrx2 activity inhibition assay
2-hydroxyethyl disulfide (HED) assay was used to test if HNE inhibits hGrx2. hGrx2 was pre-reduced by incubation with 10 m_M_ GSH at 37°C for 30 min. Excess GSH were removed by desalting on a NAP-5 Sephadex G-25 column equilibrated with PBS. Six micromolars of pre-reduced hGrx2 was incubated with 36 μ_M_ HNE. At different time intervals, aliquots of the incubation were added to a cuvette containing TE buffer (pH 7.5) with 0.2 M NADPH, 1 m_M_ GSH, 6 μg/ml GR, and 50 μg/ml BSA. 0.7 m_M_ HED was added to initiate the reaction. The activity was determined by monitoring the absorbance at 340 nm. Background of HED reaction was subtracted from the reaction curves.
Mass spectrometry analyses
6 μ_M_ pre-reduced Grx2 was incubated with 36 μ_M_ HNE for 3 h. The excess HNE was removed by ultrafiltration, and the HNE-modified Grx2 was subjected to matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry (MALDI-TOFMS) analysis (by the PK/KI facility, Karolinska Institutet). Briefly, the sample was spotted on a MALDI target plate, mixed with MALDI matrix prepared by dissolving 10 mg of Sinapinic acid in 1 ml of 30% (v/v) acetonitrile/water with 0.1% trifluoroacetic acid (TFA). In the linear positive mode, HNE-modified Grx2 was identified using a MALDI-TOF mass spectrometer (Voyager-DE PRO; Applied Biosystems). The instrument was equipped with a Nitrogen laser with a Rep Rate of 3.0 Hz. Each spectrum was the sum of 50 laser shots. The ion signal of myoglobin was used for external calibration.
To identify specific amino acid residues of Grx2 that is modified by HNE, the HNE-treated Grx2 was reduced by DTT (20 m_M_, 4 h at 37°C), alkylated by incubation with IAM (40 m_M_) at room temperature for 2 h in the dark, and then digested by incubation with trypsin (4% weight of Grx2 protein) at 37°C for 16 h. The generated peptides were applied to Nano-LC-MS/MS analysis, using the Nano-EASY LC, Proxeon LTQ Velos Orbitrap (Thermo Fisher). First, the digest was dried under vacuum and then dissolved in 0.1% formic acid-5% acetonitrile and loaded onto a prepacked C18 reverse phase column (PK/KI homemade). Next, the peptides were separated using a gradient from 5% to 40% acetonitrile in 0.1% formic acid in 30 min. Finally, the mass spectra were acquired in positive ion mode. Quantitative data were generated using in-house developed software (PK/KI).
Abbreviations Used
AF
auranofin
AP-1
activator protein 1
ASK-1
apoptosis signal-regulating kinase 1
Cys
cysteine
DMEM
Dulbecco's modified Eagle medium
GR
glutathione reductase
Grx
glutaredoxin
GSH
glutathione
H2O2
hydrogen peroxide
HED
2-hydroxyethyl disulfide
HNE
4-hydroxynonenal
IAA
iodoacetic acid
IAM
iodoacetamide
MALDI-TOFMS
matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry
MEF
mouse embryonic fibroblast
NADPH
nicotinamide adenine dinucleotide phosphate
NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
Prx
peroxiredoxin
PTEN
phosphatase and tensin homolog
Trx
thioredoxin
TrxR
thioredoxin reductase
urea-PAGE
urea-polyacrylamide gel electrophoresis
WT
wild type
Acknowledgments
We thank Lena Ringdén for excellent secretarial work, and Prof. Elias Arnér and Dr. Qing Cheng (Karolinska Institutet) for kindly providing rrTrxR, as well as Trx2 expressing plasmid. This work was supported by the Swedish Cancer Society (961), the Swedish Research Council Medicine (13X-3529), the K. A.Wallenberg Foundation, and grants from Karolinska Institutet. All MS data generation and peptide quantitation were performed at PK/KI supported by the Karolinska Institutet.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Abarikwu SO, Pant AB, and Farombi EO. 4-Hydroxynonenal induces mitochondrial-mediated apoptosis and oxidative stress in SH-SY5Y human neuronal cells. Basic Clin Pharmacol Toxicol 110: 441–448, 2012 [DOI] [PubMed] [Google Scholar]
- 2.Anathy V, Roberson EC, Guala AS, Godburn KE, Budd RC, and Janssen-Heininger YM. Redox-based regulation of apoptosis: S-glutathionylation as a regulatory mechanism to control cell death. Antioxid Redox Signal 16: 496–505, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beer SM, Taylor ER, Brown SE, Dahm CC, Costa NJ, Runswick MJ, and Murphy MP. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: implications for mitochondrial redox regulation and antioxidant DEFENSE. J Biol Chem 279: 47939–47951, 2004 [DOI] [PubMed] [Google Scholar]
- 4.Berndt C, Hudemann C, Hanschmann EM, Axelsson R, Holmgren A, and Lillig CH. How does iron-sulfur cluster coordination regulate the activity of human glutaredoxin 2? Antioxid Redox Signal 9: 151–157, 2007 [DOI] [PubMed] [Google Scholar]
- 5.Berndt C, Lillig CH, and Holmgren A. Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: implications for diseases in the cardiovascular system. Am J Physiol Heart Circ Physiol 292: H1227–H1236, 2007 [DOI] [PubMed] [Google Scholar]
- 6.Berndt C, Lillig CH, and Holmgren A. Thioredoxins and glutaredoxins as facilitators of protein folding. Biochim Biophys Acta 1783: 641–650, 2008 [DOI] [PubMed] [Google Scholar]
- 7.Bersani NA, Merwin JR, Lopez NI, Pearson GD, and Merrill GF. Protein electrophoretic mobility shift assay to monitor redox state of thioredoxin in cells. Methods Enzymol 347: 317–326, 2002 [DOI] [PubMed] [Google Scholar]
- 8.Circu ML. and Aw TY. Glutathione and modulation of cell apoptosis. Biochim Biophys Acta 1823: 1767–1777, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conrad M, Jakupoglu C, Moreno SG, Lippl S, Banjac A, Schneider M, Beck H, Hatzopoulos AK, Just U, Sinowatz F, Schmahl W, Chien KR, Wurst W, Bornkamm GW, and Brielmeier M. Essential role for mitochondrial thioredoxin reductase in hematopoiesis, heart development, and heart function. Mol Cell Biol 24: 9414–9423, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Di Sarra F, Fresch B, Bini R, Saielli G, and Bagno A. Reactivity of auranofin with selenols and thiols–implications for the anticancer activity of gold(I) compounds. Eur J Inorg Chem 2013: 2718–2727, 2013 [Google Scholar]
- 11.Du Y, Zhang H, Lu J, and Holmgren A. Glutathione and glutaredoxin act as a backup of human thioredoxin reductase 1 to reduce thioredoxin 1 preventing cell death by aurothioglucose. J Biol Chem 287: 38210–38219, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Enoksson M, Fernandes AP, Prast S, Lillig CH, Holmgren A, and Orrenius S. Overexpression of glutaredoxin 2 attenuates apoptosis by preventing cytochrome c release. Biochem Biophys Res Commun 327: 774–779, 2005 [DOI] [PubMed] [Google Scholar]
- 13.Fang J. and Holmgren A. Inhibition of thioredoxin and thioredoxin reductase by 4-hydroxy-2-nonenal in vitro and in vivo. J Am Chem Soc 128: 1879–1885, 2006 [DOI] [PubMed] [Google Scholar]
- 14.Ferri A, Fiorenzo P, Nencini M, Cozzolino M, Pesaresi MG, Valle C, Sepe S, Moreno S, and Carri MT. Glutaredoxin 2 prevents aggregation of mutant SOD1 in mitochondria and abolishes its toxicity. Hum Mol Genet 19: 4529–4542, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gustafsson TN, Sahlin M, Lu J, Sjoberg BM, and Holmgren A. Bacillus anthracis thioredoxin systems, characterization and role as electron donors for ribonucleotide reductase. J Biol Chem 287: 39686–39697, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hashemy SI. and Holmgren A. Regulation of the catalytic activity and structure of human thioredoxin 1 via oxidation and S-nitrosylation of cysteine residues. J Biol Chem 283: 21890–21898, 2008 [DOI] [PubMed] [Google Scholar]
- 17.Hashemy SI, Johansson C, Berndt C, Lillig CH, and Holmgren A. Oxidation and S-nitrosylation of cysteines in human cytosolic and mitochondrial glutaredoxins: effects on structure and activity. J Biol Chem 282: 14428–14436, 2007 [DOI] [PubMed] [Google Scholar]
- 18.Hirota K, Matsui M, Iwata S, Nishiyama A, Mori K, and Yodoi J. AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc Natl Acad Sci U S A 94: 3633–3638, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holmgren A, Johansson C, Berndt C, Lonn ME, Hudemann C, and Lillig CH. Thiol redox control via thioredoxin and glutaredoxin systems. Biochem Soc Trans 33: 1375–1377, 2005 [DOI] [PubMed] [Google Scholar]
- 20.Johansson C, Lillig CH, and Holmgren A. Human mitochondrial glutaredoxin reduces S-glutathionylated proteins with high affinity accepting electrons from either glutathione or thioredoxin reductase. J Biol Chem 279: 7537–7543, 2004 [DOI] [PubMed] [Google Scholar]
- 21.Kanzok SM, Schirmer RH, Turbachova I, Iozef R, and Becker K. The thioredoxin system of the malaria parasite Plasmodium falciparum. Glutathione reduction revisited. J Biol Chem 275: 40180–40186, 2000 [DOI] [PubMed] [Google Scholar]
- 22.Lee SR, Kim JR, Kwon KS, Yoon HW, Levine RL, Ginsburg A, and Rhee SG. Molecular cloning and characterization of a mitochondrial selenocysteine-containing thioredoxin reductase from rat liver. J Biol Chem 274: 4722–4734, 1999 [DOI] [PubMed] [Google Scholar]
- 23.Lillig CH, Berndt C, and Holmgren A. Glutaredoxin systems. Biochim Biophys Acta 1780: 1304–1317, 2008 [DOI] [PubMed] [Google Scholar]
- 24.Lillig CH, Berndt C, Vergnolle O, Lonn ME, Hudemann C, Bill E, and Holmgren A. Characterization of human glutaredoxin 2 as iron-sulfur protein: a possible role as redox sensor. Proc Natl Acad Sci U S A 102: 8168–8173, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lillig CH. and Holmgren A. Thioredoxin and related molecules—from biology to health and disease. Antioxid Redox Signal 9: 25–47, 2007 [DOI] [PubMed] [Google Scholar]
- 26.Lillig CH, Lonn ME, Enoksson M, Fernandes AP, and Holmgren A. Short interfering RNA-mediated silencing of glutaredoxin 2 increases the sensitivity of HeLa cells toward doxorubicin and phenylarsine oxide. Proc Natl Acad Sci U S A 101: 13227–13232, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lonn ME, Hudemann C, Berndt C, Cherkasov V, Capani F, Holmgren A, and Lillig CH. Expression pattern of human glutaredoxin 2 isoforms: identification and characterization of two testis/cancer cell-specific isoforms. Antioxid Redox Signal 10: 547–557, 2008 [DOI] [PubMed] [Google Scholar]
- 28.Lundberg M, Johansson C, Chandra J, Enoksson M, Jacobsson G, Ljung J, Johansson M, and Holmgren A. Cloning and expression of a novel human glutaredoxin (Grx2) with mitochondrial and nuclear isoforms. J Biol Chem 276: 26269–26275, 2001 [DOI] [PubMed] [Google Scholar]
- 29.Mari M, Morales A, Colell A, Garcia-Ruiz C, and Fernandez-Checa JC. Mitochondrial glutathione, a key survival antioxidant. Antioxid Redox Signal 11: 2685–2700, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mari M, Morales A, Colell A, Garcia-Ruiz C, Kaplowitz N, and Fernandez-Checa JC. Mitochondrial glutathione: features, regulation and role in disease. Biochim Biophys Acta 1830: 3317–3328, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsui M, Oshima M, Oshima H, Takaku K, Maruyama T, Yodoi J, and Taketo MM. Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev Biol 178: 179–185, 1996 [DOI] [PubMed] [Google Scholar]
- 32.Matthews JR, Wakasugi N, Virelizier JL, Yodoi J, and Hay RT. Thioredoxin regulates the DNA binding activity of NF-kappa B by reduction of a disulphide bond involving cysteine 62. Nucleic Acids Res 20: 3821–3830, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meuillet EJ, Mahadevan D, Berggren M, Coon A, and Powis G. Thioredoxin-1 binds to the C2 domain of PTEN inhibiting PTEN's lipid phosphatase activity and membrane binding: a mechanism for the functional loss of PTEN's tumor suppressor activity. Arch Biochem Biophys 429: 123–133, 2004 [DOI] [PubMed] [Google Scholar]
- 34.Nonn L, Williams RR, Erickson RP, and Powis G. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol Cell Biol 23: 916–922, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rackham O, Shearwood AM, Thyer R, McNamara E, Davies SM, Callus BA, Miranda-Vizuete A, Berners-Price SJ, Cheng Q, Arner ES, and Filipovska A. Substrate and inhibitor specificities differ between human cytosolic and mitochondrial thioredoxin reductases: Implications for development of specific inhibitors. Free Radic Biol Med 50: 689–699, 2011 [DOI] [PubMed] [Google Scholar]
- 36.Rhee SG, Chae HZ, and Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med 38: 1543–1552, 2005 [DOI] [PubMed] [Google Scholar]
- 37.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, and Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J 17: 2596–2606, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spyrou G, Enmark E, Miranda-Vizuete A, and Gustafsson J. Cloning and expression of a novel mammalian thioredoxin. J Biol Chem 272: 2936–2941, 1997 [DOI] [PubMed] [Google Scholar]
- 39.Stanley BA, Sivakumaran V, Shi S, McDonald I, Lloyd D, Watson WH, Aon MA, and Paolocci N. Thioredoxin reductase-2 is essential for keeping low levels of H2O2 emission from isolated heart mitochondria. J Biol Chem 286: 33669–33677, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takahashi N. and Hirose M. Determination of sulfhydryl groups and disulfide bonds in a protein by polyacrylamide gel electrophoresis. Anal Biochem 188: 359–365, 1990 [DOI] [PubMed] [Google Scholar]
- 41.Ungerstedt J, Du Y, Zhang H, Nair D, and Holmgren A.In vivo redox state of human thioredoxin and redox shift by the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA). Free Radic Biol Med 53: 2002–2007, 2012 [DOI] [PubMed] [Google Scholar]
- 42.Wu H, Xing K, and Lou MF. Glutaredoxin 2 prevents H2O2-induced cell apoptosis by protecting complex I activity in the mitochondria. Biochim Biophys Acta 1797: 1705–1715, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yan MH, Wang X, and Zhu X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic Biol Med 62: 90–101, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang H, Cao D, Cui W, Ji M, Qian X, and Zhong L. Molecular bases of thioredoxin and thioredoxin reductase-mediated prooxidant actions of (−)-epigallocatechin-3-gallate. Free Radic Biol Med 49: 2010–2018, 2010 [DOI] [PubMed] [Google Scholar]
- 45.Zhang H, Go YM, and Jones DP. Mitochondrial thioredoxin-2/peroxiredoxin-3 system functions in parallel with mitochondrial GSH system in protection against oxidative stress. Arch Biochem Biophys 465: 119–126, 2007 [DOI] [PubMed] [Google Scholar]