Common regulatory control of CTP synthase enzyme activity and filament formation (original) (raw)
CTP synthase is one of many enzymes that form novel intracellular filaments/structures. A structure–function approach is used to show that the same regulatory sites that control CTP synthase enzyme activity also control filament formation. Close coupling of assembly to enzyme regulation is proposed to be a general feature of these structures.
Abstract
The ability of enzymes to assemble into visible supramolecular complexes is a widespread phenomenon. Such complexes have been hypothesized to play a number of roles; however, little is known about how the regulation of enzyme activity is coupled to the assembly/disassembly of these cellular structures. CTP synthase is an ideal model system for addressing this question because its activity is regulated via multiple mechanisms and its filament-forming ability is evolutionarily conserved. Our structure–function studies of CTP synthase in Saccharomyces cerevisiae reveal that destabilization of the active tetrameric form of the enzyme increases filament formation, suggesting that the filaments comprise inactive CTP synthase dimers. Furthermore, the sites responsible for feedback inhibition and allosteric activation control filament length, implying that multiple regions of the enzyme can influence filament structure. In contrast, blocking catalysis without disrupting the regulatory sites of the enzyme does not affect filament formation or length. Together our results argue that the regulatory sites that control CTP synthase function, but not enzymatic activity per se, are critical for controlling filament assembly. We predict that the ability of enzymes to form supramolecular structures in general is closely coupled to the mechanisms that regulate their activity.
INTRODUCTION
The past several years have seen an explosion in the identification of novel intracellular structures (Sheth and Parker, 2003; Campbell et al., 2005; An et al., 2008; Narayanaswamy et al., 2009; Ingerson-Mahar et al., 2010; Liu, 2010; Noree et al., 2010). Although the macromolecular components and putative functions of these large cytoplasmic structures are diverse, they share a common theme: each class of structure is formed from enzymes that act in a specific biochemical or regulatory pathway. For example, processing bodies are visible supramolecular complexes comprising mRNAs and many of the enzymes that regulate their translation and stability (Sheth and Parker, 2003). Similarly, purinosomes assemble from a subset of enzymes in the de novo purine biosynthetic pathway in response to purine deprivation in order to accelerate flux through the pathway via substrate channeling (An et al., 2008, 2010a,b). Strikingly, recent visual screens of the yeast green fluorescent protein (GFP) strain collection reveal that multiple metabolic enzymes self-assemble into filaments, arguing that this mode of regulation could play a role in the control of many biosynthetic pathways (Narayanaswamy_et al._, 2009; Noree_et al._, 2010). However, although the pace at which novel cytoplasmic structures are being identified continues to accelerate, little is known about how specific enzyme regulatory mechanisms impact the large cytoplasmic structures they form.
To assess whether the regulation of enzyme activity controls the assembly of such supramolecular structures, we focused our studies on a single class of novel intracellular filaments: those formed by the Saccharomyces cerevisiae enzyme CTP synthase (Ura7p). _URA7_encodes the major CTP synthase in S. cerevisiae, which catalyzes the ATP-dependent transfer of nitrogen from glutamine to UTP, generating CTP and glutamate (Figure 1A; Ozier-Kalogeropoulos_et al._, 1991, 1994). The two halves of this reaction require both the C-terminal glutamine amidotransferase (glutamine → glutamate + NH3) and the N-terminal amidoligase (ATP + UTP + NH3 → ADP + Pi + CTP) domains of the enzyme (Figure 1A).
FIGURE 1.

CTP synthase reaction, conservation of CTP synthase assembly, and the identification of two major populations of CTP synthase structures. (A) CTP synthase reaction. (B) Ura7p/CTP synthase structures in S. cerevisiae. (C) CTP synthase staining (green) and actin staining (red) in_Drosophila melanogaster_ ovary (Noree_et al._, 2010). (D) CTP synthase staining (green) and Tau staining (red) in rat hippocampal neurons (Noree_et al._, 2010). (E) Length distribution of wild-type (WT) Ura7p-GFP structures of yeast grown for 1 d in YPD. 10 random fields from each of six independent experiments were pooled and analyzed (n = 621). Two peaks were observed in the length distribution curve with the cutoff value at 0.75 μm. (F) Percentage of foci (<0.75 μm) and filaments (≥0.75 μm) for WT Ura7p-GFP structures plotted as bar graphs.
Ura7p/CTP synthase has several features that make it an ideal model system for exploring the functional principles underlying intracellular structure formation. First, extensive studies of CTP synthase regulation in yeast and other organisms have defined multiple ligands that either stimulate (ATP, GTP, and UTP) or inhibit (CTP) enzyme activity (Long and Pardee, 1967; Levitzki and Koshland, 1972a,b; Aronow and Ullman, 1987;Pappas et al., 1998; Endrizzi et al., 2005). These nucleotides regulate catalysis via three distinct mechanisms that control many enzymes: allosteric activation (GTP), tetramerization (ATP, UTP, CTP), and competitive feedback inhibition (CTP) (Long and Pardee, 1967; Levitzki and Koshland, 1972a,b; Aronow and Ullman, 1987; Pappas et al., 1998; Endrizzi et al., 2005). In addition, several phosphorylation sites have been identified that also modulate enzyme activity (Yang et al., 1996; Yang and Carman, 1996; Park_et al._, 1999, 2003; Choi et al., 2003; Choi and Carman, 2007; Chang_et al._, 2007). Mutations that disrupt each of these regulatory mechanisms have been identified and characterized (Whelan_et al._, 1993; Willemoes_et al._, 2005; Lunn_et al._, 2008). Finally, multiple prokaryotic and eukaryotic CTP synthases form filaments, arguing that this property is evolutionarily conserved (Figure 1, B–D; Ingerson-Mahar_et al._, 2010; Liu, 2010;Noree et al., 2010). Thus CTP synthase is an excellent proving ground for deciphering how classical mechanisms of enzyme regulation are connected to the assembly of supramolecular structures.
Here we use a structure-guided, site-directed mutagenesis strategy to specifically target the major regulatory sites in CTP synthase and assess their role in controlling the frequency of CTP synthase filament formation as well as filament length when altered. Mutations that perturb a regulatory loop adjacent to the putative allosteric GTP-binding cleft and the ATP-, UTP-, and CTP-binding sites stimulate the frequency of filament formation. Given that ATP, UTP, and CTP all stabilize the catalytically active tetramer of CTP synthase (Pappas_et al._, 1998), our results argue that the basic unit of CTP synthase filaments is the inactive dimeric form of the enzyme. Our studies of filament length demonstrate that there are two populations of wild-type CTP synthase filaments: very short, “foci-like” structures and long filaments. Sites of substrate binding and end-product inhibition located on the amidoligase domain, as well as allosteric activation on the glutamine amidotransferase domain, are key regulators of filament length. A phosphorylation site on the glutamine amidotransferase domain also plays a role. These data suggest that both domains of the protein contribute to polymer structure. In contrast, a nonregulatory mutation that compromises the glutamine amidotransferase active site has no effect on CTP synthase filament formation or length. In sum, CTP synthase filament formation and structure are intimately connected with the major mechanisms used to regulate enzyme activity, but not catalytic function itself.
RESULTS
CTP synthase filaments exhibit a bimodal length distribution
Previous analyses reporting the discovery of CTP synthase filament formation in yeast,Drosophila, and bacteria described filament length of the wild-type enzyme using a qualitative classification scheme or as a single parameter such as average length (Figure 1; Ingerson-Mahar_et al._, 2010; Liu, 2010;Noree et al., 2010). A detailed analysis of Ura7p-GFP filaments under our standard growth conditions for inducing filament formation—growth to saturation at 30°C in YPD (2% peptone, 1% yeast extract, 2% dextrose)—revealed a bimodal distribution of lengths. The first peak comprised short, foci-like structures (<0.75 μm in length; ∼31% of the population), and the second peak comprised clearly defined filaments (≥0.75 μm in length; ∼69% of the population; Figure 1, E and F). Thus, to provide the most quantitative assessment of the effects of mutations on filament assembly, we carefully measured two aspects of filament formation: 1) the frequency, defined as the percentage of cells possessing Ura7p-GFP filaments, and 2) the length distribution of Ura7p-GFP filaments.
Blocking UTP-mediated tetramerization increases frequency of CTP synthase filament formation without altering the length distribution
We first examined whether tetramerization of the enzyme plays a role in CTP synthase filament formation. In the absence of nucleotides, CTP synthase is a catalytically inactive, tightly associated dimer (Yang et al., 1994;Pappas et al., 1998). In the presence of ATP, UTP, or CTP, two dimers associate to form a tetramer (Figure 2A). This oligomerization event is required for proper function of both the glutamine amidotransferase- and amidoligase-active sites (Levitzki_et al._, 1971; Levitzki and Koshland, 1972a; Pappas et al., 1998). Structural studies indicate that the dimer and tetramer interfaces are stabilized almost exclusively by polar and hydrophobic contacts between the amidoligase domains of the component monomers (Endrizzi et al., 2004). In the_Escherichia coli_ enzyme, a mutation in the binding site of the substrate UTP, which lies in the amidoligase domain near the tetramer interface, has been shown to severely compromise UTP binding, tetramer formation and catalytic activity (Figure 2B; Lunn et al., 2008). Strains expressing Ura7p-GFP with the equivalent mutation (G148A) expressed from the endogenous_URA7_ locus (see Materials and Methods) displayed an ∼2.7-fold increase in the percentage of cells that form filaments as compared with wild type (Table 1 and Figure 2C). However, neither the median length nor the length distribution of the filaments was significantly altered by the G148A mutation (Table 1 and Figure 2, C and D). To determine whether the enhanced filament formation caused by disruption of UTP binding was the consequence of destabilization of the tetramer and/or blockade of catalytic activity, we exploited previous studies that indicated that mutation of the active-site cysteine of the glutamine amidotransferase domain eliminated enzyme activity (Figure 2B; Paluh_et al._, 1985). We found that a strain expressing C404G Ura7p-GFP showed no significant difference in the number of cells with observable filaments or in filament length distribution relative to those expressing wild type (Table 1 and Figure 2, C and D). Therefore, since merely inactivating CTP synthase catalytic activity had no effect on filament formation, we conclude that the increased filament formation of the G148A mutant is due to decreased UTP-stimulated tetramerization. These results also argue that Ura7p filaments comprise inactive dimers.
FIGURE 2.

Effect of disrupting UTP-mediated tetramerization or the active site of CTP synthase on filament formation. (A) The transition of CTP synthase from an inactive dimer to an active tetramer is regulated by ATP, UTP, and CTP binding. (B) Crystal structure of E. coli CTP synthase (PDB:2AD5; Endrizzi et al., 2005), highlighting residues involved in Mg2+ATP binding/tetramerization (D72, E140; green), UTP binding/tetramerization (G142; magenta), CTP binding/tetramerization (E155; orange), GTP binding (R356/G357; cyan), and catalysis (C379; magenta). The amidoligase domain (residues 1–266) and glutamine amidotransferase domain (residues 287–544)/interdomain linker (267–286) are colored light and dark blue, respectively, and the L11 lid is colored cyan. The bound ADP and CTP molecules in the structure are colored yellow and pink, respectively. The numbers in parentheses represent the corresponding amino acids in Ura7p. A multiple sequence alignment noting the targeted residues is also provided for reference (Supplemental Figure S2). (C) Representative images of yeast strains expressing wild-type (WT), catalytic mutant, or tetramerization mutant Ura7p-GFP. The average percentage and SEM of cells with Ura7p-GFP structures are indicated below each image. (D) Percentages of foci (<0.75 μm) and filaments (≥0.75 μm) for Ura7p-GFP structures are graphed for WT Ura7p-GFP and each mutant for comparison. Protein levels were uncorrelated with the effects on filament frequency or length (Supplemental Table S1 and Supplemental Figure S1).
TABLE 1:
Frequency of CTP synthase assembly, fractions of foci and filaments, average and median lengths of structures, and total number of structures used for analysis.
| Mutants | Description | Cells with structures (%) | Percentage foci | Percentage filaments | **Average length of structures (**μm) | **Median length of structures (**μm) | Number of structures analyzed |
|---|---|---|---|---|---|---|---|
| Wild type | 26.03 ± 1.72 | 30.63 ± 3.24 | 69.37 ± 3.24 | 1.053 ± 0.039 | 1.059 | 620 | |
| E161K | CTP-binding site | 99.86 ± 0.09 | 98.47 ± 0.59 | 1.53 ± 0.59 | 0.363 ± 0.018 | 0.359 | 784 |
| E161K-C404G | CTP-binding site and catalytic site | 64.31 ± 4.43 | 99.41 ± 0.30 | 0.59 ± 0.30 | 0.355 ± 0.007 | 0.347 | 322 |
| E146A | ATP-binding site | 56.91 ± 3.98 | 26.99 ± 3.46 | 73.01 ± 3.46 | 1.171 ± 0.027 | 1.233 | 431 |
| D70A | ATP-binding site | 54.69 ± 4.08 | 36.40 ± 4.24 | 63.60 ± 4.24 | 0.893 ± 0.048 | 0.895 | 402 |
| R381M | GTP-binding site | 86.37 ± 2.69 | 13.90 ± 3.13 | 86.10 ± 3.13 | 1.370 ± 0.036 | 1.407 | 415 |
| R381P | GTP-binding site | 81.91 ± 6.61 | 17.92 ± 1.59 | 82.08 ± 1.59 | 1.202 ± 0.022 | 1.216 | 388 |
| G382A | GTP-binding site | 58.16 ± 4.80 | 46.00 ± 9.86 | 54.00 ± 9.86 | 0.828 ± 0.067 | 0.789 | 480 |
| C404G | Catalytic-site | 29.13 ± 2.57 | 29.11 ± 0.93 | 70.89 ± 0.93 | 1.030 ± 0.016 | 1.043 | 385 |
| G148A | Tetramerization | 72.08 ± 9.90 | 27.80 ± 3.36 | 72.20 ± 3.36 | 1.150 ± 0.018 | 1.179 | 519 |
| S330A | Phosphorylation site | 28.18 ± 1.86 | 39.86 ± 3.50 | 60.14 ± 3.50 | 0.905 ± 0.042 | 0.887 | 315 |
| S354A | Phosphorylation site | 27.77 ± 2.70 | 46.20 ± 2.39 | 53.80 ± 2.39 | 0.806 ± 0.027 | 0.778 | 431 |
| S424A | Phosphorylation site | 24.33 ± 3.57 | 36.50 ± 3.54 | 63.50 ± 3.54 | 0.911 ± 0.040 | 0.887 | 404 |
| S36A | Phosphorylation site | 13.90 ± 3.92 | 36.20 ± 0.16 | 63.80 ± 0.16 | 0.913 ± 0.031 | 0.905 | 395 |
| S36D | Phosphorylation site | 6.084 + 1.04 | N/A | N/A | N/A | N/A | N/A |
| S36E | Phosphorylation site | 5.346 ± 1.37 | N/A | N/A | N/A | N/A | N/A |
Increased catalytic activity is not responsible for the block in filament formation observed in feedback-resistant mutants of CTP synthase
Our findings with the UTP-binding-site mutant led us to reexamine the role of CTP-binding-site mutations in regulating filament assembly. We previously found that the amidoligase domain mutation, E161K, which blocks feedback inhibition of yeast CTP synthase by CTP, abrogates the formation of full-length filaments while also causing an increase in the formation of foci-like structures (Noree et al., 2010). Given this effect, we decided to use our new quantitative definition of foci and filaments to examine how this mutation affects the different types of structures that can be formed by CTP synthase. Analysis of the length distribution of the structures formed by E161K Ura7p-GFP revealed that when feedback inhibition is blocked, CTP synthase filament formation is completely disrupted and the enzyme can only form foci (Figure 3, A and B). This strongly argues that feedback inhibition regulates the distribution of CTP synthase between filaments and foci.
FIGURE 3.

Disruption of feedback inhibition blocks CTP synthase filament formation independent of catalytic activity. (A) Representative images of yeast strains expressing wild-type (WT), CTP-binding mutant, or CTP-binding and catalytic double mutant Ura7p-GFP. Average percentage and SEM of cells with Ura7p-GFP structures are indicated below each image. (B) Percentages of foci (<0.75 μm) and filaments (≥0.75 μm) for Ura7p-GFP structures are graphed for WT Ura7p-GFP and each mutant for comparison.
The E161K mutation decreases the affinity of the enzyme for CTP, which 1) negatively affects tetramerization (Long and Pardee, 1967; Pappas et al., 1998) and 2) increases enzyme activity, since CTP is a competitive inhibitor of the enzyme (Ostrander et al., 1998). This apparently paradoxical behavior is highly dependent upon CTP concentration. CTP synthase activity is stimulated by low concentration of CTP due to increased tetramerization, whereas it is inhibited at high concentration due to the overlap of the CTP- and UTP-binding sites (Long and Pardee, 1967; Pappas et al., 1998). To separate these two effects, we once again leveraged the properties of the C404G active-site mutation (Figure 3, A and B). If the effects of blocking feedback inhibition on filament formation/length are primarily due to an increase in CTP synthase activity, we would predict that a Ura7p that is defective for both feedback inhibition and enzymatic function would form primarily filaments at the wild-type frequency. Therefore we constructed a strain that expresses Ura7p-GFP bearing the feedback resistance mutation, E161K, and the inactivating C404G mutation. Analysis of the length distributions of E161K-C404G Ura7p-GFP filaments indicated that the double mutant can still only form foci (99% of structures <0.75 μm; Table 1 and Figure 3, A and B). The only difference between E161K Ura7p and the double mutant was a decrease in the frequency of foci formation to a value that was still significantly greater than that of wild type (99% for E161K vs. 64% for E161K-C404G). This suggested that product binding rather than catalytic activity was the main driver of foci formation. However, during our analysis we noted one additional point of concern: the E161K mutation might cause only foci due to the extremely low expression level of the E161K Ura7p-GFP. This interpretation is inconsistent with our observation that both E161K Ura7p-GFP and E161K-C404G Ura7p-GFP formed only foci in spite of the fact that the E161K-C404G Ura7p was expressed at the normal levels. We also found that when E161K Ura7p-GFP is expressed at levels close to endogenous Ura7p-GFP, it forms only foci, arguing that expression levels do not contribute to the failure of the E161K-Ura7p to form filaments (Noree et al., 2010; Supplemental Figure S3).
On the basis of this analysis, we conclude that product binding regulates the distribution of CTP synthase between foci and filaments and that this effect is not dependent on competitive inhibition of catalytic activity or the expression level of the protein. Further, the increased propensity of the E161K Ura7p to form foci, like that of the G148A mutant to form filaments, suggests that these mutations increase structure formation frequency via their common ability to inhibit nucleotide-stimulated tetramerization of the amidoligase domain.
Mutations in the ATP-binding site of CTP synthase increase filament formation
We next investigated the role of the final nucleotide known to modulate tetramer formation, the substrate ATP. In the crystal structure of the ADP/CTP bound form of the _E. coli_CTP synthase (Endrizzi et al., 2005), the carboxylates of D72 and E140 (located in the amidoligase domain) chelate a magnesium ion that binds the β-phosphate of ADP. Hence we predict that mutation of the equivalent residues in Ura7p (D70 and E146) should compromise ATP binding and hydrolysis. Strains expressing D70A and E146A Ura7p-GFP exhibited approximately twofold increase in the number of cells containing filaments (Table 1 and Figure 4A). In addition, the D70A and E146A mutant filaments were 15% shorter and 16% longer, respectively, than wild type (Table 1 and Figure 4B). Not only do these results further support the role of tetramerization in filament formation frequency, they also highlight another region of the amidoligase domain involved in filament structure.
FIGURE 4.

Mutations in the ATP-binding site of CTP synthase increase filament formation. (A) Representative images of yeast strains expressing wild-type (WT), E146A, and D70A Ura7p-GFP. Average percentage and SEM of cells with Ura7p-GFP structures are indicated below each image. (B) Percentages of foci (<0.75 μm) and filaments (≥0.75 μm) for Ura7p-GFP structures are graphed for WT Ura7p-GFP and each mutant for comparison.
The allosteric GTP-binding site regulates both the frequency of filament formation and the length of CTP synthase filaments
GTP is unique among the four nucleotide regulators of CTP synthase activity in that it is neither a substrate nor a product of CTP synthase. Instead, GTP is a positive allosteric regulator that acts to increase the rate of catalysis (_k_cat) of the glutamine hydrolysis reaction (Levitzki and Koshland, 1972b; Willemoes et al., 2005). Multiple mutations that alter allosteric regulation of the enzyme have been identified in the L11 loop of CTP synthase, a mobile segment of the protein adjacent to the allosteric GTP-binding cleft. Based on structural homology to the small GTP-binding proteins EF-Tu and EF-G, as well as other related glutamine amidotransferases, this loop has been proposed to form a “lid” that closes over the active site to enhance catalysis (Endrizzi_et al._, 2005). To ask whether the L11 lid plays a role in Ura7p filament formation, we first focused on analyzing the effects of two L11 lid mutants, R381M and R381P, which inhibit GTP binding and activation of CTP synthase from Lactococcus lactis (Figure 5A; Willemoes et al., 2005). Strikingly, yeast strains that express either R381M or R381P Ura7p-GFP exhibited an ∼3.1-fold increase in the number of cells forming filaments compared with strains expressing wild-type Ura7p-GFP (Table 1 and Figure 5B). Furthermore, the filaments formed by both mutants were significantly longer than those formed by wild-type Ura7p-GFP. Indeed, the R381M mutation caused a 33% increase in median filament length, whereas the R381P mutation caused a 15% increase (Table 1). Indeed, 86 and 82% of structures formed by the R381M and R381P mutant CTP synthases, respectively, were longer than 0.75 μm (both percentages were greater than the 69% of structures that are ≥0.75 μm in wild type; Figure 5B).
FIGURE 5.
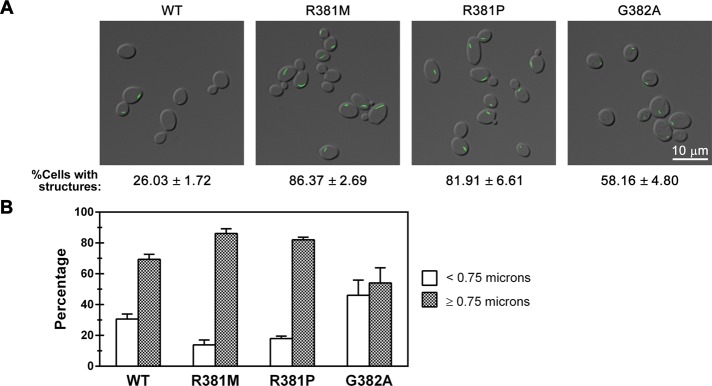
Effect of mutations in the allosteric regulatory domain on CTP synthase structure formation. (A) Representative images of yeast strains expressing wild-type (WT) or GTP-binding-site mutant Ura7p-GFP. The average percentage and SEM of cells with Ura7p-GFP structures are indicated below individual image. (B) Percentages of foci (<0.75 μm) and filaments (>0.75 μm) for Ura7p-GFP structures are graphed for WT and each GTP-binding-site mutant for comparison.
To further explore the importance of the L11 lid in filament formation, we characterized a third mutant, G382A, which in L. lactis CTP synthase increases the capacity of GTP to stimulate glutamine amidotransferase activity (Willemoes_et al._, 2005). Of interest, when this mutation was introduced into Ura7p, it, like the R381 mutations, caused an ∼2.2-fold increase in the number of cells with filaments (Figure 5B), however the median length of the filaments was shortened by 25% (Table 1 and Figure 5B). This change in median length was also reflected in a shift in the distribution of structures from long filaments to short foci, with only 54% of the structures formed by G382A Ura7p-GFP having a length ≥0.75 μm as compared with 69% of structures formed by wild-type Ura7p-GFP. We conclude that the L11 lid contributes to regulation of both the frequency of filament formation and filament length. Moreover, activating and inactivating mutations in the allosteric control region of CTP synthase have opposing effects on filament length.
Phosphorylation is not a major regulator of CTP synthase filament formation
Yeast CTP synthase filament formation is potently stimulated by the kinase inhibitor staurosporine (Noree et al., 2010). This suggested to us that phosphorylation of CTP synthase might play a direct role in regulating filament formation. Previous studies identified four major phosphorylation sites in yeast that affect its catalytic activity: Ura7p, S36, S330, S354, and S424 (Choi et al., 2003; Park_et al._, 2003). Specifically, phosphorylation at S36, S354, or S424 stimulates Ura7p catalytic activity, whereas phosphorylation at S330 inhibits enzyme activity (Choi et al., 2003; Park et al., 2003). To determine whether any of these phosphorylation sites plays a role in regulating filament formation, we generated yeast strains expressing mutant forms of Ura7p-GFP in which single phosphorylation sites were inactivated by changing the serine in the site to alanine.
The S36A mutation caused an approximately twofold decrease in the frequency of filament formation (13.9 vs. 26% for wild type) but had little effect on the length of the filaments formed (Figure 6, A and B). This result suggested that phosphorylation at S36, in particular, might be required for efficient filament assembly. To test this possibility, we changed S36 to either aspartate or glutamate, two amino acid changes that are often used to mimic phosphorylation at serine. If phosphorylation of S36 were required for efficient nucleation of filaments, we would expect these mutations to increase the frequency of filament formation. However, both the S36D and S36E mutations caused approximately fivefold decrease in the frequency of filament formation (Table 1). This result suggests that either the S36D and S36E mutations do not properly mimic phosphorylation at S36 or that S36 is merely an important residue for initiating filament formation independent of its phosphorylation state. In the_E. coli_ CTP synthase crystal structure (Endrizzi_et al._, 2004), the equivalent residue, I38, is completely buried behind the ATP-binding site. Therefore it is likely that the S36 mutations are perturbing the structure of CTP synthase (and filament formation) via alterations in hydrophobic packing (S36A mutation) or the introduction of a buried charge (S36D and S36E mutations).
FIGURE 6.

Phosphorylation is not a major regulator of CTP synthase filament. (A) Representative images of yeast strains expressing wild-type (WT) Ura7p-GFP or Ura7p-GFP where specific phosphorylation sites are mutated. The average percentage and SEM of cells with Ura7p-GFP structures are indicated below each image. (B) Percentages of foci (<0.75 μm) and filaments (≥0.75 μm) for Ura7p-GFP structures are graphed for WT and each mutant for comparison.
In contrast, changing serine S354, S424, or S330 to alanine had no effect on the frequency of cells showing filament formation (Figure 6A). However, the S354A mutation caused a significant shift in the length distribution toward shorter structures, suggesting that S354 might play a role in length control (Table 1 and Figure 6B).
DISCUSSION
The discovery of a large number of enzymes that assemble into distinct intracellular structures in response to specific metabolic conditions suggests that the formation of these structures is connected to the regulation of their enzyme activity. To address this, we used the highly conserved filament-forming behavior of CTP synthase as a test case to determine how enzyme activity is connected to filament formation. Yeast CTP synthase is activated by GTP-induced allosteric changes and ATP/UTP/CTP-induced tetramerization and is inhibited by CTP via feedback repression (Pappas et al., 1998). Phosphorylation has also been found to both positively and negatively regulate enzyme activity (Choi et al., 2003; Park et al., 2003). Here we systematically mutated sites required for these forms of enzyme regulation to assess their role in controlling CTP synthase polymerization. These studies revealed that the regulation of CTP synthase activity is tightly coupled to the control of filament formation and/or filament length. Furthermore, our results argue that CTP synthase filaments comprise an inactive form of the enzyme. Because many enzymes that form foci/filaments are regulated by mechanisms similar to those that control CTP synthase, our work suggests that the close coupling of enzyme activity to filament assembly may be a general feature of this class of metabolic enzymes.
Strikingly, our studies found that a mutation in the UTP-binding site that blocks tetramerization increases the frequency of filament formation without altering the length distribution of the filaments. Because only the tetrameric form of CTP synthase is active, this is strong evidence that CTP synthase filaments comprised the inactive form of the enzyme. The finding that an active-site mutation that blocks catalytic activity has no effect on filament length or frequency suggests that it is not the loss of catalytic activity that drives polymerization, but that the shift toward dimers is responsible for the observed effects. These data are consistent with the fact that two mutations engineered to cripple binding of ATP (D70A and E146A) and hence tetramerization also increase filament formation but have little effect on filament length (Figure 7). Although we focused on the simplest model in which dimers polymerize directly due to the fact that only dimeric and tetrameric forms of CTP synthase exist, it is still possible that the dimers could form a novel oligomer before polymerization. Structural studies of the CTP synthase filament will be necessary to resolve this issue.
FIGURE 7.
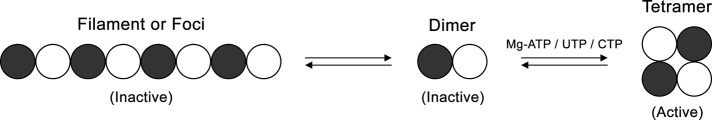
Model for CTP synthase filament formation. The transition of CTP synthase from an inactive dimer to an active tetramer is regulated by ATP, UTP, and CTP binding. We propose that the dimer can also partition into filaments in an inactive state.
Although the model of a CTP synthase filament comprising dimers is an attractive one, our studies of the L11 lid suggest that additional conformational changes are required for efficient polymerization. All of the mutations in the L11 lid that we analyzed increased the frequency of filament formation, suggesting that the conformation of this domain clearly contributes to the regulation of filament formation. Both mutations that prevent allosteric regulation by GTP, as well as one that causes increased activation by GTP, increase filament assembly. However, they have opposite effects on filament length. Of interest, the glutamine analogue 6-diazo-5-oxo-l-norleucine, which binds to the glutamine amidotransferase domain, has been found to eliminate CTP synthase filaments in E. coli and increase filament formation in Drosophila and human cells (Ingerson-Mahar et al., 2010; Carcamo_et al._, 2011; Chen_et al._, 2011). Our results, together with the previous inhibitor experiments, suggest that there are specific conformational changes within the L11 lid and likely other parts of the glutamine amidotransferase domain that affect the ability of CTP synthase to form either long filaments or short foci.
Another major site for controlling the length distribution of CTP synthase filaments is the feedback inhibition site. An E161K mutation blocks CTP binding to CTP synthase, causing a corresponding increase in enzyme activity due to the loss of feedback inhibition by CTP, as well as a decreased tendency to tetramerize. Our quantitative analysis of this mutation revealed that it completely eliminates filament assembly, producing only foci and further causing an increase in the frequency of foci formation. Furthermore, our analysis of an Ura7p double mutant that both has lost feedback inhibition (E161K) and is catalytically defective (C404G) found that catalytic activity is not required for the loss of filament formation, arguing that the increase in enzyme activity is not responsible for the block in filament assembly. Thus, like the UTP/ATP substrate binding sites, the product feedback inhibition site probably increases the frequency of structure formation via effects on tetramer formation. Further, like the allosteric L11 lid, this site also plays a critical role in controlling filament length/structure.
Of interest, although disruption of tetramerization, allosteric regulation, and feedback inhibition all appear to have strong effects on filament formation, only one of the four phosphorylation-site mutations, S354A, had effects on filament assembly that could not be attributable to the likely disruption of the CTP synthase structure. S354 appears to contribute to the control of filament length, since the S354A mutation caused an increase in foci relative to filaments while leaving the frequency of filament formation unaltered. Intriguingly, this residue is located on a surface loop ∼10–15 Å from the L11 lid on the same face of the glutamine amidotransferase domain, further suggesting that this domain plays a critical role in filament structure.
In sum, our results argue that the mechanisms that control CTP synthase activity, allosteric changes, tetramerization, feedback inhibition, and phosphorylation are also regulators of both the length of CTP synthase filaments and number of CTP synthase filaments that form. These data would be consistent with a model of a CTP synthase polymer in which inactive dimers of the enzyme interacted via the surface of the glutamine amidotransferase domain containing the L11 lid and the surface of the amidoligase domain near the CTP-binding site (Figure 7). Further structural studies will be required to confirm this proposal. The regulation of supramolecular complex formation in other enzymes may be similarly coupled to the known mechanisms for regulating the activity of those enzymes.
One of the most surprising results of our study is the finding that Ura7p can form two distinct supramolecular complexes: foci and filaments. It is unclear whether these two structures represent two distinct regulatory structures or whether foci formation is a critical step on the path to assembling a full-length filament. It is worth noting, however, that we observe primarily long filaments in older cultures (overnight to 5 d) and that acute shift to low glucose yields structures that are more like foci (unpublished data; Noree_et al._, 2010). This suggests that these two structures may be kinetically related to foci forming first, followed by maturation into a filament. This model is also consistent with the opposite effects of the E161K and G148A mutations. Both disrupt nucleotide-stimulated tetramerization and would be predicted to promote filament formation, but the E161K mutation forms only foci, whereas the G148A mutation promotes filament formation. If foci were a precursor to filaments, this would explain why both mutations increase the number of structures (foci or filaments). This also suggests that the E161 residue is necessary for the foci-to-filament transition. The possible existence of a precursor structure that matures into a filament would make these filaments distinct from classical cytoskeletal filaments and might explain why protein expression level is poorly correlated with filament assembly (Supplemental Figure S1). In vitro reconstitution and structural analysis of both foci and filaments will be necessary to firmly establish the relationship between these two structures.
Although we find that there is a close coupling between the regulation of enzyme activity and filament assembly, the question of why CTP synthase polymerizes remains unanswered. Given that CTP synthase is already regulated by feedback repression, allosteric activation, phosphorylation, and substrate-induced tetramerization, adding regulation via polymerization would seem superfluous. The fact that CTP synthase polymerizes in growth conditions in which nutrients are limiting, such as stationary phase, suggests that filament formation is used to regulate enzyme activity in response to a specific metabolic state. Feedback repression, allosteric activation, and substrate-induced tetramerization all respond rapidly to changes in metabolite levels. In contrast, a metabolic polymer that has nucleation-limited assembly and end-limited disassembly might be less responsive to metabolite levels and allow transient changes in metabolites to be ignored due to the lag in assembly and the fact that exiting the polymer will be dependent on the number of filament ends. This might be particularly important when cells exit states, such as stationary phase, where the cell might not want to activate metabolic pathways unless there is a clear, sustained change in the energy status of the cell. Given the recent progress in metabolomics, it should be possible to test this proposed role of supramolecular enzyme structures in regulating metabolic network activity.
MATERIALS AND METHODS
Media and yeast strains
All yeast strains were derived from a parent strain with the genotype MATa his3∆1 leu2∆0 met15∆0 ura3∆0 (BY4741). Strains with GFP-tagged genes were from the yeast GFP collection (Howson_et al._, 2005). All yeast strains were grown at 30°C in YPD unless otherwise indicated.
URA7-GFP plasmids were constructed with standard molecular biology techniques. A DNA cassette containing URA7::GFP plus 501 base pairs upstream of the_URA7_ start codon was amplified by PCR from genomic DNA isolated from yeasts containing URA7::GFP (from the yeast GFP collection; Invitrogen) using JW1064 and JW925 (sequences available on request). This URA7-GFP cassette was then subcloned into a pRS403 plasmid (a gift from Randy Hampton, University of California, San Diego). The resulting plasmid, named JW206 (created by Brian Sato, University of California, Irvine), was then used as a base plasmid for generating mutations in the URA7 gene by PCR-based site-directed mutagenesis, which were then validated by sequencing.
To introduce URA7::GFP variants into the endogenous URA7 locus in yeasts, the mutant plasmids were used as templates to PCR amplify a cassette containing coding region of ura7::GFP, a copy of HIS3 sequence (selectable marker), and a sequence homologous to 50 base pairs downstream of the URA7 stop codon (required for homologous recombination at the endogenous URA7 locus). Yeasts were transformed with the purified PCR product of the ura7::GFP cassette via the heat shock method (Noree et al., 2010), incubated overnight at 30°C, and then replica plated onto histidine-dropout plates. All yeast mutants were verified by DNA sequencing (Eton Bioscience and Retrogen).
Quantitation of URA7-encoded CTP synthase foci/filaments
Wild-type or mutated URA7::GFP strains were grown in 5 ml of YPD at 30°C with shaking for 1 d. Cells were fixed by adding 100 μl of 37% (wt/vol) formaldehyde (Fisher Scientific) to 1 ml of yeast liquid culture, incubated on a rotating platform for at least 15 min at room temperature, collected by centrifugation at 6000 rpm for 1 min, and washed once with sterile water. The cells were then resuspended in 1 M sorbitol (US Biologicals). A slide was prepared by pipetting a few microliters of the cell suspension onto a slide, which was then covered by a coverslip and inverted, with some pressure applied on the slide to allow excess liquid to be removed from the sample to improve imaging.
To determine the percentage of cells containing Ura7p-GFP structures, five different areas were selected (∼50 cells/area) for counting using a Zeiss Axiovert 200M microscope with a 100× Plan-Apochromat 100×/1.40 oil objective lens. The total number of cells and the number of cells with Ura7p-GFP structures were determined and reported as a percentage of cells showing Ura7p-GFP foci/filaments. Experiments were repeated five times for graphing and statistical analysis (mean ± SEM).
For analysis of the length distribution of Ura7p-GFP foci and filaments, imaging was performed using a DeltaVision system with an Olympus IX70 microscope, Olympus PlanApo 60×/1.40 Oil objective, and softWoRx software, version 2.5 (Applied Precision). At least 10 areas on the slide were randomly picked. For each, images in the _Z_-axis were taken every 0.2 μm over ∼1–2 μm. Each was then deconvolved and compressed into a single image. The processed images were then quantified using ImageJ (National Institutes of Health, Bethesda, MD). Each image was transformed into 8-bit format, adjusted to the threshold with a setting at 30, 255. The foci and filaments in each image were computed via the function Analyze Particles, excluding structures with size <0.01 μm2. The value of the “major axis” of each Ura7p-GFP structure was collected for preparing a graphic distribution of Ura7p-GFP foci and filaments (∼100–300 structures were analyzed for each mutant). Three independent experiments were done for each condition or mutation. Statistical analysis was performed using Prism 5 (GraphPad Software). One-way analysis of variance was used to test whether there was a statistically significant difference between wild type and each mutant in the length distribution of Ura7p-GFP structures.
Protein sample preparation and Western blot analysis
Whole-cell extracts were obtained by growing yeast cells in the indicated conditions. The cells with OD600 of 2.5 were harvested by centrifugation at 6000 rpm for 1 min and resuspended in 100 μl of sterile water. Next this suspension was treated with an additional 100 μl of 0.2 N sodium hydrochloride (Fisher Scientific). After a 5-min incubation at room temperature, cells were collected by centrifugation at 6000 rpm for 1 min. SDS–PAGE loading buffer (with 1× Protease Inhibitor Cocktail; Sigma-Aldrich) was added to the cell pellet. After vortexing vigorously and boiling for 5 min, the sample was spun down at 10,000 rpm for 1 min and resolved by 10% SDS–PAGE. Proteins were transferred to a nitrocellulose membrane (BioRad) by electroblotting (Owl HEP-1; Thermo Scientific). Then standard protocol for Western blot was performed. To detect GFP-tagged proteins, 1:5000 rabbit anti-GFP (Torrey Pines Biolabs) was used as a primary antibody and 1:10,000 ECL donkey anti-rabbit immunoglobulin G (IgG), horseradish peroxidase–linked whole antibody (GE Healthcare UK) as a secondary antibody. For internal loading control detection, 1:10,000 mouse anti–3-phosphoglycerate kinase (yeast) IgG1 monoclonal antibody (Invitrogen) was used as a primary antibody and 1:2500 ECL sheep anti-mouse IgG, horseradish peroxidase–linked whole antibody (GE Healthcare UK) as a secondary antibody. Quantitation of each band was done using ImageJ.
Supplementary Material
Supplemental Materials
Acknowledgments
We thank D. Forbes for critical reading of the manuscript. This work was supported by National Science Foundation Grant IOS-1144409 and San Diego Foundation Blasker Science and Technology Grant C-2011-0218 (to J.E.W.).
Abbreviations used:
CTP
cytidine 5’-triphosphate
GFP
green fluorescent protein
GTP
guanosine 5’-triphosphate
UTP
uridine 5’-triphosphate
YPD
yeast extract-peptone-dextrose–containing medium
Footnotes
*These authors contributed equally to this work.
REFERENCES
- An S, Deng Y, Tomsho JW, Kyoung M, Benkovic SJ. Microtubule-assisted mechanism for functional metabolic macromolecular complex formation. Proc Natl Acad Sci USA. 2010a;107:12872–12876. doi: 10.1073/pnas.1008451107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An S, Kumar R, Sheets ED, Benkovic SJ. Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science. 2008;320:103–106. doi: 10.1126/science.1152241. [DOI] [PubMed] [Google Scholar]
- An S, Kyoung M, Allen JJ, Shokat KM, Benkovic SJ. Dynamic regulation of a metabolic multi-enzyme complex by protein kinase CK2. J Biol Chem. 2010b;285:11093–11099. doi: 10.1074/jbc.M110.101139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronow B, Ullman B. In situ regulation of mammalian CTP synthetase by allosteric inhibition. J Biol Chem. 1987;262:5106–5112. [PubMed] [Google Scholar]
- Campbell SG, Hoyle NP, Ashe MP. Dynamic cycling of eIF2 through a large eIF2B-containing cytoplasmic body: implications for translation control. J Cell Biol. 2005;170:925–934. doi: 10.1083/jcb.200503162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carcamo WC, et al. Induction of cytoplasmic rods and rings structures by inhibition of the CTP and GTP synthetic pathway in mammalian cells. PLoS One. 2011;6:e29690. doi: 10.1371/journal.pone.0029690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YF, Martin SS, Baldwin EP, Carman GM. Phosphorylation of human CTP synthetase 1 by protein kinase C: identification of Ser(462) and Thr(455) as major sites of phosphorylation. J Biol Chem. 2007;282:17613–17622. doi: 10.1074/jbc.M702799200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Zhang J, Tastan OY, Deussen ZA, Siswick MY, Liu JL. Glutamine analogs promote cytoophidium assembly in human and Drosophila cells. J Genet Genomics. 2011;38:391–402. doi: 10.1016/j.jgg.2011.08.004. [DOI] [PubMed] [Google Scholar]
- Choi MG, Carman GM. Phosphorylation of human CTP synthetase 1 by protein kinase A: identification of Thr455 as a major site of phosphorylation. J Biol Chem. 2007;282:5367–5377. doi: 10.1074/jbc.M610993200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi MG, Park TS, Carman GM. Phosphorylation of Saccharomyces cerevisiae CTP synthetase at Ser424 by protein kinases A and C regulates phosphatidylcholine synthesis by the CDP-choline pathway. J Biol Chem. 2003;278:23610–23616. doi: 10.1074/jbc.M303337200. [DOI] [PubMed] [Google Scholar]
- Endrizzi JA, Kim H, Anderson PM, Baldwin EP. Crystal structure of Escherichia coli cytidine triphosphate synthetase, a nucleotide-regulated glutamine amidotransferase/ATP-dependent amidoligase fusion protein and homologue of anticancer and antiparasitic drug targets. Biochemistry. 2004;43:6447–6463. doi: 10.1021/bi0496945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endrizzi JA, Kim H, Anderson PM, Baldwin EP. Mechanisms of product feedback regulation and drug resistance in cytidine triphosphate synthetases from the structure of a CTP-inhibited complex. Biochemistry. 2005;44:13491–13499. doi: 10.1021/bi051282o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howson R, Huh WK, Ghaemmaghami S, Falvo JV, Bower K, Belle A, Dephoure N, Wykoff DD, Weissman JS, O'Shea EK. Construction, verification and experimental use of two epitope-tagged collections of budding yeast strains. Comp Funct Genomics. 2005;6:2–16. doi: 10.1002/cfg.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingerson-Mahar M, Briegel A, Werner JN, Jensen GJ, Gitai Z. The metabolic enzyme CTP synthase forms cytoskeletal filaments. Nat Cell Biol. 2010;12:739–746. doi: 10.1038/ncb2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitzki A, Koshland DE., Jr Ligand-induced dimer-to-tetramer transformation in cytosine triphosphate synthetase. Biochemistry. 1972a;11:247–253. doi: 10.1021/bi00752a016. [DOI] [PubMed] [Google Scholar]
- Levitzki A, Koshland DE., Jr Role of an allosteric effector. Guanosine triphosphate activation in cytosine triphosphate synthetase. Biochemistry. 1972b;11:241–246. doi: 10.1021/bi00752a015. [DOI] [PubMed] [Google Scholar]
- Levitzki A, Stallcup WB, Koshland DE., Jr Half-of-the-sites reactivity and the conformational states of cytidine triphosphate synthetase. Biochemistry. 1971;10:3371–3378. doi: 10.1021/bi00794a009. [DOI] [PubMed] [Google Scholar]
- Liu JL. Intracellular compartmentation of CTP synthase in Drosophila. J Genet Genomics. 2010;37:281–296. doi: 10.1016/S1673-8527(09)60046-1. [DOI] [PubMed] [Google Scholar]
- Long CW, Pardee AB. Cytidine triphosphate synthetase of Escherichia coli B. I. Purification and kinetics. J Biol Chem. 1967;242:4715–4721. [PubMed] [Google Scholar]
- Lunn FA, Macleod TJ, Bearne SL. Mutational analysis of conserved glycine residues 142, 143 and 146 reveals Gly(142) is critical for tetramerization of CTP synthase from Escherichia coli. Biochem J. 2008;412:113–121. doi: 10.1042/BJ20071163. [DOI] [PubMed] [Google Scholar]
- Narayanaswamy R, Levy M, Tsechansky M, Stovall GM, O'Connell JD, Mirrielees J, Ellington AD, Marcotte EM. Widespread reorganization of metabolic enzymes into reversible assemblies upon nutrient starvation. Proc Natl Acad Sci USA. 2009;106:10147–10152. doi: 10.1073/pnas.0812771106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noree C, Sato BK, Broyer RM, Wilhelm JE. Identification of novel filament-forming proteins in Saccharomyces cerevisiae and Drosophila melanogaster. J Cell Biol. 2010;190:541–551. doi: 10.1083/jcb.201003001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrander DB, O'Brien DJ, Gorman JA, Carman GM. Effect of CTP synthetase regulation by CTP on phospholipid synthesis in Saccharomyces cerevisiae. J Biol Chem. 1998;273:18992–19001. doi: 10.1074/jbc.273.30.18992. [DOI] [PubMed] [Google Scholar]
- Ozier-Kalogeropoulos O, Adeline MT, Yang WL, Carman GM, Lacroute F. Use of synthetic lethal mutants to clone and characterize a novel CTP synthetase gene in Saccharomyces cerevisiae. Mol Gen Genet. 1994;242:431–439. doi: 10.1007/BF00281793. [DOI] [PubMed] [Google Scholar]
- Ozier-Kalogeropoulos O, Fasiolo F, Adeline MT, Collin J, Lacroute F. Cloning, sequencing and characterization of the Saccharomyces cerevisiae URA7 gene encoding CTP synthetase. Mol Gen Genet. 1991;231:7–16. doi: 10.1007/BF00293815. [DOI] [PubMed] [Google Scholar]
- Paluh JL, Zalkin H, Betsch D, Weith HL. Study of anthranilate synthase function by replacement of cysteine 84 using site-directed mutagenesis. J Biol Chem. 1985;260:1889–1894. [PubMed] [Google Scholar]
- Pappas A, Yang WL, Park TS, Carman GM. Nucleotide-dependent tetramerization of CTP synthetase from Saccharomyces cerevisiae. J Biol Chem. 1998;273:15954–15960. doi: 10.1074/jbc.273.26.15954. [DOI] [PubMed] [Google Scholar]
- Park TS, O'Brien DJ, Carman GM. Phosphorylation of CTP synthetase on Ser36, Ser330, Ser354, and Ser454 regulates the levels of CTP and phosphatidylcholine synthesis in Saccharomyces cerevisiae. J Biol Chem. 2003;278:20785–20794. doi: 10.1074/jbc.M301394200. [DOI] [PubMed] [Google Scholar]
- Park TS, Ostrander DB, Pappas A, Carman GM. Identification of Ser424 as the protein kinase A phosphorylation site in CTP synthetase from Saccharomyces cerevisiae. Biochemistry. 1999;38:8839–8848. doi: 10.1021/bi990784x. [DOI] [PubMed] [Google Scholar]
- Sheth U, Parker R. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science. 2003;300:805–808. doi: 10.1126/science.1082320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan J, Phear G, Yamauchi M, Meuth M. Clustered base substitutions in CTP synthetase conferring drug resistance in Chinese hamster ovary cells. Nat Genet. 1993;3:317–322. doi: 10.1038/ng0493-317. [DOI] [PubMed] [Google Scholar]
- Willemoes M, Molgaard A, Johansson E, Martinussen J. Lid L11 of the glutamine amidotransferase domain of CTP synthase mediates allosteric GTP activation of glutaminase activity. FEBS J. 2005;272:856–864. doi: 10.1111/j.1742-4658.2004.04525.x. [DOI] [PubMed] [Google Scholar]
- Yang WL, Bruno ME, Carman GM. Regulation of yeast CTP synthetase activity by protein kinase C. J Biol Chem. 1996;271:11113–11119. doi: 10.1074/jbc.271.19.11113. [DOI] [PubMed] [Google Scholar]
- Yang WL, Carman GM. Phosphorylation and regulation of CTP synthetase from Saccharomyces cerevisiae by protein kinase A. J Biol Chem. 1996;271:28777–28783. doi: 10.1074/jbc.271.46.28777. [DOI] [PubMed] [Google Scholar]
- Yang WL, McDonough VM, Ozier-Kalogeropoulos O, Adeline MT, Flocco MT, Carman GM. Purification and characterization of CTP synthetase, the product of the URA7 gene in Saccharomyces cerevisiae. Biochemistry. 1994;33:10785–10793. doi: 10.1021/bi00201a028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Materials