Mutations of CEP83 Cause Infantile Nephronophthisis and Intellectual Disability (original) (raw)
Abstract
Ciliopathies are a group of hereditary disorders associated with defects in cilia structure and function. The distal appendages (DAPs) of centrioles are involved in the docking and anchoring of the mother centriole to the cellular membrane during ciliogenesis. The molecular composition of DAPs was recently elucidated and mutations in two genes encoding DAPs components (CEP164/NPHP15, SCLT1) have been associated with human ciliopathies, namely nephronophthisis and orofaciodigital syndrome. To identify additional DAP components defective in ciliopathies, we independently performed targeted exon sequencing of 1,221 genes associated with cilia and 5 known DAP protein-encoding genes in 1,255 individuals with a nephronophthisis-related ciliopathy. We thereby detected biallelic mutations in a key component of DAP-encoding gene, CEP83, in seven families. All affected individuals had early-onset nephronophthisis and four out of eight displayed learning disability and/or hydrocephalus. Fibroblasts and tubular renal cells from affected individuals showed an altered DAP composition and ciliary defects. In summary, we have identified mutations in CEP83, another DAP-component-encoding gene, as a cause of infantile nephronophthisis associated with central nervous system abnormalities in half of the individuals.
Main Text
Nephronophthisis (MIM 256100) is the most frequent genetic cause of chronic renal failure in children. It is an autosomal-recessive chronic tubulointerstitial nephritis that generally progresses to end-stage renal disease (ESRD) in childhood. With regards to the age of onset of ESRD, three clinical presentations have been described: (1) the infantile form, with ESRD appearing before 5 years, (2) the juvenile form (mean age of onset 13 years), and (3) the young adult form. These different forms share histological features characterized by the development of massive interstitial fibrosis with abnormal thickness of the tubular basement membranes and at the later stage formation of cysts mainly distributed at the cortico-medullary junction. Infantile nephronophthisis is generally characterized by hyperechogenic kidneys with variable kidney sizes.1 Some affected individuals with nephronophthisis also present with extrarenal symptoms: retinitis pigmentosa, intellectual disability, cerebellar ataxia, bone anomalies, or liver fibrosis, defining syndromes referred as nephronophthisis-related ciliopathies (NPHP-RCs).2
Ciliopathies are clinically and genetically heterogeneous disorders associated with dysfunction of cilia, either primary and/or motile cilia (primary ciliary dyskinesia [MIM 244400]). Primary cilia, sensory generally nonmotile cilia, are present on most cells in vertebrates and control key signaling pathways during development and tissue homeostasis.2,3 In total, more than 17 genes have been associated with the NPHP-RCs and causative mutations in these genes account for approximately 30%–50% of the NPHP-RC cases.4,5 Almost all these genes encode for proteins localized to the proximal region of the primary cilium either at the transition zone (including NPHP1 [MIM 607100], NPHP4 [MIM 6007215], and RPGRIP1L/NPHP8 [MIM 610937]) or at the inversin compartment (INVS/NPHP2 [MIM 243305], NPHP3 [MIM 608002], NEK8/NPHP9 [MIM 609799], and ANKS6/NPHP16 [MIM 615370]) where they are implicated in key cilia-dependent signaling pathways.2,4 Notably, mutations in INVS/NPHP2, NPHP3, and NEK8/NPHP9 have been associated with infantile nephronophthisis.6–8 More recently, our group and others have shown that mutations in genes encoding intraflagellar transport (IFT)-A as well as in some IFT-B subunits and IFT motors result in isolated nephronophthisis as well as in skeletal ciliopathies.2,4,5,9–11 The IFT machinery mediates the transport of the ciliary components into the cilium and along the axoneme.12
Ciliogenesis starts with the docking of the basal body to the plasma membrane. This process is mediated by structures present at the mother centriole called distal appendages (DAPs). The DAPs are projected radially from the distal end of the mother centriole and dock onto the cytoplasmic leaflet of the plasma membrane where they form the transition fibers.13 The transition fibers together with the adjacent transition zone are thought to constitute a molecular filter limiting the exchanges between the ciliary compartment and the cytoplasm.14 The molecular composition of DAPs was recently characterized and their function in ciliogenesis has been partially elucidated (see below).15–18 Mutations in genes encoding DAP components have been associated with NPHP-RC, including CEP164/NPHP15 (MIM 614848) in Joubert syndrome19 (MIM 613820) and more recently SCLT1 (MIM 611399) in one individual with a severe ciliopathy phenotype consistent with orofaciodigital syndrome type IX (MIM 258865).20
In order to identify additional genes mutated in ciliopathies, we performed two independent NGS approaches on a large cohort of affected individuals. First, we analyzed a cohort of 200 individuals with NPHP-RCs by applying exon-enriched NGS targeting up to 1,221 genes associated with cilia including all known genes associated with ciliopathies (“ciliome sequencing”)9,21,22 and consecutive barcoded NGS on a SoliD5500 (Life tech) or HiSeq 2500 (Illumina) platform. Written informed consent was obtained from all individuals enrolled in this study and/or from parents and approved by the Comité de Protection des Personnes pour la Recherche Biomédicale Ile de France II. Compound heterozygous mutations in CEP83 (CCDC41) were identified in three unrelated individuals with NPHP-RC (Table 1, Figure 1, and Figure S1 available online). All three affected individuals (NPH2036, NPH1402, and NPH1412-II1) presented with a severe renal involvement progressing to ESRD before the age of 5 years. We subsequently screened 19 unrelated individuals with early-onset nephronophthisis for CEP83 mutations in the coding exons by Sanger sequencing. Recessive mutations were identified in two additional unrelated persons, individuals NPH982 and NPH1018 (Table 1, Figures 1 and S1). Segregation of the identified mutations was confirmed whenever the parental DNA was available (Figure S1). Notably, individual NPH1412-II1 carried the same mutations as his brother, NPH1412-II2, who presents a similar phenotype (Table 1 and Figure S1). In parallel, we screened a worldwide cohort of 1,056 individuals with NPHP-RCs by exon sequencing of DAP components (CEP89 [_CCDC123/_CEP123, MIM 615470], CEP83, SCLT1, and FBF1) by a high-throughput barcoded NGS technique.23 Written informed consent was obtained from all individuals enrolled in this study and approved by the institutional review boards at the University of Michigan and the Boston Children’s Hospital. Two homozygous mutations in CEP83 were identified in two unrelated persons with early-onset nephronophthisis, individuals F374-21 and A4037-21 (Table 1, Figures 1 and S1). In these two families, the 17 genes already known to be associated with NPHP-RC were screened in order to exclude another genetic cause but no explanatory mutations were detected.
Table 1.
Genetic and Clinical Characteristics of Individuals with CEP83 Mutations
| Individual(s) | Ethnic Origin | Parental Consanguinity | Nucleotide Alteration(s)a | Deduced Protein Change | Exon (Zygosity, Segregation) | AA Conservation | PolyPhen2, Mut.taster | Nephronophthisis Age of ESRD | Additional Clinical Features |
|---|---|---|---|---|---|---|---|---|---|
| NPH2036-II1 | European (France) | no | c.121C>T | p.Arg41∗ | 3 (Het, p) | NA | NA | 3 years | mild intellectual disability, strabismus, hepatic cytolysis, cholestasis |
| c.335_352del | p.Pro112_Leu117del | 5 (Het, m) | NA | NA | |||||
| NPH1402 | European (France) | no | c.241C>T | p.Gln81∗ | 4 (Het, m) | NA | NA | 4.5 years | speech delay, hydrocephalus |
| c.2075_2077del | p.Gln692del | 17 (Het, p) | NA | NA | |||||
| F374-21 | European | no | c.260T>C | p.Leu87Pro | 4 (Hom) | D. rerio (except Xenopus) | D(0.944), DC(0.99) | ND | ND |
| NPH1018 | European (France) | no | c.625C>T | p.Arg209∗ | 7 (Het) | NA | NA | 4 years | intellectual disability, retinitis |
| c.1532G>C | p.Arg511Pro | 13 (Het) | G. gallus | D(0.915), DC(0.941) | |||||
| NPH1412-II1, -II2 | European (Poland/France) | no | c.1530C>A | p.Cys510∗ | 13 (Het, m) | NA | NA | 3 years | high blood pressure |
| c.1532G>C | p.Arg511Pro | 13 (Het, p) | G. gallus | D(0.915), DC(0.941) | |||||
| NPH982 | Turkish | yes | c.2050_2052del | p.Glu684del | 17 (Hom) | D. reriob | NA | 3 years | high blood pressure, hepatic fibrosis |
| A4037-21 | Latino | no | c.2007del | p.Glu669Aspfs∗14 | 17 (Hom) | NA | NA, DC(1.00) | 1 year | hydrocephalus, intellectual disability, dysmorphism, triple X syndrome (47, XXX), heart anomaly |
Figure 1.
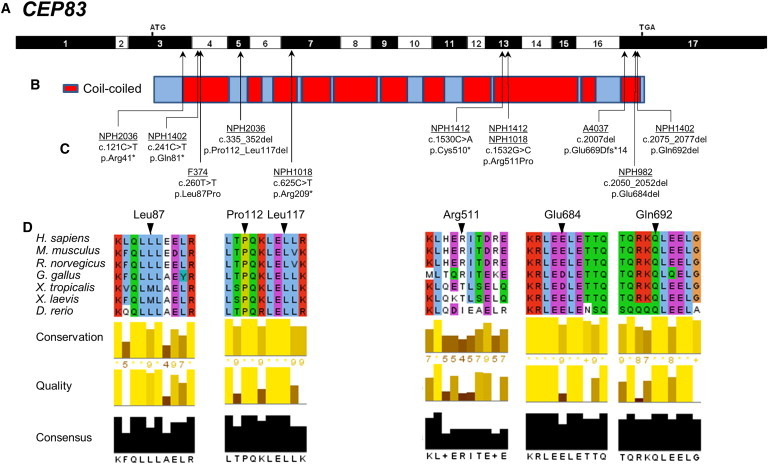
Identification of Ten Different CEP83 Mutations in Seven Families with NPHP-RC
(A) Exon structure of human CEP83 cDNA (RefSeq NM_016122.2). Positions of start codon (ATG) and of stop codon (TGA) are indicated in exons 3 and 17, respectively.
(B) Domain structure of the respective protein. CEP83 contains multiple coiled-coil (red) domains.
(C) Ten homozygous or compound-heterozygous CEP83 mutations. For the mutations detected, black arrows indicate positions in relation to exons and protein domains. Except for the c.335_352del mutation, all mutations concern amino acids found in coiled-coil domains, mostly in the N-terminal and C-terminal parts of the protein. Family numbers are underlined and predicted translational changes are indicated. Affected individuals had at least one missense mutation and a deletion.
(D) Partial protein alignment of CEP83 shows evolutionary conservation of the amino acids affected by the identified mutations (p.Leu87Pro, p.Pro112_Leu117del, p.Arg511Pro, p.Glu684del, and p.Gln692del).
In total, we identified ten different biallelic mutations in CEP83 in eight individuals with isolated (three individuals) or syndromic (five individuals) nephronophthisis. The affected individuals reached ESRD at 1 to 4 years of age (mean age of 3). Severe tubulointerstitial lesions were present in all individuals (Figure 2). Similar to what we previously reported for INVS/NPHP2 and NPHP3 mutations,8 two types of nephrohistological alterations were found in individuals with CEP83 mutations: (1) in three individuals (NPH1412, A4037-21, and NPH982), microcystic tubular dilatations were prominent and associated with tubular atrophy and interstitial fibrosis and (2) in two individuals (NPH2036 and NPH982), atrophic tubules with thickening of the basement membranes and massive interstitial fibrosis were observed. Moreover, high blood pressure was present in two individuals (Table 1) as already described in the early onset of the kidney disease.8 Four affected persons (NPH1402, NPH2036, A4037-21, NPH1018) presented with neurological alterations, including speech delay, intellectual disability, and/or hydrocephalus supported by cerebral MRI in combination with ophthalmologic defects, strabismus and retinal degeneration, in two individuals (NPH2036 and NPH1018, respectively). Two individuals presented with liver alterations, hepatic cytolysis and cholestasis (NPH2036) and severe portal septal fibrosis with mild thickening of arterial walls and increase in the number of the biliary canalicules on liver biopsy (NPH982). Moreover, individual A4037-21 exhibited a more severe phenotype with ESRD at 1 year of age, hydrocephalus, intellectual disability, facial dysmorphism, and heart anomaly complicated by triple X syndrome (47, XXX), which could not explain this clinical association (Table 1, Figures 1 and S1).
Figure 2.
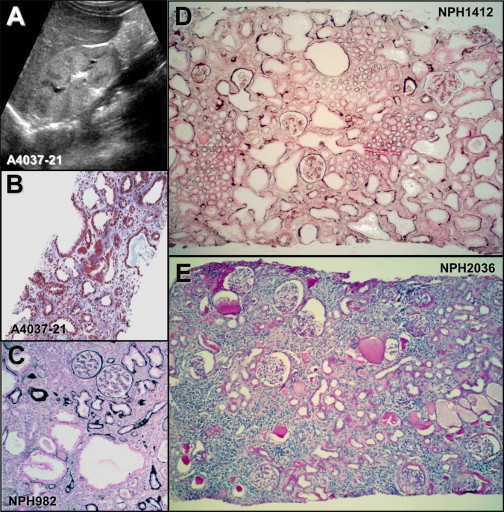
Sonographic and Histological Lesions in Individuals with CEP83 Mutations
(A) Renal ultrasound of individual A4037-21 shows corticomedullary cysts and increased echogenicity.
(B–E) Renal biopsy findings in individuals A4037-21 (B), NPH982 (C), NPH1412 (D), and NPH2036 (E).
(B) Tubular dilatations are associated with diffuse interstitial fibrosis in individual A4037-21. Trichrome light green.
(C–E) Numerous glomeruli are preserved and others show a retracted glomerular tuft surrounded by a thickened capsular basement membrane.
(C) Microcystic tubular dilatations are associated with foci of tubular atrophy and intersitial fibrosis in individual NPH982.
(D) In individual NPH1412, most cortical tubules are dilated and surrounded by interstitial fibrosis.
(C and D) Silver impregnation.
(E) Diffuse tubular atrophy, massive interstitial fibrosis, and focal interstitial infiltration are associated in individual NPH2036. PAS.
Three affected individuals carried homozygous mutations of CEP83 (RefSeq accession number NM_016122.2): one missense mutation (c.260T>C [p.Leu87Pro]), one in-frame deletion (c.2050_2052del [p.Glu684del]), and one frameshift mutation in the last exon (c.2007del [p.Glu669Aspfs∗14]) leading to a truncated protein (Table 1, Figures 1 and S1). Four families carried compound heterozygous mutations of CEP83: one truncating mutation (c.121C>T [p.Arg41∗]; c.241C>T [p.Gln81∗]; c.625C>T [p.Arg209∗]; c.1530C>A [p.Cys510∗]) in trans with a missense mutation (c.1532G>C [p.Arg511Pro]) or an in-frame deletion (c.335_352del [p.Pro112_Leu117del]; c.2075_2077del [p.Gln692del]). All the mutated or deleted amino acids are evolutionarily highly conserved and amino acid changes are predicted to be damaging (Figure 1D, Table 1). Almost all individuals carried at least one missense mutation or an in-frame deletion resulting in loss of one or a few amino acids, indicating that the function of the protein may be partially preserved (Figure 1, Table 1). Notably, only the individual A4037-21 carrying a homozygous frameshift mutation leading to a truncated protein exhibited a more severe phenotype of multiple organ involvement (Table 1, Figures 1 and S1).
CEP83 encodes Centrosomal Protein 83 kDa, CEP83, a 701-residue protein predicted to be mainly composed of coiled-coil domains (Figure 1B, red). Most of the mutations result in amino acid alteration in coiled-coil domains except for the p.Pro112_Leu117del variant (Figures 1B and 1C) and substitutions in highly conserved residues (Figure 1D). Deletion of a single amino acid (p.Glu684del and p.Gln692del) or the introduction of a proline residue (p.Leu87Pro and p.Arg511Pro) are predicted to affect the organization of coiled-coil domains through the modification of canonical repetition of hydrophobic amino acids along the alpha helix (Figure 1D).
CEP83 has been recently characterized as a key component of DAPs16,17 and is required for the recruitment of the other components, including its partner CEP164/NPHP15, to the mother centriole.16,17 As previously shown for CEP164,15,24 depletion of CEP83 affects early steps of ciliogenesis by preventing docking of the mother centriole to the primary ciliary vesicle (Figure S2).16,17 In retinal pigment epithelial (RPE1) cells, CEP83 colocalizes with CEP164 at DAPs16,17 and is also found at the Golgi apparatus.17 We observed a similar distribution in primary human skin fibroblasts of healthy controls by using a CEP83 antibody, with a predominant staining of one of the two centrioles (Figure 3A, insets, arrows). As previously shown in other cell types,15,19 CEP164 showed a centriolar ring-shaped distribution (Figure 3B, insets, arrows) and was also present in the nucleus.
Figure 3.
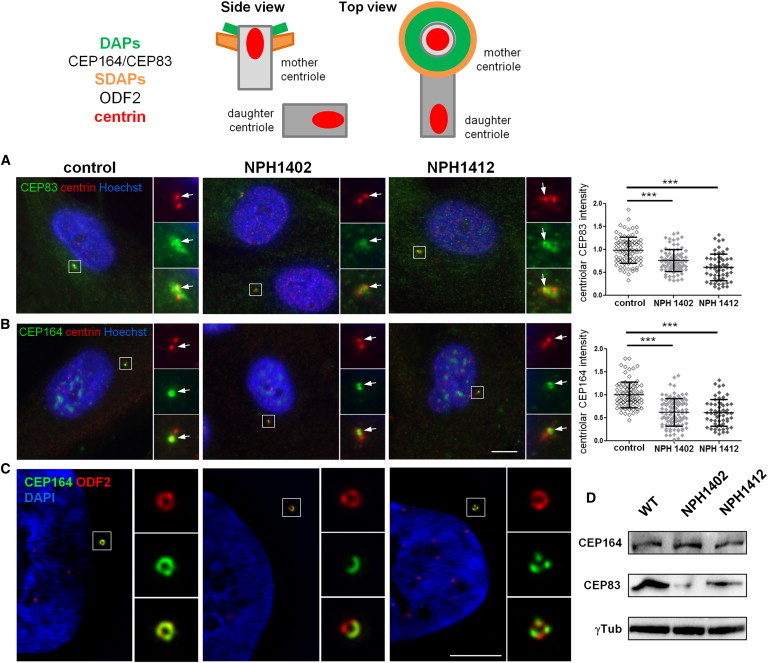
Individuals Carrying CEP83 Mutations Show Abnormal DAP Organization
Top: Schematic representation of the structural organization of the centrosome where the localization of different markers of centriolar subdomains (distal [DAPs] and subdistal [SDAPs]) are stressed.
(A–C) Fibroblasts from control or affected individuals (NPH1402 and NPH1412) were processed for immunofluorescence with antibodies against centrin (A and B, red; mouse monoclonal [Millipore, 04-1624]) to stain centrioles and CEP83 (A, green; rabbit polyclonal [Sigma, HPA038161]) or CEP164 (B, green; rabbit polyclonal [Sigma, HPA037606]) or with antibodies against ODF2 (C, red; mouse monoclonal antibody [clone 1A1, Novus Biological]) to stain SDAPs and CEP164 (C, green). Insets on the right show higher magnification of representative centrosomes indicated by a white square in the corresponding images. Arrows indicate CEP83 or CEP164 staining. The intensity of CEP83 (A) and CEP164 (B) staining at the centrosome was quantified by ImageJ (normalized arbitrary units, one representative experiment out of three [∗∗∗p < 0.0001], were calculated via Dunn’s Multiple Comparison Test after the analysis of variance ANOVA test) and shows a reduction in intensity of either CEP83 or CEP164 in affected individual cells compared to control.
(C) Samples were imaged with an n-SIM microscope (Nikon). Acquisitions were performed in 3D SIM mode, before image reconstruction with the NIS-Elements software (Nikon) based on Gustafsson et al.29
Scale bars represent 5 μm.
(D) Cell lysates from fibroblasts were collected and protein levels of CEP83, CEP164, and γ-tubulin (mouse monoclonal [Sigma, T5326]) were analyzed by immunoblotting with indicated antibodies. CEP83 level is decreased in affected individuals fibroblasts compared to control and CEP164 level remains the same.
To evaluate the impact of the identified mutations on the subcellular distribution of CEP83 and their functional consequences on DAP biogenesis, the localizations of CEP83 and CEP164 were analyzed in primary skin fibroblasts from two affected individuals (NPH1402 and NPH1412). Notably, both children carried compound heterozygous CEP83 mutations resulting in truncated protein (p.Gln81∗ and p.Cys510∗) in association with an in-frame deletion or a missense (p.Gln692del and p.Arg511Pro, respectively; Table 1 and Figure 1). Importantly, the resulting truncated proteins, if expressed, cannot be detected by the CEP83 antibody used in this study. Accordingly, CEP83 staining at the centrosome appeared fainter and dispersed around the centrioles in fibroblasts from affected individuals compared to control cells, as shown by CEP83 colabeling with centrin or γ-tubulin (Figure 3A, insets, arrows, and data not shown) and confirmed by quantification analysis (Figure 3A, graph). Remarkably, localization of CEP164 was severely altered in mutated cells with an incomplete ring-shaped centriolar distribution or even a loss of staining at the centrosome in some cells (Figures 3B, graph, and S3, arrows). The distribution of other markers, of DAPs (CEP89)18 or subdistal appendages (ODF2),16,25 did not appear to be affected by CEP83 variants (Figure S4, arrows). These results are in agreement with a partial impairment of DAP assembly in fibroblasts with CEP83 variants, sparing the overall organization of the mother centriole. This was further confirmed by high-resolution 3D fluorescence microscopy (structured illumination microscopy [SIM]; Figure 3C). Indeed, in affected individuals fibroblasts of the normal CEP164 ring-shaped organization appeared clearly defective according to this technique (Figure 3C, green), whereas ODF2 staining did not appear to be perturbed, instead presenting the typical ring-shaped organization (Figure 3C, red). Finally, immunoblotting in cells from affected individuals confirmed a reduced CEP83 level (Figure 3D), whereas CEP164 level was unchanged, indicating that CEP83 variations affect recruitment of CEP164 at the centrosome without any change in its total protein level.
We further confirmed the effects of CEP83 mutations in kidney biopsies. In control kidneys, CEP83 and CEP164 localized at one of the two centrioles (Figures S5A and S5B, insets, arrows). In kidney biopsy from individual NPH2036, CEP83 staining appeared fainter at the centrosome (Figure S5C, arrows) and CEP164 was not even detected in some centrosomes (50.1% of CEP164-positive centrosomes versus 83.6% for control; Figure S5D). Altogether, these results on tissues and cells from affected individuals suggest that both decreased CEP83 level (truncated variants) and partial impairment of the variant proteins, induced by the presence of the missense or in-frame deletions, probably account for its inability to assemble properly at the mother centriole, as well as to recruit CEP164 at the distal part of the mother centriole.
We next investigated the consequences of amino acid alterations in CEP83 on its targeting to the centrosome as well as on its interactions with known partners in vitro. In transiently transfected RPE1 cells, FLAG-CEP83 wild-type (WT) was diffusely distributed in the cytoplasm and nucleus and was also present at the centrosome where it forms a typical ring-shaped structure (Figures 4A and S6A, insets). Interestingly, endogenous CEP164 staining at the centrosome was strongly decreased in cells transfected with FLAG-CEP83 WT compared to neighboring nontransfected cells (Figures 4A, 4C, and S6A), indicating that excess of CEP83 resulted in a dominant-negative effect on DAP biogenesis. Among the variant forms of CEP83, p.Pro112_Leu117del, p.Arg511Pro, and p.Leu87Pro showed similar centrosomal localization and dominant-negative effects on CEP164 localization as WT protein (data not shown). In contrast, CEP83 p.Gln692del, p.Glu669Aspfs∗14, and p.Glu684del variants accumulated into the nucleus and failed to localize at the centrosome (Figures 4A, 4B, and S6B). CEP164 distribution was not perturbed in cells transfected with these variants compared to neighboring nontransfected cells (Figure 4A, arrows), thereby showing higher CEP164 staining intensity at the centrosome compared to cells transfected with WT CEP83 (Figures 4A and 4C). These results indicate that these variants present a decreased affinity for the mother centriole and as a result do not show dominant-negative effects on DAP formation.
Figure 4.
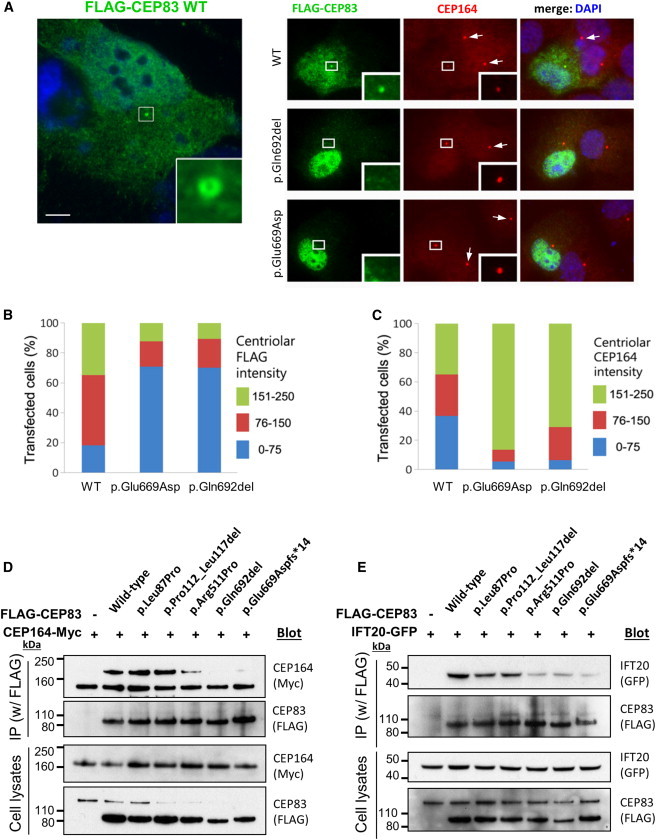
CEP83 Mutations Affect Centrosomal Localization and Interaction of CEP83 with CEP164
(A) RPE1 cells were transiently transfected (Lipofectamine LTX, Life Technologies) with plasmids encoding for FLAG-tagged wild-type (WT) or c.2007del (p.Glu669Aspfs∗14) or c.2075_2077del (p.Gln692del) CEP83 variants, fixed, and stained for FLAG (green; mouse monoclonal [Sigma, F3165]) and CEP164 (red). Scale bar represents 5 μm.
(B and C) Levels of CEP164 (C) and FLAG (CEP83, B) staining at the centrosome were quantified with ImageJ. Centriolar CEP164 intensity is decreased in cells transfected with CEP83 WT compared to cells transfected with CEP83 p.Glu669Aspfs∗14 and p.Gln692del variants (arrows).
(D and E) NIH 3T3 cells were transiently cotransfected with plasmids encoding for FLAG-tagged WT and variant forms of CEP83, Cep164-myc, and IFT20-GFP. Cell lysates were immunoprecipitated with an anti-FLAG antibody (mouse monoclonal, Sigma, M2). Cell lysates (bottom) and immunoprecipitates (IP, top) were analyzed by immunoblotting with antibodies against myc (CEP164, rabbit polyclonal [Santa Cruz Biotechnology, C-19]), FLAG (CEP83, rabbit polyclonal [Cell Signaling Technology, #2368]), and GFP (IFT20, rabbit polyclonal [Life Technologies, A-6455]) as indicated. Note that three C-terminal CEP83 variants affected the interaction with CEP164 and IFT120, whereas two N-terminal CEP83 variants did not.
We next investigated the ability of CEP83 variants to interact with its binding partners CEP164 and IFT2017 in NIH 3T3 cells by coimmunoprecipitation. As shown in Figures 4D and 4E, CEP164-myc and IFT20-GFP were efficiently coimmunoprecipitated together with FLAG-CEP83 WT, showing that these three proteins form complexes. The C-terminal CEP83 variants (p.Arg511Pro, p.Glu669Aspfs∗14, and p.Gln692del) were impaired in their ability to interact with CEP164, whereas proteins harboring p.Leu87Pro and p.Pro112_Leu117del were not (Figure 4D). Similarly, the interaction with IFT20 appeared to be decreased by C-terminal CEP83 variants although it was not affected by N-terminal CEP83 variant proteins (Figure 4E).
In summary, transfection of wild-type CEP83 results in a dominant-negative effect on the recruitment of endogenous CEP164 onto the centrosome (Figures 4A–4C). It is likely that high level of exogenous CEP83 might somehow prevent the coordinated assembly of CEP164/DAPs onto the mother centriole by a mechanism that remains to be characterized. This dominant-negative effect is conserved for all variants that are efficiently recruited to the centrosome independently of their capacity to interact with CEP164 or IFT20 (p.Leu87Pro, p.Pro112_Leu117del, and p.Arg511Pro) (Figures 4D and 4E). In contrast, variants p.Gln692del, p.Glu669Aspfs∗14, and p.Glu684del are affected for both centrosome localization and interactions with their partners CEP164 and IFT20, highlighting the important role of the last coiled-coil domain encoded by exon 17 of CEP83 (summarized in Table S1).
As mentioned above, DAPs play a crucial role during ciliogenesis (Figure S2). We confirmed that CEP83 localizes at the base of both primary and motile cilia in vitro and in vivo (Figures S7 and S8). This is consistent with the ciliary dysfunction phenotype observed in affected individuals, associated primary cilia defects (nephronophthisis and retinal degeneration), and possibly motile cilia defects as suggested by ventricular dilatation and hydrocephalus observed in some of the affected individuals (A4037-21) similar to those described for individuals with variations in SDCCAG8/NPHP10.26 We therefore examined the ability of fibroblasts from affected individuals (NPH1402 and NPH1412) to form primary cilia. Upon serum starvation, ciliogenesis was mildly but statistically significantly affected in fibroblasts from affected individuals with the percentage of ciliated cells decreased compared to control fibroblasts (Figures 5A and 5B). The formed cilia presented no obvious change in the ciliary composition because localization of the ciliary components CEP290, ANKS6, IFT140, or ARL13B were not perturbed (Figure S9 and data not shown). However, cilia present in both fibroblasts (Figures 5C) of affected individuals were significantly longer compared to controls. This phenotype was also observed in kidney biopsies from affected individuals NPH1402 and NPH2036 (Figure 5D, right panels, arrows), similar to our observations in other NPHP-RC proteins.9,27
Figure 5.
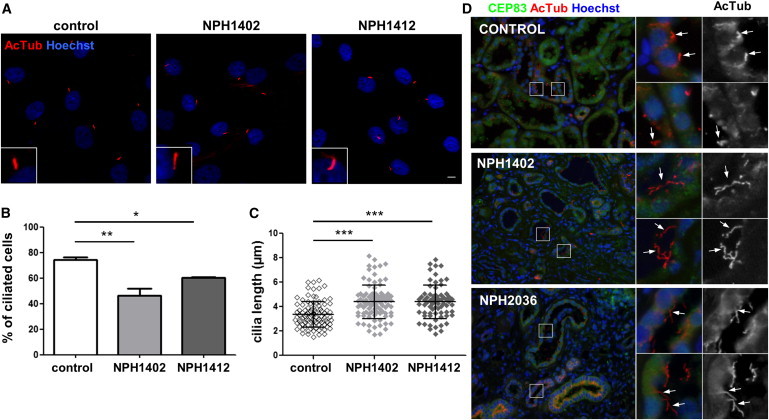
Fibroblasts from Individuals with CEP83 Mutations Show Mild Ciliary Defects
(A) Fibroblasts from control and affected individuals (NPH1402 and NPH14112) were grown on coverslips, serum starved for 48 hr, then fixed and processed for immunofluorescence with antibodies against acetylated tubulin (AcTub, red) to stain cilia. Insets show higher magnification of representative cilia. Scale bar represents 5 μm.
(B) The ability of cells to form cilia, cells with a unique rod like AcTub staining, was quantified from images obtained in (A). Graphs show the mean ± SEM of at least three independent experiments in which at least 100 cells were counted.
(C) The length of cilia was measured (μm) based on AcTub staining and the results for one representative experiment out of three are shown.
(B and C) ∗p < 0.01, ∗∗p < 0.001, and ∗∗∗p < 0.0001 were calculated via Dunn’s Multiple Comparison Test after the analysis of variance ANOVA test. Ciliogenesis is reducted in fibroblasts of affected individuals which present longer cilia.
(D) Sections of kidney biopsies from control, NPH1402, and NPH2036 individuals were stained for CEP83 (green) and AcTub (red). Nuclei were stained with Hoechst (blue). Higher magnifications of representative tubules regions are shown on the right panels and cilia are indicated by white arrows. Cilia present in the lumen of tubules are longer in affected individuals biopsies compared to control.
Together, our results show that fibroblasts and tubular renal cells from affected individuals exhibit ciliogenesis defects probably resulting from an altered assembly of DAPs, which would affect docking of altered mother centriole to the ciliary primary vesicle (Figure S2). In addition, defects in IFT20 interaction by most of the variants probably contribute to ciliary defects because IFT20 was shown to participate in ciliogenesis through vesicular trafficking from the Golgi apparatus.28
In summary, we have identified mutations in CEP83 as causing early-onset nephronophthisis and intellectual disability, adding another component of the DAPs of centrioles to the list of defective proteins in NPHP-RC. We therefore propose “NPHP18” as an alias for CEP83.
Acknowledgments
The authors thank the families who contributed to this study. We thank Vivette D. D’Agati and Michael B. Stokes (Columbia University) for a kidney biopsy report and images, as well as Gérard Pivert (Pathology Department, Necker Hospital) for kidney biopsy. We are grateful to James Sillibourne for his kind gift of the CEP89 antibody. We also thank Patrick Nitschké and the bioinformatic Plateform (Université Paris Descartes, Institut Imagine) as well as Christine Bole-Feysot, Solenn Pruvost, and Mohammed Zarhrate for their support in exome sequencing and Ludovic Lecompte and Lucie Sengmanivong from the Nikon Imaging Centre at Institut Curie-CNRS (Paris, France) for the help with the n-SIM. This research was supported by a grant from the NIH to F.H. (DK068306). H.Y.G. is a Research Fellow of the American Society of Nephrology (ASN). F.H. is an Investigator of the Howard Hughes Medical Institute and Warren E. Grupe Professor of Pediatrics at the Harvard Medical School. This work was supported by grants from the “Agence Nationale de la Recherche” (ANR) to S.S. (20100BLAN112202) and investments for the future program-ANR-10-IAHY-01 to S.S., the “Fondation pour la Recherche Médicale” (FRM) to S.S. (DEQ20071210558) and to P.K. (DEA20120624188); M.F. is supported by a fellowship from the “Région ile de France, CORDDIM” (RPH12173KKA).
Contributor Information
Friedhelm Hildebrandt, Email: friedhelm.hildebrandt@childrens.harvard.edu.
Sophie Saunier, Email: sophie.saunier@inserm.fr.
Supplemental Data
Document S1. Figures S1–S9 and Table S1
Document S2. Article plus Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
- COILS, http://www.ch.embnet.org/software/COILS_form.html
- Ensembl Genome Browser, http://www.ensembl.org/index.html
- MutationTaster, http://www.mutationtaster.org/
- NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
- Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
- PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
- RefSeq, http://www.ncbi.nlm.nih.gov/RefSeq
- UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Salomon R., Saunier S., Niaudet P. Nephronophthisis. Pediatr. Nephrol. 2009;24:2333–2344. doi: 10.1007/s00467-008-0840-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hildebrandt F., Benzing T., Katsanis N. Ciliopathies. N. Engl. J. Med. 2011;364:1533–1543. doi: 10.1056/NEJMra1010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Satir P., Christensen S.T. Overview of structure and function of mammalian cilia. Annu. Rev. Physiol. 2007;69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236. [DOI] [PubMed] [Google Scholar]
- 4.Delous M., Gaudé H.M., Saunier S. Genetic bases and pathogenic mechanisms of nephronophthisis. Drug Discov. Today Dis. Mech. 2013 Published online November 5, 2013. [Google Scholar]
- 5.Arts H.H., Knoers N.V.A.M. Current insights into renal ciliopathies: what can genetics teach us? Pediatr. Nephrol. 2013;28:863–874. doi: 10.1007/s00467-012-2259-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Otto E.A., Schermer B., Obara T., O’Toole J.F., Hiller K.S., Mueller A.M., Ruf R.G., Hoefele J., Beekmann F., Landau D. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat. Genet. 2003;34:413–420. doi: 10.1038/ng1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Otto E.A., Trapp M.L., Schultheiss U.T., Helou J., Quarmby L.M., Hildebrandt F. NEK8 mutations affect ciliary and centrosomal localization and may cause nephronophthisis. J. Am. Soc. Nephrol. 2008;19:587–592. doi: 10.1681/ASN.2007040490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tory K., Rousset-Rouvière C., Gubler M.-C., Morinière V., Pawtowski A., Becker C., Guyot C., Gié S., Frishberg Y., Nivet H. Mutations of NPHP2 and NPHP3 in infantile nephronophthisis. Kidney Int. 2009;75:839–847. doi: 10.1038/ki.2008.662. [DOI] [PubMed] [Google Scholar]
- 9.Halbritter J., Bizet A.A., Schmidts M., Porath J.D., Braun D.A., Gee H.Y., McInerney-Leo A.M., Krug P., Filhol E., Davis E.E., UK10K Consortium Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans. Am. J. Hum. Genet. 2013;93:915–925. doi: 10.1016/j.ajhg.2013.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huber C., Wu S., Kim A.S., Sigaudy S., Sarukhanov A., Serre V., Baujat G., Le Quan Sang K.-H., Rimoin D.L., Cohn D.H. WDR34 mutations that cause short-rib polydactyly syndrome type III/severe asphyxiating thoracic dysplasia reveal a role for the NF-κB pathway in cilia. Am. J. Hum. Genet. 2013;93:926–931. doi: 10.1016/j.ajhg.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmidts M., Vodopiutz J., Christou-Savina S., Cortés C.R., McInerney-Leo A.M., Emes R.D., Arts H.H., Tüysüz B., D’Silva J., Leo P.J., UK10K Mutations in the gene encoding IFT dynein complex component WDR34 cause Jeune asphyxiating thoracic dystrophy. Am. J. Hum. Genet. 2013;93:932–944. doi: 10.1016/j.ajhg.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morga B., Bastin P. Getting to the heart of intraflagellar transport using Trypanosoma and Chlamydomonas models: the strength is in their differences. Cilia. 2013;2:16. doi: 10.1186/2046-2530-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bornens M. The centrosome in cells and organisms. Science. 2012;335:422–426. doi: 10.1126/science.1209037. [DOI] [PubMed] [Google Scholar]
- 14.Reiter J.F., Blacque O.E., Leroux M.R. The base of the cilium: roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Rep. 2012;13:608–618. doi: 10.1038/embor.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graser S., Stierhof Y.-D., Lavoie S.B., Gassner O.S., Lamla S., Le Clech M., Nigg E.A. Cep164, a novel centriole appendage protein required for primary cilium formation. J. Cell Biol. 2007;179:321–330. doi: 10.1083/jcb.200707181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanos B.E., Yang H.-J., Soni R., Wang W.-J., Macaluso F.P., Asara J.M., Tsou M.-F.B. Centriole distal appendages promote membrane docking, leading to cilia initiation. Genes Dev. 2013;27:163–168. doi: 10.1101/gad.207043.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joo K., Kim C.G., Lee M.-S., Moon H.-Y., Lee S.-H., Kim M.J., Kweon H.-S., Park W.-Y., Kim C.-H., Gleeson J.G., Kim J. CCDC41 is required for ciliary vesicle docking to the mother centriole. Proc. Natl. Acad. Sci. USA. 2013;110:5987–5992. doi: 10.1073/pnas.1220927110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sillibourne J.E., Hurbain I., Grand-Perret T., Goud B., Tran P., Bornens M. Primary ciliogenesis requires the distal appendage component Cep123. Biol. Open. 2013;2:535–545. doi: 10.1242/bio.20134457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chaki M., Airik R., Ghosh A.K., Giles R.H., Chen R., Slaats G.G., Wang H., Hurd T.W., Zhou W., Cluckey A. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell. 2012;150:533–548. doi: 10.1016/j.cell.2012.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adly N., Alhashem A., Ammari A., Alkuraya F.S. Ciliary genes TBC1D32/C6orf170 and SCLT1 are mutated in patients with OFD type IX. Hum. Mutat. 2014;35:36–40. doi: 10.1002/humu.22477. [DOI] [PubMed] [Google Scholar]
- 21.Thomas S., Legendre M., Saunier S., Bessières B., Alby C., Bonnière M., Toutain A., Loeuillet L., Szymanska K., Jossic F. TCTN3 mutations cause Mohr-Majewski syndrome. Am. J. Hum. Genet. 2012;91:372–378. doi: 10.1016/j.ajhg.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perrault I., Saunier S., Hanein S., Filhol E., Bizet A.A., Collins F., Salih M.A.M., Gerber S., Delphin N., Bigot K. Mainzer-Saldino syndrome is a ciliopathy caused by IFT140 mutations. Am. J. Hum. Genet. 2012;90:864–870. doi: 10.1016/j.ajhg.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Halbritter J., Diaz K., Chaki M., Porath J.D., Tarrier B., Fu C., Innis J.L., Allen S.J., Lyons R.H., Stefanidis C.J. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. J. Med. Genet. 2012;49:756–767. doi: 10.1136/jmedgenet-2012-100973. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt K.N., Kuhns S., Neuner A., Hub B., Zentgraf H., Pereira G. Cep164 mediates vesicular docking to the mother centriole during early steps of ciliogenesis. J. Cell Biol. 2012;199:1083–1101. doi: 10.1083/jcb.201202126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakagawa Y., Yamane Y., Okanoue T., Tsukita S., Tsukita S. Outer dense fiber 2 is a widespread centrosome scaffold component preferentially associated with mother centrioles: its identification from isolated centrosomes. Mol. Biol. Cell. 2001;12:1687–1697. doi: 10.1091/mbc.12.6.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schaefer E., Zaloszyc A., Lauer J., Durand M., Stutzmann F., Perdomo-Trujillo Y., Redin C., Bennouna Greene V., Toutain A., Perrin L. Mutations in SDCCAG8/NPHP10 cause Bardet-Biedl syndrome and are associated with penetrant renal disease and absent polydactyly. Mol. Syndromol. 2011;1:273–281. doi: 10.1159/000331268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tammachote R., Hommerding C.J., Sinders R.M., Miller C.A., Czarnecki P.G., Leightner A.C., Salisbury J.L., Ward C.J., Torres V.E., Gattone V.H., 2nd, Harris P.C. Ciliary and centrosomal defects associated with mutation and depletion of the Meckel syndrome genes MKS1 and MKS3. Hum. Mol. Genet. 2009;18:3311–3323. doi: 10.1093/hmg/ddp272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Follit J.A., Tuft R.A., Fogarty K.E., Pazour G.J. The intraflagellar transport protein IFT20 is associated with the Golgi complex and is required for cilia assembly. Mol. Biol. Cell. 2006;17:3781–3792. doi: 10.1091/mbc.E06-02-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gustafsson M.G.L., Shao L., Carlton P.M., Wang C.J.R., Golubovskaya I.N., Cande W.Z., Agard D.A., Sedat J.W. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys. J. 2008;94:4957–4970. doi: 10.1529/biophysj.107.120345. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Figures S1–S9 and Table S1
Document S2. Article plus Supplemental Data