Liver Tumor Promotion by 2,3,7,8-Tetrachlorodibenzo-p-dioxin Is Dependent on the Aryl Hydrocarbon Receptor and TNF/IL-1 Receptors (original) (raw)
Abstract
We set out to better understand the signal transduction pathways that mediate liver tumor promotion by 2,3,7,8-tetrachlorodibenzo-_p_-dioxn (“dioxin”). To this end, we first employed congenic mice homozygous for either the Ahrb1 or Ahrd alleles (encoding an aryl hydrocarbon receptor (AHR) with high or low binding affinity for dioxin, respectively) and demonstrated that hepatocellular tumor promotion in response to dioxin segregated with the Ahr locus. Once we had genetic evidence for the importance of AHR signaling, we then asked if tumor promotion by dioxin was influenced by “interleukin-1 (IL-1)-like” inflammatory cytokines. The importance of this question arose from our earlier observation that aspects of the acute hepatocellular toxicity of dioxin are dependent upon IL1-like cytokine signaling. To address this issue, we employed a triple knock-out (TKO) mouse model with null alleles at the loci encoding the three relevant receptors for tumor necrosis factors α and β and IL-1α and IL-1β (i.e., null alleles at the Tnfrsf1a, Tnfrsf1b, and Il-1r1 loci). The observation that TKO mice were resistant to the tumor promoting effects of dioxin in liver suggests that inflammatory cytokines play an important step in dioxin mediated liver tumor promotion in the mouse. Collectively, these data support the idea that the mechanism of dioxin acute hepatotoxicity and its activity as a promoter in a mouse two stage liver cancer model may be similar, i.e., tumor promotion by dioxin, like acute hepatotoxicity, are mediated by the linked action of two receptor systems, the AHR and the receptors for the “IL-1-like” cytokines.
Keywords: dioxin, aryl hydrocarbon receptor (AHR), liver, tumor promotion, IL-1-like cytokine
Abbreviations
AHR
aryl hydrocarbon receptor
AP-1
activator protein 1
ARNT
AHR nuclear translocator
CA-AHR
constitutively active form of AHR
CYP
cytochrome P450
dioxin
2,3,7,8-tetrachlorodibenzo-_p_-dioxin
DEN
diethylnitrosamine
DRE
dioxin responsive enhancer element
G6Pase
glucose-6-phosphatase
H&E
hematoxylin and eosin
IL-1
interleukin-1
IL-1R
IL-1 receptor
NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
PAS
Per-Arnt-Sim
STAT3
signal transducer and activator of transcription 3
TKO
triple receptors null
TNF
tumor necrosis factor
TNFR
TNF receptor
WT
wild-type
2,3,7,8-Tetrachlorodibenzo-_p_-dioxin (“dioxin”) is the prototype for a large family of halogenated-dioxins that are commonly found as environmental pollutants (IARC, 1997). The World Health Organization's International Agency for Research on Cancer (IARC) has classified dioxin as “a known human carcinogen” (IARC, 1997). In experimental animals, dioxin is considered a “complete carcinogen,” i.e., exposure to dioxin alone can lead to an increase in tumor yields at multiple sites (IARC, 1997; Huff et al., 1994; Lucier et al., 1993). In “two-stage/initiation-promotion” models of liver, skin, lung, and ovarian cancer, dioxin is demonstrated to be a potent tumor promoter (Davis et al., 2000; Pitot et al., 1987; Poland et al., 1982). This observation, coupled to the reports that dioxin is only weakly genotoxic has led our laboratory to work under the hypothesis that dioxin carcinogenicity is mediated largely through its action as a nongenotoxic tumor promoter (Giri, 1986).
Although we do not know the mechanism by which dioxin leads to chronic diseases such as cancer, we do know that much of dioxin-induced acute toxicity is mediated by the direct interaction of this ligand with the aryl hydrocarbon receptor (AHR), a member of the basic helix-loop-helix/Per-Arnt-Sim (PAS) family of transcription factors (Hankinson, 1995; Schmidt and Bradfield, 1996). When bound by dioxin, the AHR sheds its chaperones and gains an increased affinity for the nucleus where it forms a heterodimeric complex with its partner, another PAS protein, known as the AHR nuclear translocator (ARNT). Once activated by ligand, the AHR-ARNT nuclear complex recognizes “dioxin-responsive enhancer elements” (DREs) within the genome and regulates a battery of target genes, including those encoding certain cytochromes P450 (e.g., Cyp1a1, Cyp1a2, and Cyp1b1) (Nebert and Gonzalez, 1987; Schmidt and Bradfield, 1996). In our view, none of the known DRE-driven genes has yet completely explained the toxic or carcinogenic action of dioxins. In fact, recent evidence from our own lab indicates that the Cyp1a1 and Cyp1a2 gene products provide protection from dioxin-induced acute hepatotoxicity (Nukaya et al., 2009).
We set out to test the hypothesis that dioxin's role in liver tumor promotion was similar to the mechanism underlying its acute hepatotoxicity. Specifically, we examined the hypothesis that liver tumor promotion, like acute hepatotoxicity, requires dioxin binding to the AHR and downstream signaling events that are inflammatory in nature (Pande et al., 2005). To accomplish this task, we first developed a model of dioxin liver tumor promotion in a powerful genetic model, the mouse. We then employed this tumor promotion model in two genetic proofs. First, to demonstrate a role for the AHR, we performed dioxin tumor promotion studies in mice harboring Ahr alleles encoding receptors with either high or low binding affinity for dioxin ligand (Ahrb1 vs. Ahrd alleles, respectively). Second, to provide evidence that AHR-mediated tumor promotion was dependent upon inflammatory cytokine signaling, we used mice that were null for the receptors for the “IL-1 like cytokines,” (i.e., tumor necrosis factors (TNFs) α and β and interleukin (IL)-1α and β). Our results suggest that, like acute hepatotoxicity, dioxin-promoted hepatocellular cancer in mice is dependent upon signaling by the AHR and “IL-1-like” inflammatory cytokines.
MATERIALS AND METHODS
Animals
Mice were housed in a selective pathogen-free facility on corncob bedding with food and water ad libitum following the protocol established by University of Wisconsin Medical School's Animal Care and Use Committee. Wild-type (WT) C57BL/6J mice homozygous for the high affinity Ahrb1 allele as (i.e., Ahrb1/b1) were used as controls throughout this study. In parallel, congenic C57BL/6J mice homozygous for the Ahrd allele (i.e., Ahrd/d), which encodes the lower affinity AHRd were also examined (Poland et al., 1989). The congenic _Ahrd_-C57BL/6J mice have been backcrossed >20 times to the WT C57BL/6J strain making them >99.9% WT-C57BL/6J background. The _Tnfrsf1a_−/−/_Tnfrsf1b_−/−/_Il-1r_1−/− triple null strain of mice have been previously described (Pande et al., 2005). The triple null mice have been backcrossed to the C57BL/6J strain more than six times and are thus predicted to be >99.9% C57BL/6J (Ahrb1/b1) background.
Treatment and tumor studies
At 13 days of age, male mice were injected intraperitoneally with a single dose of either 10 μl of tricaprylin or 0.1 μmol/g body weight of diethylnitrosamine (DEN) dissolved in the same solvent. At 9–10 weeks of age, the mice were treated by gavage with either 10 μg/kg body weight of dioxin or 10 ml/kg body weight of corn oil once every 2 weeks for 24 or 40 weeks (total 12 or 20 treatments, respectively). Animals were then sacrificed by CO2 euthanasia and tumors on the surface of the liver were visually counted (Moore et al., 1981). For the analysis of G-6-Pase deficient foci, two consecutive 10-μm sections were cut from the frozen liver. These sections were stained for G-6-Pase (Moore et al., 1981). Each section was carefully examined for number of foci per slide. The area of each tumor focus was estimated according to the formula A = π_r_2. The fraction of tumor foci volume was then estimated by dividing the total foci volume by the volume of the section.
Quantitation of 7-ethylguanine in mice liver DNA
DNA hydrolysis and sample enrichment and purification were carried out as reported in the literature (Chen et al., 2007). Briefly DNA was dissolved in 1 ml of 10mM sodium cacodylate buffer containing 500 fmol of [15N5]7-ethyl-Gua and heated at 100°C for 1 h. A 50 μl aliquot was removed for guanine analysis. The remaining material was loaded on a Centrifree MPS device (MW cutoff of 30,000; Amicon, Beverly, MA) to remove high molecular weight material. The filtrate was applied to a solid-phase extraction cartridge [Strata-X 33 μm, 30 mg/1 ml (Phenomenex)]. The 70% methanol fraction was collected and evaporated to dryness. The residue was dissolved in water and analyzed by liquid chromatography-nanoelectrospray-high resolution tandem mass spectrometry-selected reaction monitoring (LC-NSI-HRMS/MS-SRM). The analysis was carried out with an Eksigent NanoLC-Ultra HPLC system (Eksigent Technologies, Dublin, CA) interfaced with a ThermoScientific LTQ Orbitrap Velos (ThermoScientific, San Jose, CA) mass spectrometer. Calibration curves were constructed before each analysis using standard solutions of 7-ethyl-Gua and [15N5]7-ethyl-Gua (Balbo et al., 2011).
The 50 μl aliquot reserved for guanine analysis was diluted to 100 μl with 0.2N HCl, and heated at 80°C for 1 h. The solution was then neutralized with 10 μl NaOH 1M and diluted to 200 μl with H2O, and 20 μl was injected for HPLC-UV analysis. Calibration curves were constructed before each analysis using standard solutions of guanine (Balbo et al., 2011).
Statistical analysis
Statistical analyses were performed by Mstat 5 software (http://mcardle.oncology.wisc.edu/mstat). Intergroup comparisons of tumor incidence were performed by Fisher's exact test (Drinkwater and Denniston, 2011). Tumor multiplicities were compared using the nonparametric Wilcoxon rank sum test (Drinkwater and Denniston, 2011). Sen-Adichie test was used for comparing the dose-responses for tumor multiplicity between two mouse lines (Drinkwater and Denniston, 2011). Differences among groups were assessed statistically significant when p value was <0.05.
RESULTS
Dioxin-Induced Liver Tumor Promotion is Dependent on Binding Affinity of the AHR for Dioxin
In order to determine whether dioxin-promoted liver carcinogenesis is influenced by the binding affinity of the AHR for dioxin, we first performed an initiation-promotion assay using Ahrb1/b1 and congenic Ahrd/d mice on the C57BL/6J background (alleles encoding high and low affinity receptor, respectively). After 24 weeks of dioxin exposure (∼32 weeks of age), tumors were counted on the surface of the liver. Evidence that this protocol was measuring tumor promotion comes from both tumor incidence and tumor multiplicity data. The tumor incidence data demonstrates that mice without initiation by DEN displayed no evidence of gross liver tumor development regardless of dioxin exposure or genotype (Table 1) indicating that dioxin carcinogenesis is dependent upon an initiating event at this time point. Moreover, the significantly increased incidence of tumors in the Ahrb1/b1 mice when mice were exposed to both the initiation event (DEN) and the promotion event (dioxin), as compared with either DEN or dioxin alone, is supportive of dioxin's role as a tumor promoter in this model. Finally, the observation from Ahrd/d mice that dioxin plus DEN does not lead to a significant increase in tumor incidence as compared with DEN alone is consistent with the idea that these mice are recalcitrant to the promoting effects of dioxin because they encode an AHR with lower binding affinity for this promoter.
TabLE 1. Effect of Dioxin and Ahr Genotype on Incidence of Liver Tumors with or without DEN-Initiation.
| Genotype | DEN initiation | Tumor incidence_a_ | |||
|---|---|---|---|---|---|
| Corn-oil | Dioxin | ||||
| Ahrb1/b1 | Control | 0/10 | (0%) | 0/10 | (0%) |
| DEN | 6/9 | (67%)b | 26/26 | (100%)b,c | |
| Ahrd/d | Control | 0/10 | (0%) | 0/10 | (0%) |
| DEN | 5/11 | (45%)d | 9/11 | (82%)d |
These concepts are made even clearer by the analysis of tumor multiplicity provided in Figure 1A. We observed a >20-fold increase in the liver tumor multiplicity in DEN-initiated Ahrb1/b1 mice promoted with dioxin as compared with genotype matched vehicle control mice (DEN + corn oil;1.33 ± 0.62, DEN + dioxin; 34.31 ± 5.54, p < 0.0001) (Fig. 1A). In contrast, dioxin promotion of DEN initiated Ahrd/d mice did not significantly enhance the liver tumor multiplicity at 24 weeks (DEN + corn-oil; 1.64 ± 0.75, DEN + dioxin; 3.18 ± 0.76, p = 0.11) (Fig. 1A). To determine if the effect of dioxin on liver tumor promotion becomes significant in Ahrd/d mice with increased time of the promotion phase, we extended the dioxin exposure to 40 weeks in DEN initiated mice (Supplementary data 1). We observed that after 40 weeks of dioxin treatment, the DEN-initiated congenic Ahrd/d mice displayed a small (∼2.6-fold), but significant increase in liver tumor multiplicity (corn-oil (Ahrd/d); 25.4 ±31.1, dioxin (Ahrd/d); 65.8 ±34.0, p < 0.01) (Supplementary data 1). Taken in sum, these data are consistent with the concept that dioxin is acting as a tumor promoter in this liver model at 24 weeks of exposure and is dependent upon an initiating event.
Fig. 1.
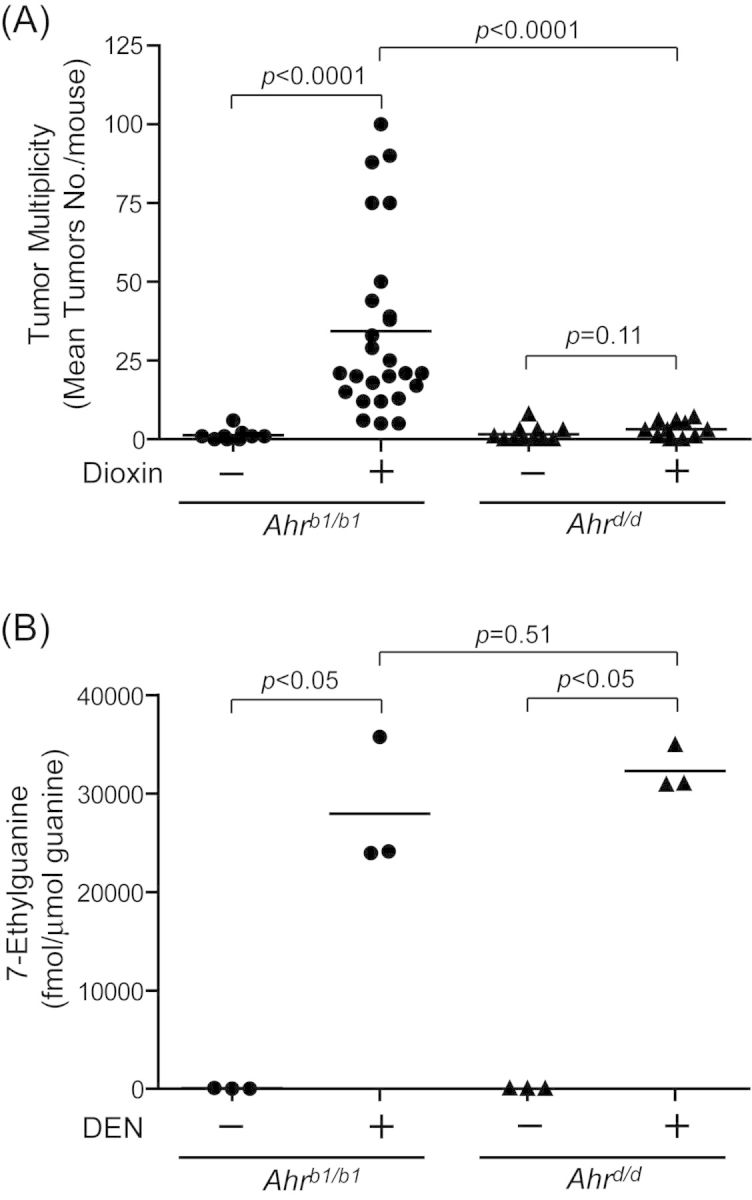
Dioxin-induced liver tumor multiplicity is dependent on the AHR-binding affinity to dioxin. (A) Dioxin-induced liver tumor multiplicity in the DEN-initiated Ahrb1/b1 and Ahrd/d mice. Mice were treated at 13 days of age with a single dose of 0.1 μmol/g body weight of DEN. At 9–10 weeks of age, the mice were treated by gavage with either 10 μg/kg body weight of dioxin or 10 ml/kg body weight of corn-oil every 2 weeks for 24 weeks. Each group contained more than nine mice (n = 9–26). Bars represent mean liver tumor multiplicities. (B) Levels of 7-methylguanine in liver DNA of Ahrb1/b1 and Ahrd/d mice. Mice were treated at 13 days of age with a single dose of 0.1 μmol/g body weight of DEN or the solvent tricaprylin. These mice were killed 24 h later. Each group contained three mice. Bars represent mean values.
Differences in Liver Tumors between Ahr Congenic Strains are not Dependent upon Differences in DEN Bioactivation
To determine whether the rates of initiation by DEN were different between our congenic Ahrb1 and Ahrd strains, we measured levels of 7-ethylguanine DNA adducts in DNA extracted from DEN treated livers of Ahrb1/b1 and Ahrd/d mice (Fig. 1B). The levels of 7-ethylguanine were 798- and 970-fold increased by treatment of DEN in the Ahrb1/b1 and Ahrd/d mice, respectively (control (Ahrb1/b1); 35 ± 43, DEN (Ahrb1/b1); 27942 ± 6758, control (Ahrd/d); 33 ± 17, DEN (Ahrd/d); 32296 ± 2298). The levels of 7-ethylguanine were not significantly different between the DEN-treated Ahrb1/b1 and Ahrd/d mice (p = 0.51).
Dioxin-Induced Preneoplastic Lesions in Livers of Ahrb1/b1 and Ahrd/d Mice
To assess the effect of DEN and/or dioxin treatment on hepatic tumor development, we monitored the number, size, and volume fraction of preneoplastic lesions, as measured by glucose-6-phosphatase (G6Pase) deficient foci, in a subset of liver sections from Ahrb1/b1 and Ahrd/d mice after 24 weeks of dioxin exposure (Table 2) (Moore et al., 1981). In the DEN-initiated Ahrb1/b1 mice, the number, size, and volume fraction of G6Pase-deficient foci were significantly increased by dioxin exposure compared with the corn oil only control (p < 0.05). However, the number, size and volume fraction of hepatic foci were not significantly altered by dioxin exposure in the DEN-initiated Ahrd/d mice compared with their genotype matched corn-oil controls. Although there was no significant increase in foci after 24 weeks in the Ahrd/d mice, again we did find differences after 40 weeks of exposure, where the size and volume fraction of foci were significantly increased in dioxin treated animals compared with those treated with corn oil (Supplementary data 2).
TabLE 2. Comparison of the Number, Size, and Volume Fraction of G-6-Pase-Deficient Foci in the Livers of Ahrb1/b1 and Ahrd/d Mice Treated with Corn-Oil or Dioxin.
| Genotype | Treatment | Number of mice | G-6-Pase-deficient foci | ||
|---|---|---|---|---|---|
| Number of foci_a_ | Focus diameter_b_ | Volume fraction_c_ | |||
| −DEN initiation | |||||
| Ahrb1/b1 | Corn-oil | 9 | 0.33 ± 0.17 | 434.7 ± 81.1 | 0.07 ± 0.03 |
| Dioxin | 10 | 0.40 ± 0.16 | 253.0 ± 19.8 | 0.03 ± 0.02 | |
| Ahrd/d | Corn-oil | 8 | 0.63 ± 0.38 | 209.1 ± 34.0 | 0.05 ± 0.04 |
| Dioxin | 9 | 0.56 ± 0.18 | 183.0 ± 23.1 | 0.03 ± 0.01 | |
| +DEN initiation | |||||
| Ahrb1/b1 | Corn-oil | 7 | 4.14 ± 0.94 | 527.1 ± 61.6 | 2.06 ± 0.59 |
| Dioxin | 23 | 12.35 ± 1.21_d_ | 1069.0 ± 47.3_d_ | 16.39 ± 3.75_d_ | |
| Ahrd/d | Corn-oil | 9 | 7.89 ± 1.83 | 498.1 ± 33.9 | 6.01 ± 1.63_d_ |
| Dioxin | 8 | 8.63 ± 2.67 | 469.9 ± 35.4 | 5.33 ± 1.94 |
Dioxin-Promoted Liver Carcinogenesis Requires IL-1-Like Cytokine Signaling
We have previously employed _Tnfrsf1a_−/−/_Tnfrsf1b_−/−/_Il-1r_1−/− triple null (TKO) mice to support the idea that aspects of dioxin-induced acute hepatotoxicity require IL-1-like cytokine signaling (Pande et al., 2005). To investigate whether these same proinflammatory cytokine receptors, and therefore their cognate cytokines themselves, are also involved in this model of dioxin-induced liver tumor promotion, the two stage initiation-promotion assay was repeated using TKO mice. We observed that the liver tumor incidence (Table 3) and multiplicity were increased by dioxin promotion in WT mice (corn-oil (WT); 6.63 ± 2.18, dioxin (WT); 57.79 ± 6.30, p < 0.0001) (Fig. 2A) and to a significantly reduced degree in TKO mice (corn-oil (TKO); 0.63 ± 0.39, dioxin (TKO); 2.29 ± 0.84, _p_ = 0.13) (Fig. 2A). This reduced tumor yield in the TKO mice was independent of DEN bioactivation as 7-ethylgluanine levels were not significantly different as compared with the WT controls (control (WT); 16 ± 21, DEN (WT); 13200 ± 1667, control (TKO); 33 ± 7, DEN (TKO); 12812 ± 1418) (Fig. 2B). Interestingly, the tumor multiplicity was reduced ∼10-fold (_p_ = 0.002) in the DEN treated TKO mice as compared with the DEN treated WT mice (Fig. 2A) indicating some endogenous tumor promotion pathways are also inhibited in TKO animals. Importantly, the 8.7-fold increase in dioxin promoted tumors of WT mice was significantly >3.6-fold increase in the corresponding TKO mice (p = 0.0005 (analyzed by Sen-Adichie test)) (Fig. 2A).
TabLE 3. Incidence of Liver Tumors by Dioxin in WT and Triple Null Mice.
| Genotype | DEN initiation | Tumor incidence_a_ | |||
|---|---|---|---|---|---|
| Corn-oil | Dioxin | ||||
| WT | DEN | 13/19 | (68%) | 19/19 | (100%)b |
| TKO | DEN | 4/19 | (21%)b | 7/17 | (41%) |
Fig. 2.

Decrease of dioxin-induced tumor multiplicity in the TNF/IL-1 receptors triple null mice. (A) Dioxin-induced liver tumor multiplicity in the DEN-initiated WT and TKO mice. Mice were treated at 13 days of age with a single dose of 0.1 μmol/g body weight of DEN. At 9–10 weeks of age, the mice were treated by gavage with either 10 μg/kg body weight of dioxin or 10 ml/kg body weight of corn-oil every 2 weeks for 24 weeks. Each group contained >17 mice (n = 17–19). Bars represent mean liver tumor multiplicities. (B) Levels of 7-methylguanine in liver DNA of WT and triple-null (TKO) mice. Mice were treated at 13 days of age with a because dose of 0.1 μmol/g body weight of DEN or the solvent tricaprylin. These mice were killed 24 h later. Each group contained three mice. Bars represent mean values.
We also examined the effect of dioxin on G6Pase-deficient liver tumor foci development in the TKO mice at 24 weeks dioxin exposure. The results are presented in Table 4. Although we observed a significant increase in numbers of foci in both WT and TKO mice treated with dioxin, we found fewer numbers of G6Pase-deficient foci in the DEN/corn-oil-treated TKO mice compared with DEN/corn-oil-treated WT mice (p < 0.05). However, no significant difference in mean diameter and volume fraction in the TKO mice treated with dioxin as compared with those treated with corn oil.
TabLE 4. Comparison of the Number, Size, and Volume Fraction of G-6-Pase-Deficient Foci in the Livers of WT and Triple-Null Mice Treated with Corn-Oil or Dioxin.
| Genotype | Treatment | Number of mice | G-6-Pase-deficient foci | ||
|---|---|---|---|---|---|
| Number of foci_a_ | Focus diameter_b_ | Volume fraction_c_ | |||
| +DEN initiation | |||||
| WT | Corn-oil | 19 | 2.58 ± 0.44 | 625.8 ± 93.9 | 3.28 ± 0.81 |
| Dioxin | 19 | 5.42 ± 0.48_d_ | 1704.0 ± 394.6_d_ | 15.44 ± 2.64_d_ | |
| TKO | Corn-oil | 19 | 0.74 ± 0.28_d_ | 721.8 ± 201.1 | 1.22 ± 0.77 |
| Dioxin | 16 | 1.86 ± 0.33_e_ | 944.2 ± 113.7 | 2.12 ± 0.58 |
DISCUSSION
Due to their environmental persistence and their potency as carcinogens, teratogens, and acute toxicants, dioxins and related halogenated aromatic pollutants pose a health risk to both humans and wildlife (Kransler et al., 2007; Wells et al., 2010). Although the carcinogenic activity of dioxin is supported by data from experimental animal systems as well as human epidemiology (Huff et al., 1994; IARC, 1997), its mechanism of carcinogenic, teratogenic, or toxic action is poorly understood. In particular, an understanding of how dioxin exposure leads to cancer in mammalian systems is an important objective, as insights into its underlying mechanism have the potential to inform our assessment of carcinogenic risk for exposed populations. For example, a formal proof for the role of the AHR in dioxin carcinogenesis will add support for receptor mediated risk assessment models (Demby and Lucier, 1996; Green, 1991; Rudmann and Durham, 1999). Moreover, if a role for inflammation can be clearly demonstrated for this tumor promoter, it may provide us with a central mechanism by which many tumor promoters and nongenotoxic carcinogens act. Finally, if inflammatory signaling ultimately proves to be a well-documented central mechanism for many tumor promoters, then anti-inflammatory interventions become a possible treatment paradigm.
These tumor promotion studies, employing the murine model, as well as congenic mice differing primarily at specific genetic loci provide evidence to support a number of important conclusions. The first conclusion is that dioxin is acting as a tumor promoter in this model system. This is based on the observation that an increase in hepatocellular tumor multiplicity after 24 weeks of dioxin exposure occurs only after an initiating dose of DEN (Fig. 1A) and the observation that there are no differences in DEN initiation, as measured by DNA alkylation, in the _Ahrb1_-Ahrd model (Fig. 1B). A second conclusion is that tumor promotion is dependent upon the binding of dioxin to the AHR protein. This is based upon the observation that sensitivity to promotion segregated with the high affinity Ahrb1 allele. A third conclusion is that inflammatory cytokines play an important role in mouse hepatocellular tumor promotion in this model. This is based on the marked reduction in dioxin promoted tumors observed in TKO mice as compared with WT controls.
Our results using the Ahrb1/Ahrd congenic system are supportive of the potential for dioxin to be a human tumor promoter. Although we were unable to demonstrate a significant increase in dioxin promoted tumors at 24 weeks of dioxin exposure in Ahrd/d mice (Fig. 1, Table 2), we were able to demonstrate a significant effect at a longer exposure of 40 weeks (Supplementary data 1 and 2). The Ahrd allele encodes a receptor that harbors the Ala375-Val 375 polymorphism that is thought to be a primary molecular explanation for the decreased binding affinity of this receptor isoform for dioxin (Chang et al., 1993; Ema et al., 1994; Poland et al., 1989, 1991; Ramadoss and Perdew, 2004). Moreover, the mouse AHRd receptor is structurally more similar to the human AHR at its C-terminal residues and overall molecular size (Ema et al., 1994; Ramadoss and Perdew, 2004). Thus, although the AHRd receptor isoform may bind dioxin with decreased affinity, as compared with the AHRb1 isoform, our results suggest that the AHRd receptor can still mediate tumor promotion by dioxin, albeit with lower relative sensitivity and/or longer response time. Given the structural and functional similarities, these data support the idea that the human receptor can also mediate the tumor promotion signal transduction events reported here for the Ahrd allele.
It is important to compare previous mouse dioxin liver cancer studies with our results. In one earlier study, transgenic mice expressing a constitutively active form of AHR (CA-AHR) were shown to display an increased number of liver tumors after a single injection of DEN, as compared with nontransgenic controls (Moennikes et al., 2004). This observation supports the idea that the activated AHR can mediate tumor promoting events in the liver. In another earlier study, an attempt was made to provide a genetic proof for a role for the AHR in dioxin-mediated carcinogenesis (Beebe et al., 1995). Although this earlier study concluded a role for the Ahr in this process, the study was confounded by the use of two highly polymorphic strains (C57BL/6J and DBA/2) to represent these two Ahr alleles. As was noted in the earlier study, the inability to clearly demonstrate segregation of dioxin promotion with the Ahr locus was probably an indication that additional polymorphic alleles in the mouse are influencing tumor yield (Bennett et al., 1995; Bilger et al., 2004). Based upon the earlier results and those we report here, we conclude that congenic strains, differing at a singular Ahr locus, provide a more robust approach to delineating the role of the AHR in complex mammalian systems.
Given the dependence of both acute (hepatic necrosis) and chronic liver effects (tumor promotion) on the Ahr locus, we examined the idea that additional pathways are shared by these endpoints. To this end, we examined the tumor phenotype in a mouse model that is a “triple knock-out” (TKO) for the genes encoding the three “IL-1-like” cytokine receptors; TNFR1, TNFR2, and IL-1R (Pande et al., 2005). The IL-1-like inflammatory pathway is intriguing as a potential player in dioxin tumor promotion. First, the cytokines TNF and IL-1 have been shown in a number of models to be tumor promoting cytokines (Ben-Neriah and Karin, 2011; He and Karin, 2011). Second, a considerable body of evidence is emerging to support the idea that the inflammatory response plays critical roles in carcinogenesis (Ben-Neriah and Karin, 2011; Grivennikov et al., 2010; He and Karin, 2011; Kuraishy et al., 2011). Third, with regard to liver tumor promotion, there is evidence that production of a target of the IL-1-like cytokines, IL-6, by resident macrophages contributes to the development of DEN-induced tumors in mice (Naugler et al., 2007). Finally, the TNF and IL-1 cytokines bind to the receptors TNFR1, TNFR2, and IL-1R and stimulate the activation of transcriptional factors NF-κB, STAT3, and AP-1 (Ben-Neriah and Karin, 2011; Grivennikov et al., 2010; He and Karin, 2011), three factors known to play important roles in inducing tumorigenic genes related to cell proliferation and survival (Ben-Neriah and Karin, 2011; Grivennikov et al., 2010; He and Karin, 2011; Kuraishy et al., 2011).
The observation that the TKO mouse model was highly resistant to the tumor promoting effects of dioxin supports the concept that AHR activation leads to subsequent inflammatory signaling through IL-1-like cytokines and that this axis that may lie at the heart of dioxin's hepatocarcinogenic and hepatotoxic action (Fig. 2A, Tables 3 and 4). This concept is in keeping with findings from other laboratories. For example, in rodent liver, Tnf-α and Il-1β gene expression has been shown to be induced by dioxin (Fan et al., 1997). Moreover, inhibition of TNF-α activity through the administration of a TNF-antibody resulted in a 54% reduction in mortality induced by acute TCDD-mediated toxicity in mice (Taylor et al., 1992). Although it is formally possible that these cytokines genes are directly induced by AHR through the DREs within their enhancer region (Schmidt and Bradfield, 1996), it is also possible that the induction of the cytokines is indirectly regulated by AHR. Although we do not provide any evidence to distinguish between these possibilities, we offer these points. Arguing against a direct DRE mechanism: First, the induction of Tnf-α and Il-1β mRNA by dioxin shows a temporal response that is delayed from the induction of well-known DRE-driven genes such as Cyp1a1 (Fan et al., 1997; Hayes et al., 2007). Second, functional DREs within the Tnf-α and Il-1β genes have been not been reported (Narayanan et al., 2012). Finally, the TNF and IL-1 cytokines are commonly produced by innate immune cells such as Kupffer cells (Laskin et al., 2011), whereas the TNF/IL-1-dependent acute hepatic inflammation induced by dioxin exposure is mediated through AHR in hepatocytes (Walisser et al., 2005). Arguing for a direct regulatory mechanism is a recent study that reports that Il-6 gene expression is regulated through functional DREs within its promoter region (DiNatale et al., 2010). Because the secretion of IL-6 can be considered a consequence of IL1/TNF signaling and because this cytokine is critically involved in the development of hepatic injury and hepatocellular carcinoma (Naugler et al., 2007), IL-6 is one candidate for a DRE-driven gene related to the dioxin liver toxicity and tumor promotion.
Our working model going forward is that dioxin induces expression of DRE-driven genes within the hepatocytes and that these genes somehow signal to the resident macrophages to release IL-1-like cytokines which leads to both acute and chronic dioxin effects. What we find equally exciting is the prospect that this inflammatory pathway may be important in the action of a wide variety of tumor promoters. In this regard, we note that liver tumor multiplicity was decreased in the TKO mice after DEN treatment alone, even in the absence of dioxin promotion (Fig. 2A). This was confirmed by histologic analysis (Table 4) and may indicate that much of liver tumor development in the mouse is dependent upon an inflammatory response mediated by the IL-1 like cytokines (Lin et al., 2013). This observation could be an indication that tumor promotion by a variety of agents, including endogenous ones, may lie at the heart of murine hepatocarcinogenesis (Mezrich et al., 2010; Nguyen and Bradfield, 2008).
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
National Institutes of Health (K08-ES-017283 to G.D.K., R01 ES020668 to C.A.B., P01-CA-022484 to C.A.B, and P30-CA-014520).
Supplementary Material
Supplementary Data
Acknowledgments
We thank Anna L. Shen for reviewing the manuscript. Conflict of interest: C.A.B. has served as a consultant to DOW Chemical Company on issues related to dioxin toxicology.
REFERENCES
- Balbo S., Villalta P. W., Hecht S. S. Quantitation of 7-ethylguanine in leukocyte DNA from smokers and nonsmokers by liquid chromatography-nanoelectrospray-high resolution tandem mass spectrometry. Chem. Res. Toxicol. 2011;24:1729–1734. doi: 10.1021/tx200262d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beebe L. E., Fornwald L. W., Diwan B. A., Anver M. R., Anderson L. M. Promotion of N-nitrosodiethylamine-initiated hepatocellular tumors and hepatoblastomas by 2,3,7,8-tetrachlorodibenzo-p-dioxin or Aroclor 1254 in C57BL/6, DBA/2, and B6D2F1 mice. Cancer Res. 1995;55:4875–4880. [PubMed] [Google Scholar]
- Ben-Neriah Y., Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat. Immunol. 2011;12:715–723. doi: 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- Bennett L. M., Farnham P. J., Drinkwater N. R. Strain-dependent differences in DNA synthesis and gene expression in the regenerating livers of CB57BL/6J and C3H/HeJ mice. Mol. Carcinog. 1995;14:46–52. doi: 10.1002/mc.2940140109. [DOI] [PubMed] [Google Scholar]
- Bilger A., Bennett L. M., Carabeo R. A., Chiaverotti T. A., Dvorak C., Liss K. M., Schadewald S. A., Pitot H. C., Drinkwater N. R. A potent modifier of liver cancer risk on distal mouse chromosome 1: Linkage analysis and characterization of congenic lines. Genetics. 2004;167:859–866. doi: 10.1534/genetics.103.024521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C., Smith D. R., Prasad V. S., Sidman C. L., Nebert D. W., Puga A. Ten nucleotide differences, five of which cause amino acid changes, are associated with the Ah receptor locus polymorphism of C57BL/6 and DBA/2 mice. Pharmacogenetics. 1993;3:312–321. doi: 10.1097/00008571-199312000-00005. [DOI] [PubMed] [Google Scholar]
- Chen L., Wang M., Villalta P. W., Hecht S. S. Liquid chromatography-electrospray ionization tandem mass spectrometry analysis of 7-ethylguanine in human liver DNA. Chem. Res. Toxicol. 2007;20:1498–1502. doi: 10.1021/tx700147f. [DOI] [PubMed] [Google Scholar]
- Davis B. J., McCurdy E. A., Miller B. D., Lucier G. W., Tritscher A. M. Ovarian tumors in rats induced by chronic 2,3,7,8-tetrachlorodibenzo-p-dioxin treatment. Cancer Res. 2000;60:5414–5419. [PubMed] [Google Scholar]
- Demby K. B., Lucier G. W. Receptor-mediated carcinogenesis: The role of biological effect modeling for risk assessment of dioxin and tamoxifen. Prog. Clin. Biol. Res. 1996;394:113–129. [PubMed] [Google Scholar]
- DiNatale B. C., Schroeder J. C., Francey L. J., Kusnadi A, Perdew G. H. Mechanistic insights into the events that lead to synergistic induction of interleukin 6 transcription upon activation of the aryl hydrocarbon receptor and inflammatory signaling. J. Biol. Chem. 2010;285:24388–24397. doi: 10.1074/jbc.M110.118570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drinkwater N. R., Denniston C. Statistical Problems in Genetics and Molecular Biology. CreatSpace Independent Publishing Platform; 2011. pp. 89–163. [Google Scholar]
- Ema M., Ohe N., Suzuki M., Mimura J., Sogawa K., Ikawa I., Fujii-Kuriyama Y. Dioxin binding activities of polymorphic forms of mouse and human aryl hydrocarbon receptors. J. Biol. Chem. 1994;269:27337–27343. [PubMed] [Google Scholar]
- Fan F., Yan B., Wood G., Viluksela M., Rozman K. K. Cytokines (IL-1beta and TNFalpha) in relation to biochemical and immunological effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in rats. Toxicology. 1997;116:9–16. doi: 10.1016/s0300-483x(96)03514-7. [DOI] [PubMed] [Google Scholar]
- Giri A. K. Mutagenic and genotoxic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin, a review. Mutat. Res. 1986;168:241–248. doi: 10.1016/0165-1110(86)90022-9. [DOI] [PubMed] [Google Scholar]
- Green S. Receptor-mediated non-genotoxic carcinogenesis: New models for risk assessment? Hum. Exp. Toxicol. 1991;10:390–395. doi: 10.1177/096032719101000602. [DOI] [PubMed] [Google Scholar]
- Grivennikov S. I., Greten F. R., Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hankinson O. The aryl hydrocarbon receptor complex. Annu. Rev. Pharmacol. Toxicol. 1995;35:307–340. doi: 10.1146/annurev.pa.35.040195.001515. [DOI] [PubMed] [Google Scholar]
- Hayes K. R., Zastrow G. M., Nukaya M., Pande K., Glover E., Maufort J. P., Liss A. L., Liu Y., Moran S. M., Vollrath A. L., et al. Hepatic transcriptional networks induced by exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Chem. Res. Toxicol. 2007;20:1573–1581. doi: 10.1021/tx7003294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G., Karin M. NF-kappaB and STAT3—Key players in liver inflammation and cancer. Cell Res. 2011;21:159–168. doi: 10.1038/cr.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff J., Lucier G., Tritscher A. Carcinogenicity of TCDD: Experimental, mechanistic and epidemiologic evidence. Annu. Rev. Pharmacol. Toxicol. 1994;34:343–372. doi: 10.1146/annurev.pa.34.040194.002015. [DOI] [PubMed] [Google Scholar]
- IARC. IARC Monogr. Eval. Carcinog. Risks Hum. Vol. 69. Lyon, France: 1997. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans: Polychlorinated Dibenzo-Para-Dioxins and Polychlorinated Dibenzofurans; pp. 1–631. 4–11 February 1997. [PMC free article] [PubMed] [Google Scholar]
- Kransler K. M., McGarrigle B. P., Olson J. R. Comparative developmental toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in the hamster, rat and guinea pig. Toxicology. 2007;229:214–225. doi: 10.1016/j.tox.2006.10.019. [DOI] [PubMed] [Google Scholar]
- Kuraishy A., Karin M., Grivennikov S. I. Tumor promotion via injury- and death-induced inflammation. Immunity. 2011;35:467–477. doi: 10.1016/j.immuni.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskin D. L., Sunil V. R., Gardner C. R., Laskin J. D. Macrophages and tissue injury: Agents of defense or destruction? Annu. Rev. Pharmacol. Toxicol. 2011;51:267–288. doi: 10.1146/annurev.pharmtox.010909.105812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H., Yan J., Wang Z., Hua F., Yu J., Sun W., Li K., Liu H., Yang H., Lv Q., et al. Loss of immunity-supported senescence enhances susceptibility to hepatocellular carcinogenesis and progression in Toll-like receptor 2-deficient mice. Hepatology. 2013;57:171–182. doi: 10.1002/hep.25991. [DOI] [PubMed] [Google Scholar]
- Lucier G., Clark G., Hiermath C., Tritscher A., Sewall C., Huff J. Carcinogenicity of TCDD in laboratory animals: Implications for risk assessment. Toxicol. Ind. Health. 1993;9:631–668. doi: 10.1177/074823379300900406. [DOI] [PubMed] [Google Scholar]
- Mezrich J. D., Fechner J. H., Zhang X., Johnson B. P., Burlingham W. J., Bradfield C. A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010;185:3190–3198. doi: 10.4049/jimmunol.0903670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moennikes O., Loeppen S., Buchmann A., Andersson P., Ittrich C., Poellinger L., Schwarz M. A constitutively active dioxin/aryl hydrocarbon receptor promotes hepatocarcinogenesis in mice. Cancer Res. 2004;64:4707–4710. doi: 10.1158/0008-5472.CAN-03-0875. [DOI] [PubMed] [Google Scholar]
- Moore M. R., Drinkwater N. R., Miller E. C., Miller J. A., Pitot H. C. Quantitative analysis of the time-dependent development of glucose-6-phosphatase-deficient foci in the livers of mice treated neonatally with diethylnitrosamine. Cancer Res. 1981;41:1585–1593. [PubMed] [Google Scholar]
- Narayanan G. A., Murray I. A., Krishnegowda G., Amin S., Perdew G. H. Selective aryl hydrocarbon receptor modulator-mediated repression of CD55 expression induced by cytokine exposure. J. Pharmacol. Exp. Ther. 2012;342:345–355. doi: 10.1124/jpet.112.193482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naugler W. E., Sakurai T., Kim S., Maeda S., Kim K., Elsharkawy A. M., Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- Nebert D. W., Gonzalez F. J. P450 genes: Structure, evolution, and regulation. Annu. Rev. Biochem. 1987;56:945–993. doi: 10.1146/annurev.bi.56.070187.004501. [DOI] [PubMed] [Google Scholar]
- Nguyen L. P., Bradfield C. A. The search for endogenous activators of the aryl hydrocarbon receptor. Chem. Res. Toxicol. 2008;21:102–116. doi: 10.1021/tx7001965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nukaya M., Moran S., Bradfield C. A. The role of the dioxin-responsive element cluster between the Cyp1a1 and Cyp1a2 loci in aryl hydrocarbon receptor biology. Proc. Natl. Acad. Sci. U.S.A. 2009;106:4923–4928. doi: 10.1073/pnas.0809613106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pande K., Moran S. M., Bradfield C. A. Aspects of dioxin toxicity are mediated by interleukin 1-like cytokines. Mol. Pharmacol. 2005;67:1393–1398. doi: 10.1124/mol.105.010983. [DOI] [PubMed] [Google Scholar]
- Pitot H. C., Goldsworthy T. L., Moran S., Kennan W., Glauert H. P., Maronpot R. R., Campbell H. A. A method to quantitate the relative initiating and promoting potencies of hepatocarcinogenic agents in their dose-response relationships to altered hepatic foci. Carcinogenesis. 1987;8:1491–1499. doi: 10.1093/carcin/8.10.1491. [DOI] [PubMed] [Google Scholar]
- Poland A., Palen D., Glover E. Tumour promotion by TCDD in skin of HRS/J hairless mice. Nature. 1982;300:271–273. doi: 10.1038/300271a0. [DOI] [PubMed] [Google Scholar]
- Poland A., Palen D., Glover E. Analysis of the four alleles of the murine aryl hydrocarbon receptor. Mol. Pharmacol. 1994;46:915–921. [PubMed] [Google Scholar]
- Poland A., Teitelbaum P., Glover E., Kende A. Stimulation of in vivo hepatic uptake and in vitro hepatic binding of [125I]2-lodo-3,7,8-trichlorodibenzo-p-dioxin by the administration of agonist for the Ah receptor. Mol. Pharmacol. 1989;36:121–127. [PubMed] [Google Scholar]
- Ramadoss P., Perdew G. H. Use of 2-azido-3-[125I]iodo-7,8-dibromodibenzo-p-dioxin as a probe to determine the relative ligand affinity of human versus mouse aryl hydrocarbon receptor in cultured cells. Mol. Pharmacol. 2004;66:129–136. doi: 10.1124/mol.66.1.129. [DOI] [PubMed] [Google Scholar]
- Rudmann D. G., Durham S. K. Utilization of genetically altered animals in the pharmaceutical industry. Toxicol. Pathol. 1999;27:111–114. doi: 10.1177/019262339902700121. [DOI] [PubMed] [Google Scholar]
- Schmidt J. V., Bradfield C. A. Ah receptor signaling pathways. Annu. Rev. Cell Dev. Biol. 1996;12:55–89. doi: 10.1146/annurev.cellbio.12.1.55. [DOI] [PubMed] [Google Scholar]
- Taylor M. J., Lucier G. W., Mahler J. F., Thompson M., Lockhart A. C., Clark G. C. Inhibition of acute TCDD toxicity by treatment with anti-tumor necrosis factor antibody or dexamethasone. Toxicol. Appl. Pharmacol. 1992;117:126–132. doi: 10.1016/0041-008x(92)90227-j. [DOI] [PubMed] [Google Scholar]
- Walisser J. A., Glover E., Pande K., Liss A. L., Bradfield C. A. Aryl hydrocarbon receptor-dependent liver development and hepatotoxicity are mediated by different cell types. Proc. Natl. Acad. Sci. U.S.A. 2005;102:17858–17863. doi: 10.1073/pnas.0504757102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells P. G., Lee C. J., McCallum G. P., Perstin J., Harper P. A. Receptor- and reactive intermediate-mediated mechanisms of teratogenesis. Handb. Exp. Pharmacol. 2010;196:131–162. doi: 10.1007/978-3-642-00663-0_6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data