Up-Regulation of PD-L1, IDO, and Tregs in the Melanoma Tumor Microenvironment Is Driven by CD8+ T Cells (original) (raw)
. Author manuscript; available in PMC: 2014 Aug 18.
Published in final edited form as: Sci Transl Med. 2013 Aug 28;5(200):200ra116. doi: 10.1126/scitranslmed.3006504
Abstract
Tumor escape from immune-mediated destruction has been associated with immunosuppressive mechanisms that inhibit T cell activation. Although evidence for an active immune response, including infiltration with CD8+ T cells, can be found in a subset of patients, those tumors are nonetheless not immunologically rejected. In the current report, we show that it is the subset of T cell–inflamed tumors that showed high expression of three defined immunosuppressive mechanisms: indoleamine-2,3-dioxygenase (IDO), PD-L1/B7-H1, and FoxP3+ regulatory T cells (Tregs), suggesting that these inhibitory pathways might serve as negative feedback mechanisms that followed, rather than preceded, CD8+ T cell infiltration. Mechanistic studies in mice revealed that up-regulated expression of IDO and PD-L1, as well as recruitment of Tregs, in the tumor microenvironment depended on the presence of CD8+ T cells. The former was driven by interferon-γ and the latter by a production of CCR4-binding chemokines along with a component of induced proliferation. Our results argue that these major immunosuppressive pathways are intrinsically driven by the immune system rather than being orchestrated by cancer cells, and imply that cancer immunotherapy approaches targeting negative regulatory immune checkpoints might be preferentially beneficial for patients with a preexisting T cell–inflamed tumor microenvironment.
Introduction
Despite recent developments in cancer immunotherapies, clinical benefit occurs in a minority of patients. This has been observed in the case of interleukin-2 (IL-2) for melanoma and kidney cancer (1), experimental cancer vaccines (2), and the recently U.S. Food and Drug Administration–approved agents Provenge for prostate cancer (3) and anti–CTLA-4 monoclonal antibody (mAb) (ipilimumab) for melanoma (4). Recent work has suggested that one explanation for tumor resistance to immunotherapies might be due to immunosuppressive events that act at the level of the tumor microenvironment (5). Key mechanisms that have been observed in clinical samples and validated as functionally important in mouse models include extrinsic suppression of CD8+ effector cells by CD4+CD25+FoxP3+ regulatory T cells (Tregs) (6), metabolic deregulation via tryptophan catabolism by indoleamine-2,3-dioxygenase (IDO) (7), and engagement of the inhibitory receptor PD-1 by the ligand PD-L1/B7-H1 (8, 9). Clinical strategies to counter these immunosuppressive pathways are currently being evaluated, already with encouraging early-phase clinical trial results (10–12). However, the mechanisms by which these immunosuppressive pathways become recruited and functionally operational within the tumor microenvironment are not clear, and which subsets of patients might express these pathways and theoretically benefit from targeting them are incompletely understood.
We and others recently have analyzed a series of melanoma metastases by gene expression profiling and confirmatory assays, and found that some samples contain abundant CD8+ T cell infiltrates and some do not (13–16). Spontaneously primed CD8+ T cells specific for defined melanoma antigens have also been identified in the peripheral blood in a subset of patients (2, 17, 18). The T cell–inflamed subset also expresses chemokines for T cell recruitment (13) and a type I interferon (IFN) transcriptional profile that appears to participate in innate immune sensing (19, 20). Clinical responders to melanoma vaccines and to ipilimumab appear to be enriched in the T cell–inflamed subset of tumors, suggesting that an ongoing dialogue between the tumor and the host immune response may be predictive of clinical benefit (14, 21). However, even if one were to enrich for patients having the inflamed tumor phenotype, fewer than half of the patients would still be estimated to respond, suggesting that additional barriers might need to be overcome to maximize therapeutic efficacy. With this notion in mind, more detailed analysis of our gene expression profiling data was performed and revealed that the T cell–inflamed subset of melanomas included those tumors showing high expression of the inhibitory factor IDO. Further interrogation of those samples revealed high expression of PD-L1/B7-H1 and also abundant FoxP3+ Tregs. Mechanistic studies in mice were performed to determine causal relationships, and our data indicate that up-regulated expression of IDO and PD-L1/B7-H1, as well as accumulation of Tregs, in the melanoma tumor microenvironment depended on CD8+ T cells. IDO and PD-L1/B7-H1 up-regulation was dependent on IFN-γ. Treg accumulation was not due to CD8+ T cells promoting conversion from FoxP3− CD4+ cells, but rather was largely due to the production of CCR4-binding chemokines with an additional contribution of induced proliferation. Collectively, these results suggest that the presence of these immunosuppressive factors in melanoma metastases is immune-intrinsic and driven by CD8+ T cells. Regarding clinical application of checkpoint blockade, these data imply that Treg depletion, PD-1/PD-L1 blockade, and IDO inhibitors may be beneficial preferentially in the subset of patients already showing a T cell–inflamed tumor microenvironment, and that alternative therapeutic strategies might be required for patients showing absence of spontaneous inflammation and adaptive immunity.
Results
Melanoma metastases that contain activated T cells have highest expression of the immunosuppressive pathways IDO, PD-L1, and Tregs
Affymetrix gene expression profiling and confirmatory assays recently revealed that some melanoma metastases contain abundant CD8+ T cells, a broad chemokine signature, and a type I IFN transcriptional profile (13, 14, 19). On the basis of parallel work suggesting dysfunction of CD8+ T cells when analyzed directly from the tumor microenvironment ex vivo (22–24), those tumors were also interrogated for the presence of putative negative regulatory mechanisms that might inhibit effector T cell function. Reanalysis of the gene array data revealed that the inflamed melanoma subset also contained the tumors with elevated IDO expression (GSE12627). IDO is a tryptophanmetabolizing enzyme that has been shown to contribute to peripheral immunologic tolerance (25). Examination of a second immunoregulatory metabolic enzyme, arginase I, revealed expression only in a subset of non–T cell–inflamed tumors, arguing that it may be less functionally relevant for inhibiting the function of T cells present in the tumor microenvironment. Because of the presence of IDO transcripts, these tumors were also interrogated for expression of other molecules indicative of distinct mechanisms of immune suppression. Expression of PD-L1/B7-H1, an inhibitory ligand that engages the negative regulatory receptor PD-1 on activated T cells (8), and FoxP3, a marker for Tregs (26), was examined. Indeed, quantitative reverse transcription polymerase chain reaction (qRT-PCR) showed higher expression of IDO, PD-L1, and FoxP3 in the tumors that contained CD8+ T cells. A strong positive correlation between expression of IDO, PD-L1, and FoxP3 transcripts (correlation coefficient = 0.999 for each paired relationship; Pearson correlation), suggesting a coordinated up-regulation of at least three immunosuppressive mechanisms in the microenvironment of inflamed tumors (Fig. 1A).
Fig. 1. Correlation of FoxP3 expression with CD8+ cell infiltration and IDO and PD-L1 expression in melanomas.
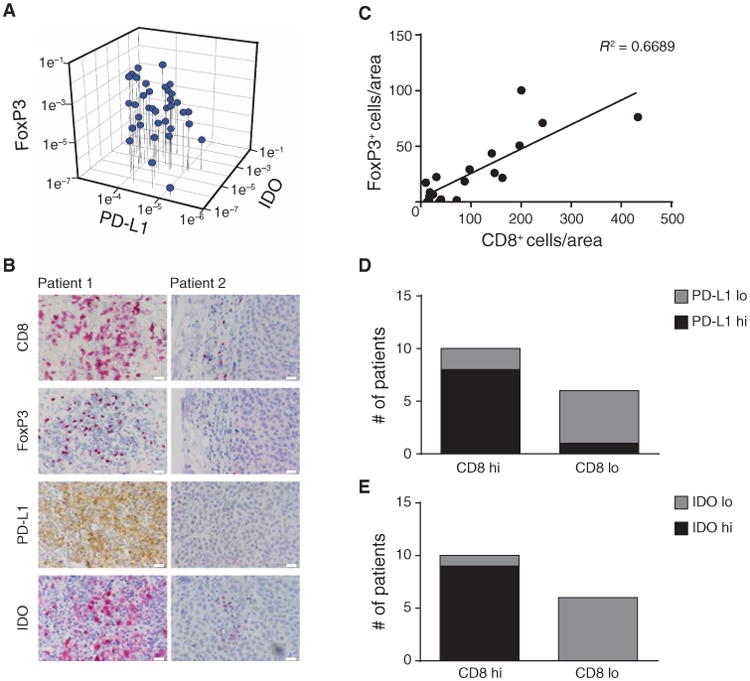
(A) From a series of metastatic melanoma patient tumor biopsies, FoxP3, PD-L1, and IDO mRNA expression levels were determined using real-time RT-PCR. 18S was used as an internal control. (B) IHC analysis was performed on a representative subset of tumors. CD8 was developed using Vulcan Red, and FoxP3 was developed using horseradish peroxidase. Similar results were observed for three independent tumors of each category. Scale bars, 20 μm. (C) Number of CD8+ and FoxP3+ cells was assessed in three visual fields (depicted as area). Correlation studies were performed with _R_2 = 0.6689 (Pearson correlation; total of 16 patients). (D and E) Tumors were grouped in CD8high and CD8low and analyzed for the amount of PD-L1 (D) or IDO (E) staining. The number of CD8+ T cells correlated significantly with the level of PD-L1 (p = 0.035) and IDO (p = 0.002) expression when tested with a two-sided χ2 in combination with Fisher's exact test.
A subset of melanoma metastases from which we had sufficient material for immunohistochemical (IHC) analysis was interrogated in more detail for expression of these markers at the protein level. Tumors with the inflamed gene expression profile showed the presence of CD8+ T cells by IHC (Fig. 1B). These tumors also showed the presence of lymphocytes with nuclear FoxP3 staining, consistent with the presence of Tregs. Double staining revealed that FoxP3 was expressed by CD4+ T cells and not by CD8+ cells (fig. S1). In addition, PD-L1 protein was detected at high levels, as was expression of IDO (Fig. 1B). PD-L1 and IDO appeared to be expressed at least by tumor cells based on morphology but also could be expressed by some stromal cells. In contrast, melanoma metastases that lacked a CD8+ T cell infiltrate showed minimal expression of FoxP3, PD-L1, or IDO (Fig. 1B). Statistical analysis over a panel of samples revealed a positive correlation (Pearson correlation, P < 0.0001) between the number of CD8+ T cells and the number of FoxP3+ Tregs (Fig. 1C), as well as with the expression of PD-L1 and IDO (Fig. 1, D and E). Together, these data indicate that the abundance of these three immune inhibitory factors is correlated with the degree of infiltration of melanoma metastases with CD8+ T cells.
Up-regulation of IDO and PD-L1 in the tumor microenvironment in vivo is dependent on CD8+ T cells and IFN-γ
On the basis of the above correlative data, we turned to murine in vivo models to consider mechanisms by which CD8+ T cells might promote induction of PD-L1 and IDO, as well as accumulation of Tregs, in the tumor microenvironment in vivo. The fact that IDO and PD-L1 can be induced by proinflammatory cytokines such as IFN-γ in vitro (8, 27) suggested that cytokines produced by activated CD8+ Tcells in the tumor microenvironment could explain the elevated expression of these factors preferentially in inflamed tumors in vivo. B16 melanoma expressed low levels of IDO mRNA and of PD-L1/B7-H1 protein in vitro (fig. S2, A and B). However, implantation of B16. SIY melanoma cells subcutaneously into immunocompetent C57BL/6 mice followed by reanalysis after 7 days ex vivo revealed marked up-regulation of both IDO and PD-L1/B7-H1 mRNA and also PD-L1/B7-H1 protein expression (Fig. 2, A and B, and fig. S2, A and B, for B16 in vivo). When B16. SIY melanoma cells were implanted in mice depleted of CD8+ T cells, or into IFN-γ–deficient mice, this up-regulation failed to occur (Fig. 2, A and B). Increased expression of class I and II major histocompatibility complex (MHC) molecules on the tumor cells also required CD8+ T cells (fig. S2, C and D). Therefore, up-regulated expression of both IDO and PD-L1/B7-H1 in the melanoma tumor microenvironment in vivo depends on CD8+ T cells and IFN-γ.
Fig. 2. Dependence of IDO, PD-L1, and FoxP3+ Tregs in the B16 tumor microenvironment on host CD8+ T cells and IFN-γ.

B16.SIY melanoma cells were implanted subcutaneously in wild-type (WT) C57BL/6 mice treated with isotype control antibody (n = 2; gray) or CD8-depleting antibody (n = 3; dashed line) or in IFN-γ−/− mice (n = 2; solid line). (A to D) At day 7, the tumors were subjected to ex vivo RT-PCR and fluorescence-activated cell sorting (FACS) analysis for IDO (A), PD-L1 (B), and FoxP3 (C and D). FACS data for PD-L1 are shown as geometric mean within the GFP+CD45− population, and the Tregs were identified as GFP−CD8−CD4+CD25+FoxP3+ cells. Shown is the mean ± SEM and tested for significance using one-way analysis of variance (ANOVA) (Kruskal-Wallis) test (WT = 5; IFN-γ = 6; CD8-depleted = 5) with P = 0.0016 for IDO expression, P =0.01 for PD-L1 expression, and P =0.0003 for FoxP3 expression (qRT-PCR and FACS). The results shown are representative of two independent experiments, which were combined for statistical purposes. KO, knockout; n.s., not significant.
This model also was used to explore a role for CD8+ T cells in Treg accumulation within the tumor microenvironment. Tregs were confirmed to accumulate in B16. SIY melanoma tumors after 7 days of implantation in vivo, both by FoxP3 RT-PCR analysis and using flow cytometry to assess the presence of CD4+CD25+FoxP3+ cells among dissociated tumor cells ex vivo (Fig. 2, C and D). This Treg accumulation was markedly reduced in mice depleted of CD8+ T cells but not significantly altered in IFN-γ−/− mice (Fig. 2, C and D). These results confirm that CD8+ T cells are required for maximal accumulation of Tregs in the melanoma tumor microenvironment in vivo, but largely via a mechanism independent of IFN-γ.
CD8+ T cells do not support Treg accumulation through induced conversion from FoxP3− CD4+ T cells
Three hypothetical mechanisms were considered that could explain a functional role for CD8+ T cells in Treg accumulation in the tumor microenvironment. The first consideration was promotion of conversion of CD4+FoxP3− T cells into induced Tregs. To test this notion, Rag2−/− mice received green fluorescent protein (GFP)–negative T cells from FoxP3-GFP reporter mice followed by implantation of B16 melanoma. Seven days later, tumors were removed, lymphocytes were harvested, and the percentage of Tregs was analyzed by flow cytometry. As shown in Fig. 3, no CD4+CD25+FoxP3+ T cells were detected in the tumor microenvironment in this model, arguing against conversion of induced Tregs. Similar results were obtained when CD25-depleted T cells were used rather than the FoxP3-GFP reporter mice (fig. S3). Inasmuch as CD8+ T cells were indeed present in these hosts, we conclude that CD8+ T cells are not facilitating conversion of CD4+FoxP3− T cells into induced Tregs.
Fig. 3. Lack of detectable conversion of FoxP3− CD4+ T cells to induced Tregs in B16 melanoma in vivo.
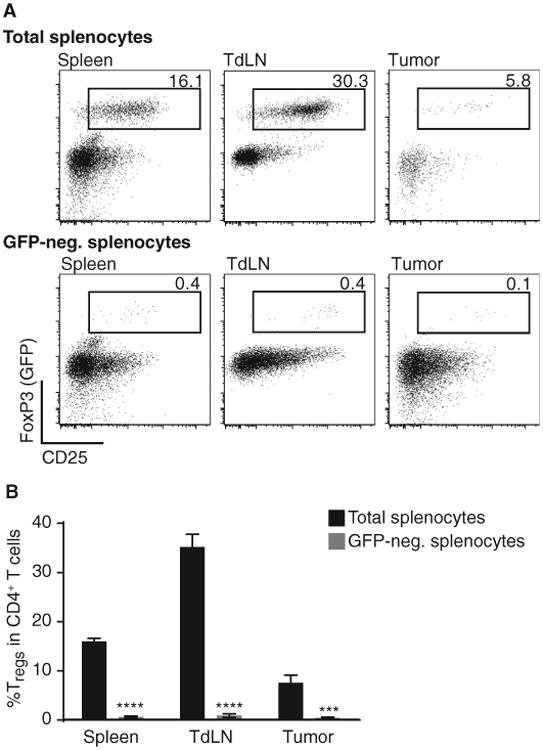
After tumor inoculation of B16 cells, Rag2−/− mice were injected with either 3 × 106 total splenocytes or GFP-depleted splenocytes of a FoxP3-GFP reporter mouse. On day 7 tumor, tumor-draining lymph node (TdLN) and spleen were analyzed for the amount of GFP+/CD25+/FoxP3+ cells within their CD4+ T cell compartment. (A) Representative example for results obtained with total splenocytes (top row) or GFP-depleted splenocytes injected in vivo (lower row). Cells were pregated on living cells and CD3+CD4+ cells. (B) Statistical analysis of six mice shown as mean ± SEM and two-sided Mann-Whitney U test to determine significance. ****P < 0.0001; ***P < 0.001).
Treg accumulation in the tumor microenvironment is mediated via a combination of CD8+ T cell–dependent chemokine-mediated recruitment and induced proliferation
Two other potential mechanisms by which CD8+ T cells could support accumulation of Tregs in the tumor microenvironment are induced proliferation of Tregs and production of chemokines that promote Treg recruitment. To determine whether the proliferative rate of Tregs in vivo was influenced by CD8+ T cells, C57BL/6 mice were depleted of CD8+ T cells or treated with an isotype control antibody, and B16 melanoma was implanted subcutaneously. On day 7, a single dose of 5-bromo-2′-deoxyuridine (BrdU) was injected intraperitoneally, and 1 day later, the tumor and lymph nodes were harvested for flow cytometric analysis of the Treg population. As shown in Fig. 4, about 26% of Tregs in the draining lymph nodes were BrdU-positive in tumor-bearing mice, and this fraction was not altered with CD8+ T cell depletion. In contrast, in the tumor microenvironment, the total number of Tregs was reduced when CD8+ T cells were eliminated, consistent with what we had observed previously. In addition, the proportion of remaining Tregs incorporating BrdU was modestly reduced, from about 27 to 10% (Fig. 4). Although the absolute number of Tregs in tumors from CD8-depleted mice was small (mean of 200 Tregs per tumor) and therefore subject to the caveat of difficulty quantifying small frequencies, this reduction was statistically significant (P < 0.0001). Therefore, CD8+ T cells may support Treg accumulation in the tumor microenvironment, at least in part, through induced proliferation of Tregs in situ.
Fig. 4. CD8+ T cells contribute to Treg proliferation in the tumor site in vivo.

B16 cells were injected subcutaneously in WT mice treated with CD8-depleting antibody or isotype control. Twenty-four hours after BrdU pulse, given on day 7 after tumor inoculation, tumor and tumor-draining lymph node were analyzed for the amount of proliferated CD4+FoxP3+ Tregs. (A) PercentageofFoxP3+ cells detected within the CD4+ T cell fraction either BrdU− (black) or BrdU+ (gray) (n = 6 out of two independent experiments; shown as mean ± SEM). (B) Fraction of proliferated (BrdU+) cells detected within the total Treg fraction (CD3+CD4+FoxP3+) in tumor (left) and tumor-draining lymph node (right). Using a two-sided Mann-Whitney U test to compare the two proliferated fractions, we confirmed a significantly reduced proliferation rate within the tumor-infiltrating Tregs under CD8-depleting conditions. *P =0.002; **P = 0.0094.
Because the reduction in the proliferating fraction of Tregs was modest compared to the reduction in total Tregs in the tumor micro-environment when CD8+ T cells were depleted, it seemed likely that Treg recruitment also might be critically regulated. Because previous work suggested that chemokines could contribute to Treg accumulation in tissues (28), we evaluated whether CD8+ effector T cells might produce any chemokines capable of Treg recruitment and whether chemokine-mediating trafficking participated in vivo. Rag2−/− mice were implanted with B16 melanoma tumors and reconstituted intravenously with 1 × 106 splenic CD4+CD25+ T cells either with or without previous administration of CD8+ T cells. Two days later, the tumor was harvested, and Treg infiltration was quantitated by flow cytometry. As shown in Fig. 5A, significant accumulation of Tregs in the tumor was only observed when CD8+ T cells were present (P = 0.0087, Mann-Whitney U test; n = 6). Flow cytometric analysis confirmed that most of the Tregs expressed the chemokine receptor CCR4 (fig. S4A). To determine whether chemokine receptor signaling in Tregs was necessary for their accumulation in the tumor site, the Tregs were pretreated with pertussis toxin before in vivo transfer to block chemokine receptor signaling. This treatment completely prevented Treg accumulation in the tumor microenvironment. To assess whether CCR4 was specifically involved, Tregs were pretreated with the CCR4 antagonist C021. This treatment also prevented Treg accumulation in the tumor (Fig. 5A). In contrast, neither pertussis toxin nor C021 affected the number of Tregs detected in lymphoid organs (Fig. 5B), arguing that their distribution, but not survival, was affected. The defined chemokine ligands for CCR4 are CCL17 and CCL22. In vitro–primed CD8+ T cells were analyzed for chemokine production after restimulation in vitro, and significant production of CCL22 mRNA was detected (Fig. 5C), which was confirmed by CCL22 enzyme-linked immunosorbent assay (ELISA) (fig. S4C). Purified CD8+ Tcells harvested from the B16 tumor microenvironment were also analyzed and were similarly found to produce CCL22 (Fig. 5C) but not CCL17 (fig. S4B). Together, these data indicate that the major mechanism by which CD8+ T cells support Treg accumulation in the tumor microenvironment is through the production of CCL22 and recruitment of Tregs via CCR4.
Fig. 5. Chemokine-mediated recruitment of Tregs in the tumor micro-environment supported by CD8+ T cells in vivo.

(A and B) B16 cells were engrafted into Rag2−/− (subcutaneously) followed, 24 hours later, by injection of 3 × 106 CD8+ T cells (intravenously) (if indicated), and on day 10 after tumor inoculation, 1 × 106 Tregs (CD4+ CD25+) were given (intravenously). PTX Tregs were treated with pertussis toxin for 1.5 hours in vitro before injection, and C021 Tregs were treated with the CCR4 antagonist C021 for 2 hours before injection. Forty-eight hours after Treg injection, tumor and spleen were analyzed for the number of infiltrating Tregs (CD3+, CD4+,FoxP3+). Tregs could only be detected in the tumor site if nontreated and given in combination with pre-engrafted CD8+ T cells (A), whereas chemokine receptor inhibitory treatment did not alter homing to the spleen (B). Shown are means ± SEM of n = 6 out of two independent experiments; significance was tested using two-sided Mann-Whitney U test (**P = 0.0024; ****P < 0.0001). (C) Naïve (CD62L+) CD8 T cells were cultured for 6 hours or 7 days ± 6 hours or assayed ex vivo. After activation, mRNA expression levels of CCL22 were assessed by qRT-PCR, normalized to 18S RNA, and were relative to the ex vivo expression level of CCL22 (*) (left panel). Tumor-infiltrating CD8 T cells were sorted out of B16 tumors 12 days after engraftment and analyzed either ex vivo or after activation for 6 hours (right panel). Shown are means ± SEM of n = 3, with numbers indicating the fold change compared to ex vivo naïve or tumor-infiltrating CD8+ T cells.
Human CD8+ T cells support the recruitment of human Tregs in a xenograft system
It was of interest to confirm whether human CD8+ T cells also could support recruitment of human Tregs into the melanoma tumor micro-environment. Purified CD8+ T cells from normal human donors were stimulated with anti-CD3/anti-CD28 mAb-coated beads for 10 days to generate primed effector cells (29). Supernatants from purified activated human CD8+ T cells were therefore analyzed for chemokine production, and as with mouse cells, CCL22 was detected in T cell supernatants (Fig. 6A). To determine whether the CCL22 produced by CD8+ T cells was functionally capable of recruiting Tregs, CD4+CD25hi cells were sorted from normal donor peripheral blood cells. Flow cytometric analysis confirmed that these cells were FoxP3+ and that most expressed the chemokine receptor CCR4 (fig. S5), and in vitro suppression assays confirmed that these cells potently inhibited CD4+ and CD8+ T cell proliferation in vitro (fig. S6). Supernatants from activated CD8+ effector T cells were then used in Transwell migration assays with sorted CD4+CD25hi cells. Activated CD8+ T cell supernatants were capable of promoting strong chemotaxis of Tregs in vitro, to the same extent as recombinant CCL22 (Fig. 6B). In addition, a neutralizing anti-CCL22 mAb blocked most of the migration observed with the CD8+ T cell supernatants, down to the level of background migration detected upon treatment with pertussis toxin as an inhibitor of chemokine receptor function (Fig. 6B).
Fig. 6. Human CD8+ T cells support the recruitment of Tregs in a xenograft model in vivo.

(A) Purified CD8+ T cells were stimulated with anti-CD3/anti-CD28 mAb-coated beads for different terms, and supernatants were collected and analyzed using CCL22 ELISA. Data represent means ± SEM and are representative of two independent experiments done with two normal donors each. (B) Supernatants from activated human CD8+ effector cells or control culture medium were pretreated with anti-CCL22 mAb (aCCL22) or mouse IgG for 30 min and then placed in the bottom wells of a Transwell system. Flow cytometrically sorted CD4+CD25hi cells were added to the top wells, and 2 hours later, the cells that migrated to the bottom well were counted. Data represent means ± SEM and are representative of three independent experiments done in duplicate. PTX, pertussis toxin–treated Tregs. (C) The indicated cell subsets were injected into mice bearing human melanoma xenografts by tail vein injection. Forty-eight hours later, tumors were collected, made into single-cell suspensions, and stained for human CD8, CD4, and FoxP3. Representative FACS plots are shown for two independent experiments done with five mice in each group. P < 0.01, Treg cell injection followed by CD8+ T cell injections versus Tregs injected alone.
To determine whether the presence of human CD8+ effector T cells could support recruitment of Tregs into the tumor microenvironment in vivo, a human melanoma xenograft model was used. Non-obese diabetic/severe combined immunodeficient (NOD/SCID) mice were inoculated subcutaneously with the human melanoma cell line M537, which we previously observed expresses several chemokines capable of recruiting CD8+ effector T cells (including CCL3, CCL4, and CCL5) (13). Tumors were allowed to grow for about 4 weeks, at which time the tumor diameter was 0.5 to 1 cm. Normal donor Tregs were isolated by flow cytometric sorting, and CD8+ effector cells were generated in vitro as described above. These cell populations were injected intravenously alone or in combination, and tumors were harvested 24 hours later for analysis of infiltrating T cell populations by flow cytometry. When Tregs were transferred alone, a minimal number were detected in the tumor microenvironment. However, this number was significantly increased when CD8+ effector cells were also delivered (Fig. 6C). Thus, human CD8+ effector T cells can promote increased migration of Tregs into the melanoma tumor microenvironment in vivo.
Discussion
A major subset of human melanomas lacks evidence for innate immune activation, chemokine production, and T cell markers. Immune escape in that subset appears to be a consequence of immunologic ignorance and/or immune “exclusion” (13, 14, 30). However, for the subset of tumors that does show a strong inflammatory response including accumulation of antigen-specific CD8+ T cells, the mechanism of immune escape has been less clear, and the goal of our current study was to elucidate this process using human melanoma metastases as a foundation combined with mouse mechanistic experiments. Our results indicate that CD8+ T cell–infiltrated tumors contain high presence of CD4+CD25+FoxP3+ Tregs, as well as expression of IDO and PD-L1, arguing that immunosuppressive mechanisms dominate in these tumors and prevent optimal T cell function. In addition, and contrary to initial preconceptions, our data argue that the recruited involvement of these immunosuppressive pathways is intrinsic to the immune system and likely represents a set of physiologic negative feedback loops.
Our murine mechanistic experiments demonstrated that the induction of IDO and PD-L1 in the melanoma tumor microenvironment is mediated by CD8+ T cells and IFN-γ. In some tumor cell lines, PD-L1 expression appears to be up-regulated intrinsically via PTEN silencing and Akt activation (31). There may be a functional distinction between tumors having intrinsic PD-L1 expression and those in which PD-L1 is induced via adaptive immune cells. It has been speculated that clinical response to antibodies targeting the PD-L1/PD-1 interaction may end up being restricted to cases in which PD-L1 expression is associated with a CD8+ T cell infiltrate and therefore is likely blunting host immunity (32). It is also conceivable that in some cases, natural killer cells could contribute to up-regulation of these molecules through IFN-γ, although our mechanistic experiments demonstrated that CD8+ T cells were required.
The CD8+ Tcell–dependent accumulation of Tregs largely appeared to be driven by CCR4-binding chemokines, with a component of induced Treg proliferation contributing. In vitro, the only chemokine produced by primed human CD8+ effector T cells capable of inducing migration of Tregs was CCL22, which also was the only defined CCR4 ligand found to be produced by tumor-infiltrating lymphocytes (TILs) in vivo. These results are consistent with reports indicating a role for this chemokine in transplantation tolerance via Treg recruitment (33). Although direct CCL22 production by tumor cells in other cancer types also has been suggested (34), we did not detect CCL22 mRNA expression in a series of melanoma cell lines, arguing that in the melanoma context, it is likely derived from infiltrating nontumor stromal cells (13). Although our current results indicated that activated CD8+ T cells themselves are one source of CCL22, they do not exclude a contribution of CCL22 from other stromal cell components such as macrophages, although our mechanistic experiments indicated that CD8+ T cells were required for Treg accumulation in the tumor. In addition to CCL22, our results do not exclude a role for other chemokines contributing to Treg recruitment over the prolonged course of natural tumor development in vivo. A recent report suggested that CCL1 could contribute to Treg recruitment in a different tumor model in vivo (35).
If IDO, PD-L1, and Tregs represent immune-intrinsic negative feedback loops, then the question arises as to why tumors are not rejected if initial accumulation of CD8+ effector cells occurs unencumbered by these inhibitory pathways. One potential immune inhibitory mechanism that might be more directly attributable to the tumor itself is T cell anergy (36). Most solid tumor cells lack expression of the costimulatory molecules B7-1 and B7-2, which would favor anergy induction. Indeed, our gene expression profiling analyses revealed background levels of B7-1 and B7-2 transcripts in most of the tumors, arguing that collective expression of these molecules by tumors and their stromal cells also is likely low. Previous work has shown that antigen-presenting cells in the tumor microenvironment can be tolerogenic (37), and that some cytokines abundant in the tumor, such as IL-10, can inhibit dendritic cell activation and result in T cell anergy (38). Mechanistic studies have suggested that prevention or reversal of a T cell anergic state can promote tumor rejection in vivo (39–41). One could therefore envision that a costimulation-poor tumor microenvironment might lead to classical anergy, which would lead to defective IL-2 production and proliferation and diminished activity of other effector functions. It is noteworthy that in vitro models have demonstrated that anergic T cells produce reduced but still detectable levels of IFN-γ (36), which would be available to induce IDO and PD-L1 expression. Expressionof the latter would then cooperate to inhibit the activation of additional T cells trafficking into the tumor microenvironment. Whether anergic T cells continue to produce chemokines such as CCL22 has not been reported, but it is of interest that tumor-infiltrating CD8+ T cells (many of which are dysfunctional) did produce CCL22 in our current study.
Our study is limited in that we focused our attention on three major immune inhibitory mechanisms in this study (PD-L1, IDO, and Tregs) because they are associated with the presence of a CD8+ T cell infiltrate and therefore are candidates for inhibiting T cell function at the tumor site. Several other candidate inhibitory mechanisms have been described, including secretion of the cytokines IL-10 or TGF-β (transforming growth factor–β), or extrinsic suppression by myeloid-derived suppressor cells. Although we have not found these to be associated with a T cell infiltrate in melanoma metastases, or to be linked to clinical outcome to vaccination, these factors could nonetheless contribute to immune evasion.
Several strategies are being pursued clinically to counter the inhibitory effect of these negative regulatory mechanisms as a strategy for cancer immunotherapy. These include antibodies against PD-1 or PD-L1 (11, 42, 43), small-molecule inhibitors of IDO (44), and reagents targeting CD25 to deplete Tregs (12). Some of these approaches have already demonstrated clinical activity in subsets of melanoma patients. Our current results suggest that the efficacy of these strategies might be restricted to those patients having a T cell–inflamed tumor micro-environment and the accompanying up-regulation of the targets of these agents. Early data analyzing PD-L1 expression in tumors as a predictive indicator support this notion (32). Because multiple immune inhibitory pathways appear to be involved concurrently to facilitate immune escape, our data also imply that combination therapies may be advantageous to target two or more immunosuppressive simultaneously. Indeed, preclinical data support the notion that combinatorial manipulation of two immunoregulatory pathways can provide synergy for improved immune-mediated tumor control in vivo (6, 45, 46).
Materials and Methods
Study overview
Patient-derived samples were analyzed by IHC for immunoregulatory molecules. Data analysis was done in a blinded fashion, and counting was done three times. Mechanistic experiments were performed in mice and were carried out at least twice, in an independent way. Both experiments were expected to give similar results to generate conclusions. All experiments were designed to have at least three mice per group, with higher mouse numbers being used for experiments having a broader value distribution.
Patient samples
Melanoma metastases were pretreatment biopsies from patients participating in immunotherapy trials at the University of Chicago. All patients provided written informed consent that included tissue studies for biomarker analysis. For RNA analysis and gene expression profiling, samples were flash-frozen and stored at −80°C. When sufficient material was available, additional samples were formalin-fixed and embedded for IHC.
Reverse transcription–polymerase chain reaction
Total RNA was deoxyribonuclease (DNase) I–treated, and complementary DNA (cDNA) was synthesized from 1 μg of total RNA with M-MLV Reverse Transcriptase (Invitrogen) according to the manufacturer's directions. Reactions were run on an ABI Prism 7300 Sequence Detection System machine and analyzed. Prevalidated primers and probes specific for CD8 (Hs02621753_m1) and FoxP3 (Hs02622129_m1) were purchased from Applied Biosystems. The primer/probe sets for IDO and PD-L1 are as follows: IDO M34455 (forward: 5′-tggagaaagcccttcaagtgtt-3′, reverse: 5′-TCATACACCAGACCGTCTGAT-3′, probe: 6FAM-TCTGGCTGGAAAGGCAACCCCC-TAMRA) and PD-L1 NM_014143 (forward: 5′-tctggcacatcctccaaatg-3′, reverse: 5′-CAGTGCTACACCAAGGCATAATAAG-3′, probe: 6FAM-aaggactcacttggtaattctgggagcca-TAMRA). For mouse experiments, RT-PCR was performed with commercial Roche probes for FoxP3 (together with primers forward: 5′-aaacacccagccactcca-3′ and reverse: 5′-cttccaagtctcgtctgaagg-3′), IDO (5′-gggcttcttcctcgtctctc-3′ and 5′-tggatacagtggggattgct-3′), PD-L1 (5′-ccatcctgttgttcctcattg-3′ and 5′-tccacatctagcattctcacttg-3′), and CCL22 (5 -tcttgctgtggcaattcaga-3′ and 5′-gcagagggtgacggatgtag-3′) (Roche Applied Science).
Immunohistochemistry
Melanoma biopsies were immunostained with mouse antibodies for CD8 (NeoMarkers) and/or FoxP3 (Abcam). After tris-buffered saline (TBS) washing, secondary goat anti-mouse immunoglobulin G (IgG) conjugated to a horseradish peroxidase (Envision+ System, DAKO) or to an alkaline phosphatase (Biocare Medical) was applied. Reactions were developed with 3,3′-diaminobenzidine (DAB) chromogen or Vulcan Red, respectively, and counterstained with hematoxylin. Appropriate negative controls for the immunostaining were prepared by omitting the primary antibody step and substituting it with non-immune mouse serum.
The anti–PD-L1/B7-H1 murine IgG antibody was a gift from L. Chen. Tissue sections were deparaffinized and rehydrated through xylenes and serial dilutions of ethanol to deionized water. They were incubated in antigen retrieval buffer (pH 9, DAKO, S2367) and heated in a steamer at greater than 97°C for 20 min. Anti–B7-H1 antibody (1:1000) was applied on tissue sections for overnight incubation at 4°C temperature. After TBS wash (0.05 M tris base, 0.9% NaCl, pH 8.4), tissue sections were incubated with biotinylated anti-mouse IgG (1:100, BA-2001, Vector Laboratories) followed by Elite ABC kit for 30-min incubation at room temperature. The antigen-antibody binding was detected with the TSA Biotin System (NEL700A001KT, PerkinElmer) and DAB (DAKO, K3468).
The anti-IDO rabbit antiserum was a gift from R. Netwon (Incyte Pharmaceuticals). Tissue sections were treated as for the anti–PD-L1 antibody above. Anti-IDO antibody (1:1000) was applied on tissue sections for overnight incubation at 4°C. After TBS wash, tissue sections were incubated with biotinylated anti-rabbit IgG (1:200 dilution, BA-1000, Vector Laboratories) for 30-min incubation at room temperature. The antigen-antibody binding was detected by Alkaline Phosphatase Standard ABC kit (AK-5000, Vector Laboratories) and Vulcan Fast Red (FR850s, Biocare Medical) system. Tissue sections were briefly immersed in hematoxylin for counterstaining and were covered with cover glasses.
Antibodies and flow cytometry
If not indicated differently, all analyses using flow cytometry were performed with the FoxP3 staining kit (BD Biosciences) according to the manufacturer's instructions combined with the fixable live/death discrimination dye (eFlour 450, eBioscience). The following antibodies were used throughout the study: anti-CD3 AF700 (clone 17A2), anti-FoxP3 APC (allophycocyanin) (clone JFK-16a), anti-BrdU FITC (fluorescein isothiocyanate) (clone Bu20a), anti–PD-L1 PE (phycoerythrin) (clone MIH5) (eBioscience), anti-CD4 peridinin chlorophyll protein (PerCP)–Cy5.5 (clone RM4-5), anti-CD8 APC-Cy7 (clone 53-6.7) (BioLegend), and anti-CD25 PE (clone PC61) (BD Biosciences). Samples were acquired on the LSR2 blue (BD Biosciences) and analyzed with FlowJo software (TreeStar).
B16 melanoma in vivo tumor experiments
B16 and GFP+ B16.SIY melanoma cells were cultured as described (47). GFP+ B16.SIY cells (1 × 106) were inoculated subcutaneously in 6-week-old wild-type or IFN-γ−/− C57BL/6 mice (Jackson Laboratories). Some mice were intraperitoneally injected with 100 mg of isotype control or CD8-depleting antibody twice a week (starting at day −1; BioXCell). The CD8 depletion was confirmed by FACS. At day 7, tumors and spleens (as control) were harvested and split in two to be either saved in RNAlater (Ambion Inc.) for RT-PCR analysis or used to prepare single-cell suspensions for immediate FACS analysis for CD4, CD8, PD-L1, and FoxP3 as described (6, 48).
In vivo Treg conversion assay
B16 cells were injected subcutaneously in 6-week-old Rag2−/− (Jackson Laboratories) mice on day 0. The following day, GFP-expressing splenocytes from FoxP3-GFP reporter mice (49) were removed by sorting, and 3 × 106 GFP-depleted or total splenocytes were injected intravenously into tumor-bearing mice. On day 7 after tumor inoculation, tumor and tumor-draining lymph nodes were collected and processed into single-cell suspensions for subsequent analysis by flow cytometry. Cells were analyzed with live/dead discrimination and stained for CD3, CD4, CD8, CD25, and FoxP3. The experiment was confirmed with wild-type donor mice depleted for CD4+CD25+ cells using Miltenyi kits (according to the manufacturer's instructions).
In vivo Treg proliferation assay
B16 cells (2 × 106) were injected subcutaneously in 6-week-old wild-type mice treated with CD8-depleting antibody or isotype control as described above. On day 7 after tumor inoculation, one dose of BrdU (0.8 μg per mouse) (BD Biosciences) was given intraperitoneally, and 24 hours later, the tumor and tumor-draining lymph nodes were collected and processed into single-cell suspensions for subsequent analysis by flow cytometry. Cells were analyzed with live/dead discrimination and stained for CD3, CD4, CD8, CD25, FoxP3, and BrdU (according to the manufacturer's instructions for BrdU staining kit, eBioscience).
In vivo Treg recruitment assay
B16 cells (2 × 106) were injected subcutaneously into 6-week-old Rag2−/− (Jackson Laboratories) mice on day 0. The following day, 3 × 106 MACS-purified CD8+ T cells (Miltenyi kit according to the manufacturer's instructions) from wild-type mice were injected intravenously if indicated. On day 10 after tumor inoculation, 1 × 106 CD4+CD25+ MACS-enriched Tregs were given intravenously. If indicated, Tregs were pretreated in vitro with pertussis toxin (20 ng/ml for 1.5 hours) (Sigma-Aldrich) or C021 (60 nM for 2 hours) (Millipore), whereas untreated control Tregs were cultured for the same time in medium. Forty-eight hours later, the tumor and tumor-draining lymph nodes were collected and processed into single-cell suspensions for subsequent analysis by flow cytometry. Cells were analyzed with live/dead discrimination and stained for CD3, CD4, CD8, CD25, and FoxP3.
In vitro and ex vivo T cell stimulation
For naïve CD8+ T cells, spleens from wild-type mice were harvested, and single-cell suspension was prepared. Cells were negatively enriched for CD8 followed by positive enrichment for CD62L (both MACS Miltenyi kits, according to the manufacturer's instructions). Enriched cells were either used directly for RNA isolation and reverse transcriptase (Applied Biosystems) (ex vivo) or stimulated for the indicated time with anti-CD3–coated (5 μg/ml, clone 145-2C11, BioLegend) and anti-CD28–coated (3 μg/ml, clone EL4, BD Biosciences) plates. TILs were sorted with the FACSAria (BD Biosciences) by staining for CD3 and CD8. Isolated cells were stimulated on antibody-coated plates as described above for isolation of RNA and RT-PCR analysis.
Enzyme-linked immunosorbent assay
Human and mouse CCL22 was measured in triplicate from culture supernatants with commercially available kits from R&D Systems and analyzed with SoftMax analysis software (Molecular Devices).
Human Treg cell sorting and migration assay
CD4+ lymphocytes were positively selected from normal donor peripheral blood mononuclear cells with the MACS CD4+ kit. Selected CD4+ cells were labeled with anti-CD4 and anti-CD25 fluorescenceconjugated antibodies. Natural Tregs were then sorted by gating on bright CD25 expression. Sorted Tregs were loaded onto the top chamber of Transwells (5-mmpore size, Costar) for migration analysis. A total of 500- μl recombinant chemokine (100 ng/ml), culture supernatants, or control media were plated in the bottom wells. The anti-CCL22–blocking antibody was purchased from R&D Systems. Migration was allowed to proceed for 1.5 to 2 hours, and the cells present in the bottom well were counted. Cultures were analyzed in triplicate, and mean ± SE was determined.
NOD/SCID tumor establishment T cell injections
Six-week-old NOD/SCID mice (five mice per group) were inoculated subcutaneously with the human melanoma cell line M537 (3 × 106 cells). After tumors were allowed to grow for about 4 weeks, activated purified CD8+ effector cells (3 × 106 per mouse) were injected into one group of mice by tail vein injection. The next day, sorted human Tregs were injected into the indicated groups of mice intravenously. After an additional 48 hours, mice were sacrificed, and tumors were removed and minced into single-cell suspensions. Flow cytometric analysis was performed with antibodies specific for human CD8, CD4, and FoxP3 and then analyzed as above.
Statistical analysis
All statistical analyses were done with GraphPad Prism software (GraphPad). For all statistical analysis, we assumed non-Gaussian distribution due to sample size, and if not indicated otherwise, data are shown as means ± SEM.
Supplementary Material
S1-6
Fig. S1. Costaining of CD4 and FoxP3 in human melanoma sections.
Fig. S2. Expression level of IDO, PD-L1, and MHC I/II in B16 tumors in vitro and in vivo.
Fig. S3. Lack of Treg conversion from CD25-depleted T cells.
Fig. S4. Characterization of TILs compared to in vitro–primed CD8 T cells.
Fig. S5. CCR4 expression on human CD4 T cells and Tregs.
Fig. S6. Suppressive functional capacity of human CD25hi Tregs.
Acknowledgments
We thank J. Peyton and T. Kuna for technical assistance, H. Harlin for help with the melanoma gene array data, R. Duggan in Flow Cytometry Facility for cell sorting and phenotype analysis, R. Newton for the anti-human IDO antibody, L. Chen for the anti–PD-L1 mAb, T. Li for assistance with the IHC staining, and T. Krausz for pathology support.
Funding: This work was supported by R01 CA127475, R01 CA118153, and the Melanoma Research Alliance. S.S. is a postdoctoral fellow of the German Research Foundation.
Footnotes
Author contributions: S.S. and R.M.S. designed and performed all mouse-related experiments. Y.Z. performed experiments and analysis on patient samples. J.W. assisted with TIL sorting and chemokine analysis. Y.M. designed and performed experiments using the xenograft model system. T.T.H. assisted in pathological screening of human IHC. T.F.G. designed the overall study. T.F.G. and S.S. drafted and revised the manuscript.
Competing interests: The authors declare that they have no competing interests.
References and Notes
- 1.Atkins MB, Kunkel L, Sznol M, Rosenberg SA. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: Long-term survival update. Cancer J Sci Am. 2000;6(Suppl. 1):S11–S14. [PubMed] [Google Scholar]
- 2.Peterson AC, Harlin H, Gajewski TF. Immunization with Melan-A peptide-pulsed peripheral blood mononuclear cells plus recombinant human interleukin-12 induces clinical activity and T-cell responses in advanced melanoma. J Clin Oncol. 2003;21:2342–2348. doi: 10.1200/JCO.2003.12.144. [DOI] [PubMed] [Google Scholar]
- 3.Tanimoto T, Hori A, Kami M. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:1966. doi: 10.1056/NEJMc1009982. [DOI] [PubMed] [Google Scholar]
- 4.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbé C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gajewski TF, Meng Y, Harlin H. Immune suppression in the tumor microenvironment. J Immunother. 2006;29:233–240. doi: 10.1097/01.cji.0000199193.29048.56. [DOI] [PubMed] [Google Scholar]
- 6.Kline J, Brown IE, Zha YY, Blank C, Strickler J, Wouters H, Zhang L, Gajewski TF. Homeo-static proliferation plus regulatory T-cell depletion promotes potent rejection of B16 melanoma. Clin Cancer Res. 2008;14:3156–3167. doi: 10.1158/1078-0432.CCR-07-4696. [DOI] [PubMed] [Google Scholar]
- 7.Uyttenhove C, Pilotte L, Théate I, Stroobant V, Colau D, Parmentier N, Boon T, Van den Eynde BJ. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–1274. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 8.Dong H, Chen L. B7-H1 pathway and its role in the evasion of tumor immunity. J Mol Med. 2003;81:281–287. doi: 10.1007/s00109-003-0430-2. [DOI] [PubMed] [Google Scholar]
- 9.Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, Gajewski TF. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004;64:1140–1145. doi: 10.1158/0008-5472.can-03-3259. [DOI] [PubMed] [Google Scholar]
- 10.Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL, Gilson MM, Wang C, Selby M, Taube JM, Anders R, Chen L, Korman AJ, Pardoll DM, Lowy I, Topalian SL. Phase I study of single-agent anti–programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28:3167–3175. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rasku MA, Clem AL, Telang S, Taft B, Gettings K, Gragg H, Cramer D, Lear SC, McMasters KM, Miller DM, Chesney J. Transient T cell depletion causes regression of melanoma metastases. J Transl Med. 2008;6:12. doi: 10.1186/1479-5876-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, Gajewski TF. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69:3077–3085. doi: 10.1158/0008-5472.CAN-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gajewski TF, Louahed J, Brichard VG. Gene signature in melanoma associated with clinical activity: A potential clue to unlock cancer immunotherapy. Cancer J. 2010;16:399–403. doi: 10.1097/PPO.0b013e3181eacbd8. [DOI] [PubMed] [Google Scholar]
- 15.Messina JL, Fenstermacher DA, Eschrich S, Qu X, Berglund AE, Lloyd MC, Schell MJ, Sondak VK, Weber JS, Mulé JJ. 12-Chemokine gene signature identifies lymph node-like structures in melanoma: Potential for patient selection for immunotherapy? Sci Rep. 2012;2:765. doi: 10.1038/srep00765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Erdag G, Schaefer JT, Smolkin ME, Deacon DH, Shea SM, Dengel LT, Patterson JW, Slingluff CL., Jr Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res. 2012;72:1070–1080. doi: 10.1158/0008-5472.CAN-11-3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, Johnson D, Swetter S, Thompson J, Greenberg PD, Roederer M, Davis MM. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med. 1999;5:677–685. doi: 10.1038/9525. [DOI] [PubMed] [Google Scholar]
- 18.Valmori D, Dutoit V, Rubio-Godoy V, Chambaz C, Liénard D, Guillaume P, Romero P, Cerottini JC, Rimoldi D. Frequent cytolytic T-cell responses to peptide MAGE-A10254–262 in melanoma. Cancer Res. 2001;61:509–512. [PubMed] [Google Scholar]
- 19.Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8a+ dendritic cells. J Exp Med. 2011;208:2005–2016. doi: 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, Murphy KM, Schreiber RD. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208:1989–2003. doi: 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ji RR, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, Alaparthy S, Berman D, Jure-Kunkel M, Siemers NO, Jackson JR, Shahabi V. An immune-active tumor micro-environment favors clinical response to ipilimumab. Cancer Immunol Immunother. 2012;61:1019–1031. doi: 10.1007/s00262-011-1172-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harlin H, Kuna TV, Peterson AC, Meng Y, Gajewski TF. Tumor progression despite massive influx of activated CD8+ T cells in a patient with malignant melanoma ascites. Cancer Immunol Immunother. 2006;55:1185–1197. doi: 10.1007/s00262-005-0118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mortarini R, Piris A, Maurichi A, Molla A, Bersani I, Bono A, Bartoli C, Santinami M, Lombardo C, Ravagnani F, Cascinelli N, Parmiani G, Anichini A. Lack of terminally differentiated tumor-specific CD8+ T cells at tumor site in spite of antitumor immunity to self-antigens in human metastatic melanoma. Cancer Res. 2003;63:2535–2545. [PubMed] [Google Scholar]
- 24.Appay V, Jandus C, Voelter V, Reynard S, Coupland SE, Rimoldi D, Lienard D, Guillaume P, Krieg AM, Cerottini JC, Romero P, Leyvraz S, Rufer N, Speiser DE. New generation vaccine induces effective melanoma-specific CD8+ T cells in the circulation but not in the tumor site. J Immunol. 2006;177:1670–1678. doi: 10.4049/jimmunol.177.3.1670. [DOI] [PubMed] [Google Scholar]
- 25.Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, Brown C, Mellor AL. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281:1191–1193. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 26.Nomura T, Sakaguchi S. Naturally arising CD25+CD4+ regulatory T cells in tumor immunity. Curr Top Microbiol Immunol. 2005;293:287–302. doi: 10.1007/3-540-27702-1_13. [DOI] [PubMed] [Google Scholar]
- 27.Munn DH, Mellor AL. IDO and tolerance to tumors. Trends Mol Med. 2004;10:15–18. doi: 10.1016/j.molmed.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 28.Iellem A, Mariani M, Lang R, Recalde H, Panina-Bordignon P, Sinigaglia F, D’Ambrosio D. Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4+CD25+ regulatory T cells. J Exp Med. 2001;194:847–853. doi: 10.1084/jem.194.6.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meng Y, Harlin H, O’Keefe JP, Gajewski TF. Induction of cytotoxic granules in human memory CD8+ T cell subsets requires cell cycle progression. J Immunol. 2006;177:1981–1987. doi: 10.4049/jimmunol.177.3.1981. [DOI] [PubMed] [Google Scholar]
- 30.Gajewski T, Zha Y, Thurner B, Schuler G. Association of gene expression profile in melanoma and survival to a dendritic cell-based vaccine. J Clin Oncol. 2009;27:9002. [Google Scholar]
- 31.Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, Mischel PS, Stokoe D, Pieper RO. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13:84–88. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 32.Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL, Chen L. Colocalization of inflammatory response with B7-H1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4:127ra37. doi: 10.1126/scitranslmed.3003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee I, Wang L, Wells AD, Dorf ME, Ozkaynak E, Hancock WW. Recruitment of Foxp3+ T regulatory cells mediating allograft tolerance depends on the CCR4 chemokine receptor. J Exp Med. 2005;201:1037–1044. doi: 10.1084/jem.20041709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 35.Hoelzinger DB, Smith SE, Mirza N, Dominguez AL, Manrique SZ, Lustgarten J. Blockade of CCL1 inhibits T regulatory cell suppressive function enhancing tumor immunity without affecting T effector responses. J Immunol. 2010;184:6833–6842. doi: 10.4049/jimmunol.0904084. [DOI] [PubMed] [Google Scholar]
- 36.Schwartz RH. T cell clonal anergy. Curr Opin Immunol. 1997;9:351–357. doi: 10.1016/s0952-7915(97)80081-7. [DOI] [PubMed] [Google Scholar]
- 37.Watkins SK, Zhu Z, Riboldi E, Shafer-Weaver KA, Stagliano KE, Sklavos MM, Ambs S, Yagita H, Hurwitz AA. FOXO3 programs tumor-associated DCs to become tolerogenic in human and murine prostate cancer. J Clin Invest. 2011;121:1361–1372. doi: 10.1172/JCI44325. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Steinbrink K, Jonuleit H, Müller G, Schuler G, Knop J, Enk AH. Interleukin-10–treated human dendritic cells induce a melanoma-antigen–specific anergy in CD8+ T cells resulting in a failure to lyse tumor cells. Blood. 1999;93:1634–1642. [PubMed] [Google Scholar]
- 39.Brown IE, Blank C, Kline J, Kacha AK, Gajewski TF. Homeostatic proliferation as an isolated variable reverses CD8+ T cell anergy and promotes tumor rejection. J Immunol. 2006;177:4521–4529. doi: 10.4049/jimmunol.177.7.4521. [DOI] [PubMed] [Google Scholar]
- 40.Chen L, Ashe S, Brady WA, Hellström I, Hellström KE, Ledbetter JA, McGowan P, Linsley PS. Costimulation of antitumor immunity by the B7 counterreceptor for the T lymphocyte molecules CD28 and CTLA-4. Cell. 1992;71:1093–1102. doi: 10.1016/s0092-8674(05)80059-5. [DOI] [PubMed] [Google Scholar]
- 41.Townsend SE, Allison JP. Tumor rejection after direct costimulation of CD8+ T cells by B7-transfected melanoma cells. Science. 1993;259:368–370. doi: 10.1126/science.7678351. [DOI] [PubMed] [Google Scholar]
- 42.Sznol M, Powderly JD, Smith DC, Brahmer JR, Drake CG, McDermott DF, Lawrence DP, Wolchok JD, Topalian SL, Lowy I. Safety and antitumor activity of biweekly MDX-1106 (anti-PD-1, BMS-936558/ONO-4538) in patients with advanced refractory malignancies. J Clin Oncol. 2010;28:15s. [Google Scholar]
- 43.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K, Leffet L, Hansbury MJ, Thomas B, Rupar M, Waeltz P, Bowman KJ, Polam P, Sparks RB, Yue EW, Li Y, Wynn R, Fridman JS, Burn TC, Combs AP, Newton RC, Scherle PA. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood. 2010;115:3520–3530. doi: 10.1182/blood-2009-09-246124. [DOI] [PubMed] [Google Scholar]
- 45.Chen SH, Pham-Nguyen KB, Martinet O, Huang Y, Yang W, Thung SN, Chen L, Mittler R, Woo SL. Rejection of disseminated metastases of colon carcinoma by synergism of IL-12 gene therapy and 4-1BB costimulation. Mol Ther. 2000;2:39–46. doi: 10.1006/mthe.2000.0086. [DOI] [PubMed] [Google Scholar]
- 46.Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci USA. 2010;107:4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blank C, Brown I, Kacha AK, Markiewicz MA, Gajewski TF. ICAM-1 contributes to but is not essential for tumor antigen cross-priming and CD8+ T cell-mediated tumor rejection in vivo. J Immunol. 2005;174:3416–3420. doi: 10.4049/jimmunol.174.6.3416. [DOI] [PubMed] [Google Scholar]
- 48.Zhang L, Gajewski TF, Kline J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood. 2009;114:1545–1552. doi: 10.1182/blood-2009-03-206672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6:1142–1151. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1-6
Fig. S1. Costaining of CD4 and FoxP3 in human melanoma sections.
Fig. S2. Expression level of IDO, PD-L1, and MHC I/II in B16 tumors in vitro and in vivo.
Fig. S3. Lack of Treg conversion from CD25-depleted T cells.
Fig. S4. Characterization of TILs compared to in vitro–primed CD8 T cells.
Fig. S5. CCR4 expression on human CD4 T cells and Tregs.
Fig. S6. Suppressive functional capacity of human CD25hi Tregs.