Structure of the bacteriophage ϕ29 DNA packaging motor (original) (raw)
. Author manuscript; available in PMC: 2014 Sep 2.
Published in final edited form as: Nature. 2000 Dec 7;408(6813):745–750. doi: 10.1038/35047129
Abstract
Motors generating mechanical force, powered by the hydrolysis of ATP, translocate double-stranded DNA into preformed capsids (proheads) of bacterial viruses1,2 and certain animal viruses3. Here we describe the motor that packages the double-stranded DNA of the Bacillus subtilis bacteriophage ϕ29 into a precursor capsid. We determined the structure of the head–tail connector—the central component of the ϕ29 DNA packaging motor—to 3.2Å resolution by means of X-ray crystallography. We then fitted the connector into the electron densities of the prohead and of the partially packaged prohead as determined using cryo-electron microscopy and image reconstruction analysis. Our results suggest that the prohead plus dodecameric connector, prohead RNA, viral ATPase and DNA comprise a rotary motor with the head–prohead RNA–ATPase complex acting as a stator, the DNA acting as a spindle, and the connector as a ball-race. The helical nature of the DNA converts the rotary action of the connector into translation of the DNA.
The bacteriophage ϕ29 (Fig. 1) is a 19-kilobase (19-kb) double-stranded DNA (dsDNA) virus with a prolate head and complex structure4. The prohead (Fig. 1), into which the DNA is packaged, is about 540Å long and 450Å wide5. The ϕ29 connector, a cone-shaped dodecamer of gene product 10 (gp10), occupies the pentagonal vertex at the base of the prohead5 and is the portal for DNA entry during packaging and DNA ejection during infection6. The connector, in association with the oligomeric, ϕ29-encoded prohead RNA (pRNA) and a viral ATPase (gp16), is required for DNA packaging7–9. However, only the first 120 bases of the 174-base pRNA are essential for packaging7. The covalent adduct of the genomic dsDNA with gp3 (DNA–gp3) can be packaged into proheads in about three minutes in vitro (P.J.J., unpublished results). The connector proteins of tailed phages6 vary in relative molecular mass (_M_r) from 36,000 (36K) in ϕ29 to 83K in phage P22, and assemble into oligomers with a central channel. The structure of the isolated ϕ29 connector has been studied by atomic force microscopy10 and cryo-electron microscopy (cryo-EM) of two-dimensional arrays11, immuno-electron microscopy12 and X-ray crystallography13,14.
Figure 1.
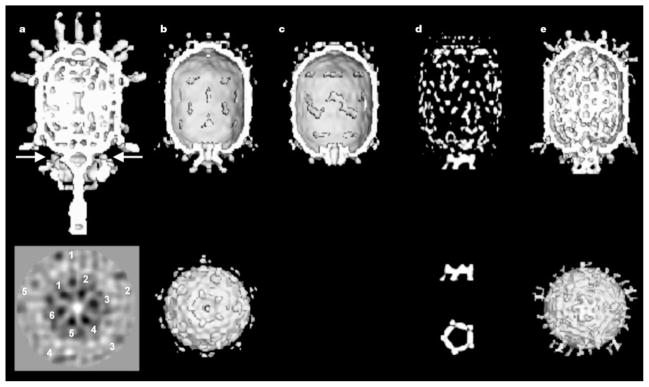
Cryo-EM reconstructions. Top and bottom rows: a, mature ϕ29; b, prohead + 120-base pRNA; c, prohead treated with RNase; d, difference map between b and c; and e, partially packaged particle (with DNA in channel). End-on views, looking along the tail towards the head, are given for b and e. Orthogonal difference pRNA densities are shown below the difference map in d. The arrow in a shows the position of the section below that was obtained by averaging particles without imposing five-fold symmetry. Note that both the five-fold symmetry of the downward-pointing head fibres (numbered 1 to 5) and six-fold symmetry (numbered 1 to 5) of the lower collar are visible.
The connector structure, as now determined by X-ray crystallography, can be divided into three, approximately cylindrical regions: the narrow end, the central part, and the wide end, having external radii (Å ) of 33, 47 and 69, respectively (Fig. 2). These regions are respectively 25, 28 and 22Å in height, making the total connector 75Å long. The internal channel has a diameter of about 36Å at the narrow end, increasing to 60Å at the wide end. Comparison with electron microscopy reconstructions5,11 shows that the narrow end protrudes from the portal vertex of the phage head, is associated with the multimeric pRNA, and binds the lower collar in the mature virus.
Figure 2.

Connector structure ribbon diagrams. a, The dodecameric connector seen from the tail looking towards the head; b, a side view with the pRNA-binding site at the bottom, showing the conical structure of the connector and the helical twist of each subunit around the 12-fold axis (white); c, a stereo diagram of a pair of monomers. One monomer is coloured red in the central domain, green in the wide-end domain that resides inside the capsid, and yellow at the narrow-end domain. The other monomers are all coloured blue. The ordered part of the polypeptide starts with helix α1 on the outside of the connector, going towards the wide end (residues 61 to 128 and 247 to 286). Helix α3 (residues 129 to 157) returns the chain to the narrow end (residues 158 to 202). The tip of the connector at the narrow end is formed by residues 164 to 170 and 185 to 196. Helix α5 (residues 208 to 226) returns the polypeptide to the wide end through the second disordered section. (Drawn with the program MOLSCRIPT25 and RASTER3D25.)
The electron density of the connector was interpreted in terms of the amino-acid sequence15 and was confirmed by the two Hg sites (see Methods section) corresponding to the only cysteine residues in the sequence. Residues 1 to 11, 229 to 246, and 287 to 309 at the carboxy terminus are not seen in the electron density. The second and third disordered regions are both located on the inside of the channel, close to the junction of the central and wide regions. The structure is dominated by three long helices (α1, α3 and α5) in each monomer that run the length of the central region, joining the two end domains of the connector (Fig. 2). These helices are arranged at an angle of about 40° with respect to the central 12-fold axis. The two end domains are composed predominantly of β-sheets and extended polypeptide chains. Immuno-electron microscopy mapping of the sequence onto the connector surface12 is consistent with the X-ray structure only as far as localization of the external amino-terminal residues with the pRNA-binding region at the narrow end of the connector. The RNA recognition motif structure, previously predicted for the N-terminal regions of the connector monomer16,17, is not present in the structure.
The surface of the monomer presents a net negative charge to one neighbour and a net positive charge to the other neighbour, possibly aiding the assembly of the dodecamer. The exterior of the connector has no significant regions of charge accumulation, implying that its rotation might be facilitated by its oily, smooth, external surface. However, the basic character of the disordered 11 amino-terminal residues could alter the surface properties to some extent and may facilitate interaction with the pRNA. In contrast, the inside of the channel has a preponderance of negative charge at its wide end, which may repel the DNA, permitting its smooth passage during packaging and ejection. The channel through which messenger RNA is translocated in reoviruses has similar properties18.
We have determined the structures of four distinct types of ϕ29 particles by cryo-EM and image reconstruction analyses (Fig. 1, Table 1): the ϕ29 prohead including pRNA, the prohead after removal of the pRNA, the partially DNA-packaged prohead including the pRNA and ATPase (gp16), and (data not shown) empty proheads found in the in vitro packaging system. A difference map between proheads with and without the pRNA locates the pRNA at the narrow end of the connector, in agreement with ref. 19. The difference map (Fig. 1d) was based on reconstructions in which the five-fold symmetry had been imposed along the long axis of the particles. If the pRNA were six-fold symmetric as suggested by genetic data8,9, the resultant averaged density would be weak and smeared. However, we observe that the pRNA has excellent density, higher by a factor of ~1.5 than the density of the head, thus demonstrating that the pRNA is consistent with the imposed five-fold symmetry. Furthermore, a reconstruction computed without imposing five-fold symmetry (Table 1, data set b′; M.C.M., Y.T., P.J.J., D.L.A., T.S.B. and M.G.R., manuscript in preparation; also see the Methods section) also showed good pRNA density with almost exact five-fold symmetry.
Table 1.
Cryo-EM data and image reconstruction
| Data set* | Prohead+pRNA | RNase-treated prohead | Prohead+pRNA+DNA−gp3+gp16 | |
|---|---|---|---|---|
| (b) | (b′) | (c) | (e) | |
| Symmetry imposed | 5 | 1 | 5 | 5 |
| Underfocus (μm)† | 2.3 | 2.3 | 2.3 | 2.3 |
| Number of particles‡ | 609 (1,153) | 680 (1,153) | 285 (4,81) | 534 (1,038) |
| Correlation coefficient§ | 0.324(0.047) | 0.477 (0.061) | 0.263 (0.056) | 0.236 (0.050) |
| Resolution (Å) | 26 | 35 | 35 | 32 |
The difference density representing the pRNA consists of a continuous ring with internal and external radii of 34 and 62Å, respectively. In addition, there are five spokes radiating slightly outwards, which are in the same orientations relative to the head in all reconstructions (Fig. 1). The alignment of the pRNA spokes with the prohead implies that either the pRNA communicates with the prohead through the connector or that the pRNA and the head directly interact. Indeed, the cryo-EM reconstructions of the prohead show density connecting the pRNA and head at a contour level well above that of noise in the background (Fig. 3a).
Figure 3.

Cryo-EM density fitted with atomic structures. a, Cross-section of the cryo-EM prohead density (red) fitted with the Cα backbone of the connector (yellow) and the cryo-EM pRNA difference map (green). Shown also is a DNA molecule placed through the central channel of the connector. The prohead capsid, one of the five contacts between the pRNA with the capsid, and the partially disordered residues 229 to 246 and 287 to 309 in the connector are indicated. b, Stereo drawing of the 120-base pRNA pentamer fitted into the cryo-EM density. Secondary structural elements27 are shown in white (A helix), blue (C helix), orange (E helix), yellow (CE loop), green (D helix) and red (D loop). Four intermolecular base pairs are shown in yellow-red. It had previously been shown that helices D, C and E participate in binding pRNA to the connector, and it was suggested that helix A binds the viral ATPase (gp16)28. (Drawn with the programs XTALVIEW25 and RASTER3D25.)
Although genetic studies8,9 had shown that the pRNA forms hexamers by intermolecular base pairing, only a pentameric model could be readily fitted into the cryo-EM density, with the five A helices fitting into the radial spokes (Fig. 3b). A hexameric ring would have had too large a radius. It is possible that a hexameric pRNA might be required to initiate pRNA binding, but that at some stage one pRNA molecule8,9 is shifted out of the ring or eliminated. As the handedness of the pRNA density is not known, the direction of the pRNA helices within the ring is uncertain.
The X-ray structure of the connector fits accurately into the portal vertex of the prohead reconstruction (Fig. 3a). The wide end of the connector contacts the inside of the head, whereas the narrow end protrudes from the portal vertex where it is encircled by the pentameric pRNA, which makes five, symmetrically disposed, interactions with the head (Fig. 3a). It appears that the orientation of the pRNA is determined by its interaction with the head rather than with the connector. Although the contact region of pRNA and connector is substantial, the oily surface of the connector and its 5–12 symmetry mismatch would aid its smooth rotation, as proposed in ref. 1. Residues 229 to 246 and 287 to 309, disordered in the crystal structure, occur in the DNA channel, roughly at the border between the central and wide regions of the connector, which corresponds to a low-density extension of the DNA channel (Fig. 3a). The partially disordered residues include the unusual amino-acid sequences Glu-Lys-Lys-Glu-Arg and Arg-Arg-Glu, which might be ideal for interacting with the charged sugar-phosphate DNA backbone.
Actively packaging prohead samples were flash-frozen 2.5 min after initiation of packaging. The resulting cryo-EM images were divided, by visual inspection, into two groups, one representing particles that appeared to be empty (data not shown) and the other those that were partially filled (Fig. 1e; Table 1, data set e). The reconstruction of the empty particles was similar to that of the prohead including the pRNA (Fig. 1b; Table 1, data set b), whereas the partially filled prohead reconstruction showed both the five-fold pRNA structure and additional density associated with each of the five rotary spokes (Fig. 1e), implying that the components corresponding to this density are involved in DNA packaging. This density, therefore, might be attributed to the viral ATPase (gp16). The partially filled particles contain less DNA in the head as compared to mature virions, indicating that the packaging process was incomplete at the time of freezing. This cryo-EM image reconstruction also shows some density inside the narrow end of the connector channel (Fig. 1e), which can be interpreted as DNA entering the prohead.
The accumulated data provide the basis for proposing different mechanisms for the DNA packaging machine. Here we discuss one proposal that is consistent with the structural observations (Fig. 4). The DNA, connector and prohead – pRNA – ATPase complex form a set of concentric structures with 101-, 12- and 5-fold symmetry, respectively. These constitute a movable central spindle, intervening ball-race, and a static outer assembly (stator), which powers the motor. The sequential firing of the ATPases (I to V) powers a series of conformational changes that translate the DNA into the head (Fig. 4). As successive ATPases occur one-fifth of a rotation around the circumference of the DNA, the DNA must either rotate by one-fifth of a turn or translate by one-fifth of its pitch to maintain the same relationship to the currently active ATPase (compare Fig. 4a and c). The large size of the DNA would make translation energetically favourable. By the time each of the five ATPases have fired, the connector will have rotated 60°, and the DNA will have been translated the length of one pitch of its helix, thus packaging 10 base pairs. This corresponds to a movement of two base pairs per ATP hydrolysis, consistent with the observed ATP consumption during packaging20.
Figure 4.

The DNA packaging mechanism. Shown is one cycle in the mechanism that rotates the connector and translates the DNA into the head. The view down the connector axis (top) is towards the head, whereas the bottom row shows side views corresponding to that seen in Fig. 2b. Eleven of the 12 subunits (A, B,…, L) of the connector are shown in green; the ‘active’ monomer is shown in red. The connector is represented as a set of small spheres at the narrow end and a set of larger spheres at the wide end connected by a line representing the central helical region. The pRNA–ATPase complexes (I, II, III, IV, V), surrounding the narrow end, are shown by a set of four blue spheres and one red sphere. The DNA base aligned with the active connector monomer is also shown in red. In a, the active pRNA–ATPase I interacts with the adjacent connector monomer (A), which in turn contacts the aligned DNA base. In b, the narrow end of the connector has moved anticlockwise by 12° to place the narrow end of monomer C opposite ATPase II, the next ATPase to be fired, causing the connector to expand lengthwise by slightly changing the angle of the helices in the central domain (white arrow with asterisk). In c, the wide end of the connector has followed the narrow end, while the connector relaxes and contracts (white arrow with two asterisks), thus causing the DNA to be translated into the phage head. For the next cycle, ATPase II is activated, causing the connector to be rotated another 12°, and so forth. (Drawn with the program RASTER3D25.)
The narrow- and wide-end domains of each connector monomer differ in their orientation by up to 2° relative to the central helices, which are twisted around the central axis and are roughly orthogonal to the DNA backbone. The flexibility of the end domains would allow the connector to rotate by a two-step mechanism (Fig. 4). Thus, the connector may have a more complex role than a ball-race because it may also be required to undergo a helical spring-like oscillation that couples the rotational motor action to the linear translocation of the DNA spindle.
The DNA packaging mechanism utilizes the symmetry mismatch and ATP hydrolysis to effect connector rotation as proposed in refs 1 and 21. However, the connector does not rotate along the DNA helix, as in a rigid nut–bolt assembly1; rather, each connector monomer occupies a different position on the DNA helix after every ATP hydrolysis, as proposed in ref. 21. The energy for DNA packaging is supplied by the external stator composed of the head, pRNA and ATPase.
There are several well characterized examples of molecular motors. These fall into two classes, the first of which consists of rotary motors, such as the F1 ATPase22 and the bacterial flagellar motor23, which turn a spindle by a rotary conformational change mechanism. The second class consists of linear motors, like those required for muscle contraction24, where conformational fluctuations undergo a translation with each step. The ϕ29 DNA packaging motor as we have described it represents a third class, where rotary motion is converted into translational movement.
Methods
X-ray crystallography
Bacteriophage ϕ29 connectors were produced in Escherichia coli and were crystallized14 into several different space groups. The crystals used for the present studies belonged to space group _C_2, with a = 177.2, b = 169.2, c = 185.4Å, β = 114.10°, consistent with one connector per crystallographic asymmetric unit. X-ray diffraction data were collected at the Cornell High Energy Synchrotron Source (CHESS), the Advanced Photon Source (APS) BioCARS beamline 14BMc, and the APS Structural Biology Center beamline 19ID. The data used had an _R_merge of 6.5% to 3.2Å resolution (24.0% in the highest resolution shell). The Hg salt, thimerosal, was used as a heavy atom derivative. The diffraction data of this derivative had an _R_merge of 10.8% (26.0%) to 3.4Å resolution and a difference in structure amplitudes of 24% with respect to the native data.
A self-rotation function showed that the connector had 12-fold symmetry with the axis perpendicular to the unique monoclinic b axis, and a Patterson self-translation function25 showed that the axis passed through one of the crystallographic dyads. A three-dimensional, low-resolution, electron density distribution was constructed of the connector, based on orthogonal views of the connector derived from cryo-EM images of two-dimensional, tilted arrays11. The position along and rotation around the 12-fold axis of the model was determined by structure factor correlation searches using the program AMoRe25 and packing considerations. The initial model phases were extended from 20Å to 3.5Å resolution (A.A.S. et al., manuscript in preparation) by 12-fold, non-crystallographic symmetry (NCS), electron density averaging25 and solvent flattening using the program DM25. The resultant map was not good enough for interpretation, but did permit the solution of the Hg sites in a 4.0Å resolution difference map. Two independent sites per monomer were found, each with 12 sites related by the NCS. The sites were refined with the program MLPHARE and used to compute phases to 3.2Å resolution, which were then improved by iterative density NCS averaging and solvent flattening. The final map could be readily interpreted in terms of the amino-acid sequence15 using the program O25.
Cryo-electron microscopy
The preparation of specimens for cryo-EM was performed as described previously5. The model-based polar-Fourier-transform method26 was used to determine the orientation of each particle. To confirm the five-fold symmetry of the pRNA, the five-fold symmetry imposed on the initial reconstruction was relaxed. Although the orientation of the particles in each image had been determined with respect to the five-fold symmetric head, a subsequent search was performed over the five different possible orientations, and the images were averaged without imposing five-fold symmetry (M.C.M., Y.T., P.J.J., D.L.A., T.S.B. and M.G.R., manuscript in preparation). This procedure was applied to images of proheads and native virus. A section through the reconstruction, perpendicular to the phage axis (Fig. 1), shows both the five-fold symmetry of the head fibres and the six-fold symmetry of the lower collar in the same section, demonstrating that the procedure did not unintentionally impose five-fold symmetry.
In their cryo-EM image reconstruction of ϕ29 proheads, Iberra et al.19 attributed the five, roughly radial, pRNA spokes protruding from the connector to accidental inclusion of proheads in an inverted orientation, causing superposition of the head fibres onto the pRNA. However, in our samples all particles were lying almost flat on the grid, which made it easy to distinguish the two ends of the head. Furthermore, the presumed pRNA features are of much higher density than the head fibres, showing that the head fibres could not give rise to the RNA spokes. Misoriented particles also should have produced an image of the connector at both ends of the head and would have resulted in ‘fibres’ on the connector in the reconstruction of the RNase-treated prohead that is missing the pRNA. None of these features was observed.
Acknowledgments
We thank J. Carrascosa for comments and discussions of this paper. We also thank the staff of CHESS, APS BioCARS and APS Structural Biology Center for assistance in data collection. This work was supported by the NSF (M.G.R.), the NIH (D.L.A. and T.S.B.), and an NSF Shared Instrumentation grant (T.S.B. and M.G.R).
Footnotes
Coordinates of the connector (accession number 1FOU) and pRNA (accession number 1FOQ) have been deposited with the Protein Data Bank.
References
- 1.Hendrix RW. Symmetry mismatch and DNA packaging in large bacteriophages. Proc Natl Acad Sci USA. 1978;75:4779–4783. doi: 10.1073/pnas.75.10.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hendrix RW. Bacteriophage DNA packaging: RNA gears in a DNA transport machine. Cell. 1998;94:147–150. doi: 10.1016/s0092-8674(00)81413-0. [DOI] [PubMed] [Google Scholar]
- 3.Zhou ZH, Chen DH, Jakana J, Rixon FJ, Chiu W. Visualization of tegument-capsid interactions and DNA in intact herpes simplex virus type 1 virions. J Virol. 1999;73:3210–3218. doi: 10.1128/jvi.73.4.3210-3218.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson DL, Hickman DD, Reilly BE. Structure of Bacillus subtilis bacteriophage ϕ29 and the length of ϕ29 deoxyribonucleic acid. J Bacteriol. 1966;91:2081–2089. doi: 10.1128/jb.91.5.2081-2089.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tao Y, et al. Assembly of a tailed bacterial virus and its genome release studied in three dimensions. Cell. 1998;95:431–437. doi: 10.1016/s0092-8674(00)81773-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valpuesta JM, Carrascosa JL. Structure of viral connectors and their function in bacteriophage assembly and DNA packaging. Q Rev Biophys. 1994;27:107–155. doi: 10.1017/s0033583500004510. [DOI] [PubMed] [Google Scholar]
- 7.Guo P, Erickson S, Anderson D. A small viral RNA is required for in vitro packaging of bacteriophage ϕ29 DNA. Science. 1987;236:690–694. doi: 10.1126/science.3107124. [DOI] [PubMed] [Google Scholar]
- 8.Zhang F, et al. Function of hexameric RNA in packaging of bacteriophage ϕ29 DNA in vitro. Mol Cell. 1998;2:141–147. doi: 10.1016/s1097-2765(00)80123-9. [DOI] [PubMed] [Google Scholar]
- 9.Guo P, Zhang C, Chen C, Garver K, Trottier M. Inter-RNA interaction of phage ϕ29 pRNA to form a hexameric complex for viral DNA transportation. Mol Cell. 1998;2:149–155. doi: 10.1016/s1097-2765(00)80124-0. [DOI] [PubMed] [Google Scholar]
- 10.Müller DJ, Engel A, Carrascosa JL, Vélez M. The bacteriophage ϕ29 head-tail connector imaged at high resolution with the atomic force microscope in buffer solution. EMBO J. 1997;16:2547–2553. doi: 10.1093/emboj/16.10.2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Valpuesta JM, Fernández JJ, Carazo JM, Carrascosa JL. The three-dimensional structure of a DNA translocating machine at 10 Å resolution. Structure. 1999;7:289–296. doi: 10.1016/s0969-2126(99)80039-2. [DOI] [PubMed] [Google Scholar]
- 12.Valle M, et al. Domain architecture of the bacteriophage ϕ29 connector protein. J Mol Biol. 1999;288:899–909. doi: 10.1006/jmbi.1999.2731. [DOI] [PubMed] [Google Scholar]
- 13.Guasch A, et al. Crystallographic analysis reveals the 12-fold symmetry of the bacteriophage ϕ29 connector particle. J Mol Biol. 1998;281:219–225. doi: 10.1006/jmbi.1998.1928. [DOI] [PubMed] [Google Scholar]
- 14.Badasso MO, et al. Purification, crystallization, and initial X-ray analysis of the head-tail connector of bacteriophage ϕ29. Acta Crystallogr D. 2000;56:1187–1190. doi: 10.1107/s0907444900009239. [DOI] [PubMed] [Google Scholar]
- 15.Vlcek C, Paces V. Nucleotide sequence of the late region of Bacillus ϕ29 completes the 19285-bp sequence of ϕ29 genome. Comparison with the homologous sequence of phage PZA. Gene. 1986;46:215–225. doi: 10.1016/0378-1119(86)90406-3. [DOI] [PubMed] [Google Scholar]
- 16.Grimes S, Anderson D. RNA dependence of the bacteriophage ϕ29DNA packaging ATPase. J Mol Biol. 1990;215:559–566. doi: 10.1016/s0022-2836(05)80168-8. [DOI] [PubMed] [Google Scholar]
- 17.Donate LE, Valpuesta JM, Mier C, Rojo F, Carrascosa JL. Characterization of an RNA-binding domain in the bacteriophage ϕ29 connector. J Biol Chem. 1993;268:20198–20204. [PubMed] [Google Scholar]
- 18.Reinisch KM, Nibert ML, Harrison SC. Structure of the reovirus core at 3.6 Å resolution. Nature. 2000;404:960–967. doi: 10.1038/35010041. [DOI] [PubMed] [Google Scholar]
- 19.Ibarra B, et al. Topology of the components of the DNA packaging machinery in the phage ϕ29 prohead. J Mol Biol. 2000;298:807–815. doi: 10.1006/jmbi.2000.3712. [DOI] [PubMed] [Google Scholar]
- 20.Guo P, Peterson C, Anderson D. Prohead and DNA-gp3-dependent ATPase activity of the DNA packaging protein gp16 of bacteriophage ϕ29. J Mol Biol. 1987;197:229–236. doi: 10.1016/0022-2836(87)90121-5. [DOI] [PubMed] [Google Scholar]
- 21.Dube P, Tavares P, Lurz R, van Heel M. The portal protein of bacteriophage SPP1: a DNA pump with 13-fold symmetry. EMBO J. 1993;12:1303–1309. doi: 10.1002/j.1460-2075.1993.tb05775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abrahams JP, Leslie AGW, Lutter R, Walker JE. Structure at 2.8Å resolution of F1-ATPase from bovine heart mitochondria. Nature. 1994;370:621–628. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- 23.Silverman MR, Simon MI. Flagellar rotation and the mechanism of bacterial motility. Nature. 1974;249:73–74. doi: 10.1038/249073a0. [DOI] [PubMed] [Google Scholar]
- 24.Vale RD, Milligan RA. The way things move: looking under the hood of molecular motor proteins. Science. 2000;288:88–95. doi: 10.1126/science.288.5463.88. [DOI] [PubMed] [Google Scholar]
- 25.Rossmann MG, Arnold E. International Table for Crystallography. F. International Union of Crystallography, Kluwer Academic; Dordrecht: (in the press) [Google Scholar]
- 26.Baker TS, Cheng RH. A model-based approach for determining orientations of biological macromolecules imaged by cryoelectron microscopy. J Struct Biol. 1996;116:120–130. doi: 10.1006/jsbi.1996.0020. [DOI] [PubMed] [Google Scholar]
- 27.Bailey S, et al. Phylogenetic analysis and secondary structure of the Bacillus subtilis bacteriophage RNA required for DNA packaging. J Biol Chem. 1990;265:22365–22370. [PubMed] [Google Scholar]
- 28.Reid RJD, Bodley JW, Anderson D. Identification of bacteriophage ϕ29 prohead RNA domains necessary for in vitro DNA-gp3 packaging. J Biol Chem. 1994;269:9084–9089. [PubMed] [Google Scholar]