Prediction of Missense Mutation Functionality Depends on both the Algorithm and Sequence Alignment Employed (original) (raw)
. Author manuscript; available in PMC: 2014 Sep 4.
Published in final edited form as: Hum Mutat. 2011 Apr 7;32(6):661–668. doi: 10.1002/humu.21490
Abstract
Multiple algorithms are used to predict the impact of missense mutations on protein structure and function using algorithm-generated sequence alignments or manually curated alignments. We compared the accuracy with native alignment of SIFT, Align-GVGD, PolyPhen-2 and Xvar when generating functionality predictions of well characterized missense mutations (n = 267) within the BRCA1, MSH2, MLH1 and TP53 genes. We also evaluated the impact of the alignment employed on predictions from these algorithms (except Xvar) when supplied the same four alignments including alignments automatically generated by (1) SIFT, (2) Polyphen-2, (3) Uniprot, and (4) a manually curated alignment tuned for Align-GVGD. Alignments differ in sequence composition and evolutionary depth. Data-based receiver operating characteristic curves employing the native alignment for each algorithm result in area under the curve of 78-79% for all four algorithms. Predictions from the PolyPhen-2 algorithm were least dependent on the alignment employed. In contrast, Align-GVGD predicts all variants neutral when provided alignments with a large number of sequences. Of note, algorithms make different predictions of variants even when provided the same alignment and do not necessarily perform best using their own alignment. Thus, researchers should consider optimizing both the algorithm and sequence alignment employed in missense prediction.
Keywords: multiple sequence alignment, SIFT, PolyPhen-2, Align-GVGD, Xvar, BRCA1, MSH2, MLH1, TP53
INTRODUCTION
Nonsynonymous or missense changes found as either single nucleotide polymorphisms (nsSNPs) or rare mutations result in amino acid substitutions in the protein product which may or may not affect protein function. Large-scale sequencing projects yield many missense mutations with unknown biological significance. Determining the pathogenicity of missense mutations is an important step in identifying and interpreting variants associated with disease.
A number of algorithms have been developed to predict the impact of missense mutations on protein structure and function including sequence and structure-based approaches. A review of available computational methods for assessing the functional effects of missense mutations has been performed (Ng and Henikoff, 2006; Karchin, 2009; Thusberg and Vihinen, 2009; Jordan et al., 2010). Many methods base their predictions on phylogenetic information implying the pathogenicity of missense mutations is assessed from the observed amino acid variation at a given residue in the multiple sequence alignment employed. Variables that affect the prediction accuracy of these algorithms include the gene examined, the number of sequences in the alignment, the evolutionary distances among species, the algorithm used, and the importance of absolute amino acid conservation versus relatively conservative missense changes (Greenblatt et al., 2003). Researchers have used multiple methods as a way to increase confidence in identifying deleterious mutations when predictive results differ between methods (Chan et al., 2007; Chun and Fay, 2009) but it has been argued these algorithms have major similarities ‘underneath the lid’ and the correlation of their outputs is the result of similarity of their inputs, which is not a cause for increased confidence (Karchin et al. 2009). Problems in comparing multiple methods extend further because there is no standard classification system used to categorize the predicted functionality of the variants, needed to provide a statistical measure of performance of the methods. Researchers have addressed this problem by grouping the predictions from the algorithms into two main categories: variants that are predicted to be deleterious or neutral (Chan et al., 2007).
Several studies have compared the prediction accuracy of sequence (Balasubramanian et al., 2005; Mathe et al., 2006) and structure-based algorithms (Bao et al., 2005; Chan et al., 2007; Chao et al., 2008) using alignments generated by the algorithms or manually curated alignments. However, it remains unclear how the predictions change when the sequence alignment provided changes, since it has been suggested that when the outputs from the algorithms differ, it is most likely due to employing different protein sequence alignments (Karchin, 2009). Past research has shown high predictive values for methods that use evolutionary sequence conservation, surprisingly with or without protein structural information (Chan et al., 2007). Since sequence alignments influence sequence-based methods which ultimately generate measures of pathogenicity (Kryukov et al., 2007), it is important to determine which types of sequence alignments will lead to better in silico assessments (Tavtigian et al., 2008).
In this study we employed four commonly used algorithms:
- SIFT (http://sift.jcvi.org/). The method ‘Sorts Intolerant From Tolerant’ is a sequence homology-based tool that predicts variants in the query sequence as “neutral” or “deleterious” using normalized probabilities calculated from the input multiple sequence alignment (Ng and Henikoff, 2001). SIFT obtains this multiple sequence alignment by internally generating it or by allowing the user to submit their own FASTA-formatted alignment. The alignment built by SIFT contains homologous sequences with a medium conservation measure of 3.0 where conservation is represented by information content (Schneider et al., 1986) to minimize false positive and false negative error. The authors mention better results may be obtained using only ortholog sequences because including paralogs can confound predictions at residues conserved only among the orthologs (Ng and Henikoff, 2002). Variants at a position with normalized probabilities less than 0.05 are predicted deleterious and predicted neutral with a probability greater than or equal to 0.05.
- Align-GVGD (http://agvgd.iarc.fr/). This method predicts variants in the query sequence based on a combination of Grantham Variation (GV), which measures the amount of observed biochemical evolutionary variation at a particular position in the alignment, and Grantham Deviation (GD), which measures the biochemical difference between the reference and amino acid encoded by the variant (Mathe et al., 2006). The original classifier uses a set of five criteria based on GV and GD which classifies variants as “neutral”, “unclassified' or “deleterious” (Mathe et al., 2006). For example, in the extreme case of GV = 0, the alignment is completely conserved at that position and any other variant will be considered deleterious. The new classifier provides ordered grades ranging from the most pathogenic to least likely pathogenic (Tavtigian et al., 2008). The algorithm has primarily been used for a few clinically relevant tumor suppressor genes such as BRCA1, TP53 and the author provides highly manually curated alignments which may cause a favorable bias towards this algorithm when applied to the class of genes studied here. These alignments contain a small number of full-length ortholog sequences with a long range of evolutionary depth. The author argues the alignment should not only be restricted to true orthologs due to the biological phenomenon of functional diversification among paralogs (Abkevich et al., 2004), but also should sample enough sequences at sufficient evolutionary distance from each other (alignment depth) for the best accuracy of the algorithms (Tavtigian et al., 2008). The experimentalist can provide his or her own alignment for other genes.
- PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/). This method is the latest tool developed by the authors of the original PolyPhen (Ramensky et al., 2002). Its novel features include the set of predictive features, the alignment pipeline and the probabilistic classifier based on machine-learning methods. PolyPhen-2 predicts variants as “benign”, “possibly damaging” or “probably damaging” based on eight sequenced-based and three structure-based predictive features which were selected by an iterative greedy algorithm. Another useful feature is the algorithm calculates a Bayes posterior probability that a given mutation is deleterious (Adzhubei et al., 2010). The web-based version requires use of the built in alignment pipeline, but the user may download and install the latest version of PolyPhen-2 to submit their own alignment. The alignment pipeline used in PolyPhen-2 selects homologous sequences using a clustering algorithm and then constructs and refines the alignment yielding an alignment containing both orthologs and paralogs that may or may not be full length, which yields a wider breadth of sequences but decreased depth compared with the Align-GVGD alignment. The authors argue this leads to more accurate predictions because a majority of deleterious variants affect protein structure compared to specific protein function (Adzhubei et al., 2010).
- Xvar (http://xvar.org/). This recently developed web-based algorithm (Reva et al., 2010) by the same authors of the original algorithm combinatorial entropy optimization (Reva et al., 2007) cannot accept user-defined multiple sequence alignments from the investigator as input. The Xvar server can map the variant to both the Uniprot (http://www.uniprot.org/) and NCBI Reference Sequence (Refseq) protein (http://www.ncbi.nlm.nih.gov/refseq/) and to the 3D structure in Protein Data Bank (PDB) (http://www.pdb.org/pdb/) if available. Once the Uniprot IDs are identified, they are used to build local sequence alignments and extract information about the domain boundaries, annotated functional regions and protein-protein interaction instead of using full-length sequences as in the other three algorithms. Xvar predicts variants as “neutral”, “low”, “medium” or “high”.
Sets of missense mutations with known functionality are needed to compare the algorithms. Locus-specific databases (LSDBs), which are “curated collections of sequence variants in genes associated with disease” (Greenblatt et al., 2008), can be used as a ‘gold standard’ containing both neutral and deleterious variants. The variants from the LSDBs are evaluated by the algorithms which allow a comparison of the algorithms for sensitivity, specificity and receiver operating curves. Tavtigian et al. (2008) argue the best data sets for comparing these algorithms are LSDBs which are curated by individuals or groups specialized in the analysis of each specific gene (Chan et al., 2007; Chao et al., 2008). A description of LSDBs for cancer susceptibility genes available on the internet can be found in Greenblatt et al. (2008). We evaluated the algorithms on four sets of variants with known functionality from BRCA1 (MIM# 113705), MSH2 (MIM# 609309), MLH1 (MIM# 120436) and TP53 (MIM# 191170) cancer associated genes.
MATERIALS AND METHODS
Multiple Sequence Alignments
The four multiple sequence alignments used as input to compare the variability of predictions from each algorithm include:
- An automatically generated alignment from SIFT
- An automatically generated alignment from PolyPhen-2 with a wide breadth of sequences (http://genetics.bwh.harvard.edu/pph2/). After downloading and installing the latest version of PolyPhen-2, the alignment automatically generated by the alignment pipeline can be obtained in a FASTA-formatted alignment.
- A small highly curated alignment with long evolutionary depth ideal for Align-GVGD (http://agvgd.iarc.fr/). The BRCA1, MSH2, MLH1 and TP53 curated protein alignments can be directly obtained from the website and formatted into a FASTA-formatted alignment.
- An uncurated alignment (http://www.uniprot.org/) automatically generated in Uniprot using the built in ClustalW feature with sequences included based on a criteria of 50% identity. This alignment was used as an unbiased alignment in the sense that none of the programs were built or trained on this type of alignment, which makes it suitable for testing the variability of the predictions from these algorithms.
All field tests using the four algorithms were run during July 23-27, 2010 on a Mac OS X 10.5.8. Safari 5.0 was used to access the web-based methods.
SIFT
The web-based method SIFT version 4.0.3 was used with all default settings. The alignment built by SIFT was created using the SIFT Sequence option. The other three multiple sequence alignments were each submitted under the SIFT Aligned Sequences option. Variants were predicted as “neutral” or “deleterious”.
Align-GVGD
The web-based method Align-GVGD was used with all default settings. The Align-GVGD alignment for BRCA1, MSH2, MLH1 and TP53 are freely available on the website. Each of the other three types of multiple sequence alignments were also submitted. Variants were predicted as “neutral”, “unclassified” or “deleterious”. For this study, the variants predicted as “neutral” and “unclassified” were grouped together as neutral variants.
PolyPhen-2
The latest version of PolyPhen-2 version 2.0.22 and helper programs were downloaded and installed on 3.06 GHz Intel Core 2 Duo processor with 6GB L2 Cache memory computer at Rice University. The standard output reported by the downloaded algorithm uses the default classifier model HumDiv, but predictions may also be obtained using the HumVar model reporting only minor differences between the two models (data not shown). The alignment built by PolyPhen-2 was automatically generated. The other three multiple sequence alignments could be submitted because the downloaded algorithm allows the user to submit their own FASTA-formatted alignment. Variants were predicted as “benign”, “possibly damaging” or “probably damaging”. For this study, the variants predicted as “possibly damaging” and “probably damaging” were grouped together as deleterious variants.
Xvar
The web-based method Xvar version 0.75 beta was used with all default settings. The variants were submitted along with their Uniprot accession IDs. They were predicted as “neutral”, “low”, “medium” and “high”. For this study, the variants predicted as “neutral” and “low” were grouped together as neutral variants and the variants predicted as “medium” and “high” were grouped together as deleterious variants.
Mutation Databases
To test the algorithms we used existing sets of missense mutations with curated functionality available either in the literature or curated locus specific databases for four cancer associated genes: BRCA1, MLH1, MSH2 and TP53.
- The online Breast Cancer Information Core (BIC) (Szabo et al., 2000) mutation database (http://research.nhgri.nih.gov/bic/) was used to identify neutral (n = 16) and deleterious (n = 17) mutations from BRCA1. The steering committee of the BIC has manually reviewed available data from the literature and likelihood ratios (Goldgar et al., 2004) to define clinically relevant or benign missense changes using three categorizations of clinically relevant (yes, no or unknown).
- The MLH1 missense mutations were obtained from two papers (Raevaara et al., 2005; Chao et al., 2008). The first paper performed mismatch repair functional assays to identify neutral (n = 10) mutations with wild-type activity and deleterious (n = 18) mutations with impaired mismatch repair activity. These MLH1 mutations were compared in a previous paper (Chan et al., 2007) that used the three algorithms SIFT, Align-GVGD and PolyPhen, but they did not compare the results using different alignments. The second paper compiled a list of all known MLH1 missense mutations from several LSDBs with supporting data which was used as rigorous criteria to classify the variants as neutral (n = 18) or deleterious (n = 37). Subtracting the overlap between the papers yielded a total of (n = 21) neutral and (n = 39) deleterious variants.
- The MSH2 variants were obtained from the same two papers as the MLH1 variants above (Raevaara et al., 2005; Chao et al., 2008). The first paper identified neutral (n = 3) mutations with wild-type activity and deleterious (n = 11) mutations with impaired mismatch repair activity. These MSH2 mutations were also compared in the paper (3). The second paper compiled classified neutral (n = 8) or deleterious (n = 13) variants with the same criteria as above. Subtracting the overlap between the papers yielded a total of (n = 11) neutral and (n = 19) deleterious variants.
- The online TP53 database from the International Agency for Research on Cancer (IARC) was used to identify neutral (n = 4) and deleterious (n = 140) mutations from the TP53 gene (http://www-p53.iarc.fr/). A description of inclusion criteria for the polymorphisms and germline deleterious variants is given (Olivier et al., 2002).
Statistics
Several statistical measures of performance were used to compare the performance of the algorithms for each of the four sets of mutations with known functionality. Using the notation of true positives (TP), true negatives (TN) false positives (FP) and false negatives (FN), we compute sensitivity as TP / (TP + FN) (probability of identifying true deleterious mutations) and specificity as TN / (TN + FP) (probability of identifying true neutral mutations). Some algorithms provide more than two prediction categories, e.g. neutral, possibly and probably damaging for Polyphen-2. Therefore, as described above for each method, we grouped the output into two categories “deleterious” and “neutral” based on similar groupings in previous studies (Chan et al., 2007).
A receiver operating characteristic (ROC) curve (Fawcett, 2006) is a technique that allows combining the mutation data and visualizing the performance of the algorithm with the native alignment and the three additional alignments provided (treated as a probabilistic classifier in this case). The ROC graph is a two dimensional graph that plots sensitivity against 1 -specificity depicting the relative tradeoffs between the true positives and false positives. ROC curves can be based on discrete or continuous classifiers. An algorithm that only reports a finite set of prediction categories, such as “neutral” or “deleterious” is a discrete classifier. However, if the algorithm reports a continuous score, being the degree or probability of a mutation belonging to a prediction category then it is a continuous classifier. We have reported the ROC curves in Figure 3 using the associated continuous scores available for any mutation tested in each algorithm (in fact, these scores underlie the discrete classifications). [[COPYEDITOR AND TYPESETTER: PLEASE ALLOW THIS EARLY CALL OUT OF FIGURE 3]] Accuracy is measured by the area under the ROC curve (AUC); an area of 1 corresponds to a perfect prediction, whereas an area of 0.5 corresponds to a “pure chance” prediction. AUC less than 0.5 may be interpreted as a systematically incorrect prediction. The AUC of a given classifier can be represented as the probability that given an alignment the algorithm will rank a randomly chosen deleterious mutation higher than a randomly chosen neutral mutation (Fawcett, 2006). In our case, we use an AUC formula equivalent to the Wilcoxon test of ranks (Hanczar et al., 2010). The confidence intervals of the estimated AUC values are identical with the confidence intervals of the Wilcoxon rank statistic (Hogg and Tanis, 2006). The ROC curves and AUC values for all algorithm/alignment pairs were computed using the ROCR package in R (Sing et al., 2005).
Figure 3.
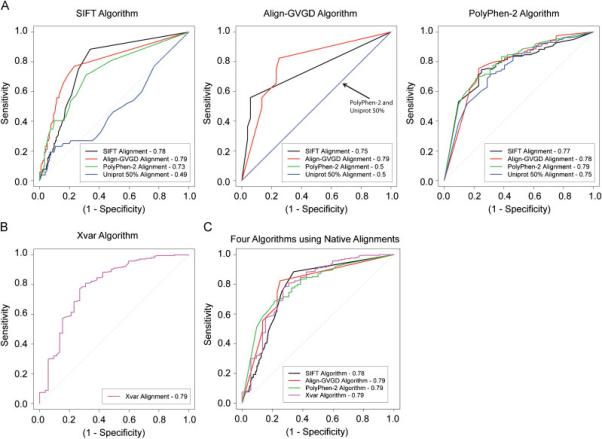
A) Receiver operating characteristic (ROC) curves using probabilities and scores associated with each prediction for each of the three algorithms SIFT, Align-GVGD, and PolyPhen-2. For each algorithm, four colored lines (black, red, green blue) are drawn representing the four alignments used in each algorithm. The area under the curve (AUC) is reported in the legend. B) Receiver operating characteristic (ROC) curve using the four genes BRCA1, MSH2, MLH1, and TP53 from the Xvar algorithm. The pink line drawn represents the Xvar alignment. C) ROC curves comparing the performance of the four algorithms using their own native alignments.
RESULTS
In this analysis we compare the predicted functionality of the same set of curated missense mutations in tumor suppressor genes using existing algorithms SIFT, Align-GVGD, PolyPhen-2 and Xvar. In addition, we provided SIFT, Align-GVGD and Polyphen-2 the same four sequence alignments for each gene analyzed to determine the impact of the alignment on prediction. Xvar is excluded from this latter analysis because it currently does not accept multiple sequence alignments as input. Specificity was measured by the performance on correctly calling neutral variants neutral while sensitivity was measured by the performance on correctly calling deleterious variants deleterious.
BRCA1 Tumor Suppressor Gene
The native BRCA1 sequence alignments were built for the four algorithms as described in Methods. In addition, the three non-native alignments were used as inputs in each of the three algorithms, SIFT, Align-GVGD and PolyPhen-2 (see Table 1 for the number of sequences in each alignment). A description of the set of the well-characterized neutral (n = 16) and deleterious (n = 17) BRCA1 variants from the BRCA1 LSDB is described in Methods.
Table 1.
Specificity and sensitivity summary for all four genes BRCA1 (n = 16 neutral, n = 17 deleterious), MSH2 (n = 11 neutral, n = 19 deleterious), MLH1 (n = 21 neutral, n = 39 deleterious) and TP53 (n = 4 neutral, n = 140 deleterious) using the same four alignments
| Algorithms | ||||||||
|---|---|---|---|---|---|---|---|---|
| SIFT | Align-GVGD | PolyPhen-2 | ||||||
| Genes | Alignments | No. Sequences in Alignment | Spec (%) | Sens (%) | Spec (%) | Sens (%) | Spec (%) | Sens (%) |
| BRCA1 | SIFT | 360 | 31.3 | 94.1 | 93.8 | 64.7 | 56.3 | 94.1 |
| Align-GVGD | 13 | 75.0 | 70.6 | 93.8 | 70.6 | 31.3 | 82.4 | |
| PolyPhen-2 | 279 | 43.8 | 47.1 | 100.0 | 0.0 | 37.5 | 76.5 | |
| Uniprot 50% | 275 | 50.0 | 47.1 | 100.0 | 0.0 | 25.0 | 76.5 | |
| MSH2 | SIFT | 45 | 45.5 | 89.5 | 81.8 | 21.1 | 27.3 | 94.7 |
| Align-GVGD | 14 | 18.2 | 89.5 | 54.5 | 89.5 | 27.3 | 89.5 | |
| PolyPhen-2 | 56 | 45.5 | 94.7 | 100.0 | 0.0 | 36.4 | 89.5 | |
| Uniprot 50% | 53 | 18.2 | 94.7 | 100.0 | 0.0 | 18.2 | 94.7 | |
| MLH1 | SIFT | 29 | 52.4 | 71.8 | 81.0 | 48.7 | 42.9 | 66.7 |
| Align-GVGD | 11 | 57.1 | 97.4 | 52.4 | 97.4 | 42.9 | 97.4 | |
| PolyPhen-2 | 191 | 61.9 | 89.7 | 100.0 | 0.0 | 66.7 | 89.7 | |
| Uniprot 50% | 45 | 52.4 | 82.1 | 100.0 | 0.0 | 42.9 | 97.4 | |
| TP53 | SIFT | 72 | 75.0 | 84.3 | 100.0 | 57.9 | 75.0 | 87.1 |
| Align-GVGD | 9 | 75.0 | 85.7 | 100.0 | 82.1 | 25.0 | 92.1 | |
| PolyPhen-2 | 93 | 100.0 | 73.6 | 100.0 | 0.0 | 100.0 | 84.3 | |
| Uniprot 50% | 113 | 100.0 | 32.1 | 100.0 | 0.0 | 75.0 | 90.0 |
Figure 1A shows the output of each algorithm using the neutral (n = 16) BRCA1 variants when given the same four alignments. We found the algorithm PolyPhen-2 to be the least sensitive to the varying alignments. The Align-GVGD algorithm was the most sensitive to the varying alignments because the algorithm will predict all variants neutral, regardless of pathogenicity, when provided an alignment with a large number of sequences such as the PolyPhen-2 and Uniprot 50% alignments. Although this translates to an apparently high specificity for the Align-GVGD algorithm, this feature of the algorithm leads to the prediction of neutrality being solely based on the number of sequences in the alignment. Surprisingly, we see that algorithms do not necessarily perform best using their own alignment (Table 1). For example, the SIFT algorithm has the highest specificity using the Align-GVGD alignment (compared to its own) possibly because the Align-GVGD alignment is only made up of orthologs (Ng and Henikoff, 2002). We also found that the SIFT algorithm overcalls neutral variants as deleterious, low specificity, as previously noted by others (Mathe et al., 2006; Karchin et al., 2008). Using its own or native alignment, the Xvar algorithm has specificity (Table 2) that is equal to or smaller than the specificities of the other three algorithms using their optimal alignment (Table 1).
Figure 1.
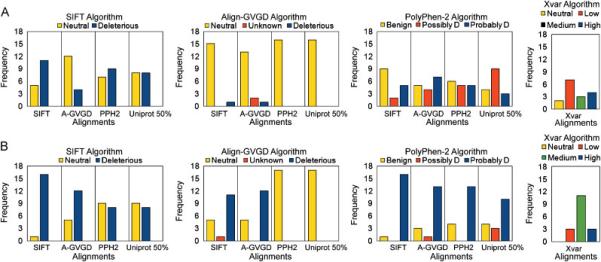
A) Predictions of neutral (n = 16) BRCA1 missense mutations using three algorithms with four alignments each. The four alignments are represented by SIFT (SIFT), Align-GVGD (A-GVGD), PolyPhen-2 (PPH2), and Uniprot 50% (Uniprot 50%). The prediction categories for PolyPhen-2 Possibly Damaging and Probably Damaging have been abbreviated to ‘Possibly D’ and ‘Probably D’. The algorithm Xvar employs its own alignment. B) Predictions of deleterious (n = 17) BRCA1 missense mutations using three algorithms with four alignments each.
Table 2.
Specificity and sensitivity summary using the Xvar default alignment for the four genes BRCA1, MSH2, MLH1 and TP53
| Xvar | ||
|---|---|---|
| Genes | Spec (%) | Sens(%) |
| BRCA1 | 56.3 | 82.4 |
| MSH2 | 27.3 | 100 |
| MLH1 | 33.3 | 100 |
| TP53 | 50.0 | 95.7 |
The results in Figure 1B show the output of each algorithm for the deleterious (n = 17) BRCA1 variants when given the same four alignments. The Align-GVGD algorithm shows a poor sensitivity, again because it incorrectly predicts all 17 deleterious variants as neutral when provided large number of sequences, as it is the case for PolyPhen-2 and Uniprot 50% alignments. The algorithm PolyPhen-2 has the highest sensitivity using the SIFT alignment which is another example of an algorithm performing best with an alignment other than its own (Table 1). When comparing the Xvar algorithm using its native alignment to the other algorithms, we see Xvar has a high sensitivity (Table 2) that is similar to the sensitivities reported from the other algorithms using their optimal alignment (Table 1).
MSH2, MLH1 Mismatch Repair Genes and TP53 Tumor Suppressor Gene
The native MSH2, MLH1 and TP53 sequence alignments were built for the four algorithms as described in Methods. In addition, the three non-native alignments for each gene were used as inputs in each of the three algorithms, SIFT, Align-GVGD and PolyPhen-2 (see Table 1 for the number of sequences in each alignment). The three sets of variants from MSH2 (n = 11 neutral, n = 19 deleterious), MLH1 (n = 21 neutral, n = 39 deleterious) and TP53 (n = 4 neutral, n = 140 deleterious) are described in Methods. Overall we found similar results to BRCA1 for variants from these three cancer genes (Table 1). However, SIFT algorithm reports higher sensitivities using the MSH2 and MLH1 variants compared to the BRCA1 variants. We also note that although for BRCA1 the highest specificity of the SIFT algorithm was seen by using the Align-GVGD alignment this was not true for the other three genes. The results for MSH2, MLH1 and TP53 using the Xvar algorithm are given in Table 2 which again demonstrates the algorithm using its native alignment reports similar specificity values to the other algorithms. When comparing sensitivity, Xvar using its native alignment reports higher sensitivities (Table 2) that is equal to or greater than the sensitivities of the other algorithms using their best alignment (Table 1) for all three genes.
Overall Sensitivity and Specificity
A boxplot summary of the sensitivity and specificity values for the three algorithms, which combines the gene- and alignment-specific information, illustrates how much variation is caused by employing different alignments (Figure 2). For each alignment, the four sensitivity values are computed by grouping the mutations within each of the four genes BRCA1, MSH2, MLH1 and TP53, yielding a total of 16 sensitivity values for each algorithm. We note that there are only four neutral TP53 variants which may inflate the specificity values; therefore we excluded TP53 specificity values in the figure and used only 12 specificity values for each algorithm. We also computed confidence intervals for the sensitivity and specificity estimates (see Supp. Tables S1-S4), using the Wilson score method (Agresti, 2002). The results from Figure 2 show PolyPhen-2 and SIFT both have a high median sensitivity of 0.90 and 0.85, respectively. We note PolyPhen-2 and SIFT both have similar median specificity values, 0.40 and 0.52, respectively, highlighting that the specificities are significantly lower than the sensitivities. Thus, these algorithms are more likely to make mistakes by calling neutral variants deleterious. For both sensitivity and specificity PolyPhen-2 has a smaller interquartile range (IQR) than SIFT, which means that PolyPhen-2 is less sensitive to the sequence alignment employed. As noted previously, Align-GVGD is very sensitive to the algorithm employed. The high specificity seen in Align-GVGD is misleading because the algorithm predicts all variants neutral, regardless of pathogenicity, when using alignments with a large number of sequences. This feature of the algorithm also results in low sensitivity with large IQR. Thus, even though Align-GVGD performs well using its own alignment, it is more dependent on the alignment employed than either SIFT or PolyPhen-2. The results of this analysis are that only PolyPhen-2 and SIFT are appropriate for use with non-native alignments that are not manually curated, with PolyPhen-2 modestly outperforming SIFT. The corresponding boxplot for the Xvar algorithm is not provided because Xvar requires the use of its native alignment; however, the median sensitivity is 0.98 and the median specificity (excluding TP53) is 0.33.
Figure 2.
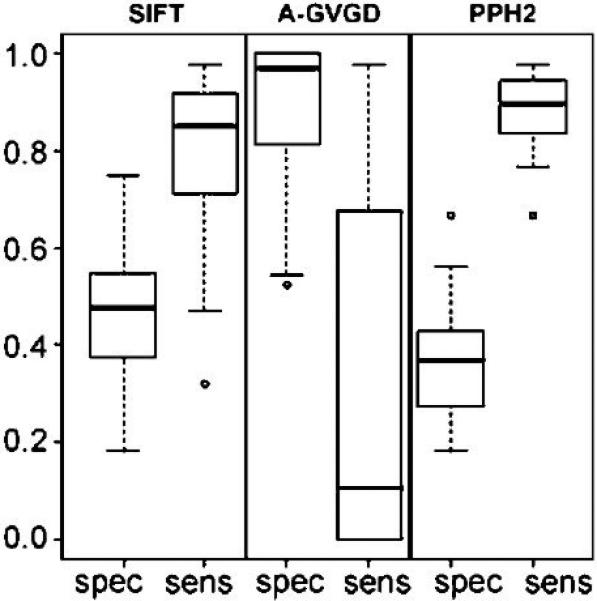
Boxplots of specificity (spec) and sensitivity (sens) for each algorithm as given in Table 1. Sensitivity values are reported using all four genes BRCA1, MSH2, MLH1 and TP53, but TP53 is excluded in specificity values to account for potential bias given that there are only 4 neutral variants. The three algorithms are represented by SIFT (SIFT), Align-GVGD (A-GVGD) and PolyPhen-2 (PPH2).
Receiver Operating Characteristic curves
Each algorithm provides a quantitative probability or score as output as well as a prediction category, e.g. “probably damaging” for Polyphen-2. This enabled us to compare alignment-specific information for each algorithm using the concept of receiver operating characteristic (ROC) curves to provide a succinct graphical summary of all four algorithms, treated as continuous classifiers (Figure 3A and Figure 3B; also, see Statistics Section in Materials and Methods). Align-GVGD and PolyPhen-2 algorithms performed best using their native alignment, but the SIFT algorithm had a higher AUC when using an alignment, manually curated Align-GVGD, other than its own. When employing the optimal alignment for each algorithm, the AUC values are 79% for all four algorithms. The PolyPhen-2 algorithm is shown to be the least dependent on the alignment employed as seen by the nearly overlapping ROC curves and similar AUC values for all four alignments provided. In comparison the SIFT and Align-GVGD algorithms show much greater variation in their ROC curves employing different alignments. As seen in Figure 2, Figure 3 shows PolyPhen-2 and SIFT are the only two methods appropriate for use with non-native algorithm-generated alignments. The area under the ROC curve (AUC) values are reported in Table 3 with the associated confidence intervals. When we rank the averaged AUC values for each alignment strategy we obtain 0.790, 0.766, 0.674, and 0.579 for the Align-GVGD, SIFT, PolyPhen-2 and Uniprot alignments, respectively.
Table 3.
Area under the curve (AUC) from receiver operating curves for each algorithm using each alignment using probabilities and scores associated with each mutation prediction
| Algorithms | Alignments | AUC | CI |
|---|---|---|---|
| SIFT | SIFT | 0.777 | (0.690, 0.865) |
| Align-GVGD | 0.790 | (0.702, 0.877) | |
| PolyPhen-2 | 0.730 | (0.643, 0.818) | |
| Uniprot 50% | 0.487 | (0.400, 0.575) | |
| AGVGD | SIFT | 0.747 | (0.659, 0.835) |
| Align-GVGD | 0.791 | (0.703, 0.878) | |
| PolyPhen-2 | 0.500 | (0.412, 0.588) | |
| Uniprot 50% | 0.500 | (0.412, 0.588) | |
| PPH2 | SIFT | 0.773 | (0.686, 0.861) |
| Align-GVGD | 0.779 | (0.692, 0.867) | |
| PolyPhen-2 | 0.792 | (0.704, 0.879) | |
| Uniprot 50% | 0.750 | (0.662, 0.838) | |
| Xvar | Xvar | 0.790 | (0.703, 0.878) |
Given that most investigators will utilize online tools where the algorithm employs its native alignment we compared the ROC curves for the four algorithms using their native alignments (Figure 3C). This analysis demonstrates no significant differences in the shape of the curve or AUC values (between 78-79%) for all four algorithms. The same analysis utilizing the optimal alignment for each algorithm results in only a small difference in the AUC for the SIFT algorithm increasing from 78 to 79%.
DISCUSSION
Accurately predicting the impact of missense mutations on protein function depends on the algorithm used, the type of sequence alignment provided, and on the number of sequences in the alignment. In addition to problems of interpretation there are technical difficulties as well. In our experience, when simply submitting a list of missense mutations to an algorithm the user must be able to: (1) manipulate the input format specified by each algorithm, (2) build an optimal protein sequence alignment, if required, (3) be knowledgeable of Unix system commands, (4) interpret server error messages, and (5) transform the output to a working format for further studies. Standard input and output formats are needed to alleviate the burden on the user. Also tools to create informative protein sequence alignments for each protein are necessary to accurately predict the impact of missense mutations on protein function. An additional source of error in prediction when analyzing sequence variants identified through disease status is that all algorithms focus on the missense change encoded by the variant when in reality the sequence variant may also impact gene expression for example through alternative splicing of the messenger RNA.
Chan et al. (2007) compared four methods: SIFT, Align-GVGD, PolyPhen and the BLOSUM62 matrix, using the native alignments supplied by the program or manually curated for Align-GVGD. In the paper each method individually had a limited overall predictive value (72.9-82.0%), but when all four methods agree (62.7%), the overall predictive value increased to 88.1%. Karchin et al. (2009) argued these algorithms have major similarities ‘underneath the lid’ of each method and the correlation of their outputs is the result of similarity of their inputs, which is not a cause for increased confidence. Chun and Fay (2009) suggested differences between missense predictions from the algorithms may be due to differences in the sequences and/or alignments used to identify evolutionary conserved mutations. We directly tested this idea by comparing the predictions of the SIFT, Align-GVGD and PolyPhen-2 algorithms by supplying the same four alignments to each algorithm. Surprisingly we found a given algorithm did not necessarily perform best using the alignment provided by the creator of the algorithm. For example, the PolyPhen-2 algorithm reported higher sensitivities in all four genes using alignments other than its own and SIFT had a slightly higher AUC when provided the Align-GVGD alignment containing only orthologs as originally predicted by Ng and Henikoff (2002). The three algorithms SIFT, PolyPhen-2 and Xvar all had a high sensitivity, but low specificity implying these algorithms may overcall neutral variants deleterious. This feature was most pronounced for Xvar with higher sensitivities and lower specificities than most of the other algorithms. We showed Align-GVGD was the most affected by alignment employed, performing well when using manually curated alignments, but calling all variants neutral when alignments contain a large number of sequences. Thus, for large-scale sequencing experiments Align-GVGD would require development of alignments with orthologous sequences through evolution for all genes. Conversely, the PolyPhen-2 algorithm was shown to be the least sensitive to alignment provided with nearly overlapping ROC curves. The ROC analysis resulted in AUC values of 78-79% for all four algorithms using native alignments and 79% when using the optimal alignment for each algorithm which shows despite the differences in predictions from the algorithms and alignments the overall performance of these four commonly used methods is similar.
Karchin et al. (2009) further argued that when the outputs from the algorithms SIFT and PolyPhen differ, it is more likely due to using different protein sequence alignments compared to the differences in scores used to classify the variants. From our experimental design, we were able to directly test this hypothesis. Using a Venn diagram we depict the disjoint classification of variants predicted deleterious and neutral, respectively by different algorithms all employing the Align-GVGD alignment (as all three algorithms performed well with this alignment) (Figure 4). When considering the predicted deleterious mutations the four algorithms agree on 195 mutations (77%) using the Align-GVGD alignment, but the SIFT, Align-GVGD and PolyPhen-2 algorithms agree on an additional three mutations for a total of 198 mutations (79%). Of this 195 only 181 are actually classified as deleterious by the LSDB. Interestingly, Chun and Fay (2009) compared the predicted deleterious mutations from the three algorithms SIFT, PolyPhen and Likelihood Ratio Test (LRT) resulting in a very low overlap of 5%, but when we perform a similar analysis employing the algorithms' own alignment, we see a much higher overlap of 70%. When considering the predicted neutral mutations, the four algorithms only agree on 15 mutations (20%); excluding Xvar results in agreement for another 13 mutations for a total of 28 mutations (39%) which again demonstrates problems in predicting variants to be neutral. Only 11 of the 15 variants are classified as neutral by the LSDB implying even when provided the same alignment the algorithms make different predictions. Further research is needed to understand the underlying differences in these algorithms. Thus, in order to predict missense mutation functionality, the researcher should consider optimizing both the algorithm and sequence alignment employed.
Figure 4.
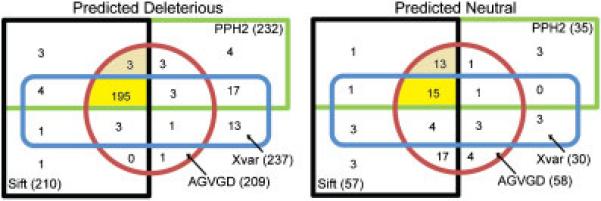
Predictions of neutral and deleterious mutations with the SIFT, Align-GVGD and PolyPhen-2 algorithms using the Align-GVGD alignment and the Xvar algorithm using its own alignment. We also depict the exclusive overlap of the predictions between the four algorithms to show their agreement (dark yellow) and between the three algorithms SIFT, Align-GVGD and PolyPhen-2 (light yellow).
Supplementary Material
Supp Table S1-S4
ACKNOWLEDGMENTS
We thank Shamil Sunyaev, Ph.D., and Ivan Adzhubey, Ph.D. for providing guidance on using the PolyPhen-2 algorithm. We also thank Sean Tavtigian, Ph.D. for providing guidance on the Align-GVGD alignments. This work was supported by CPRIT grant RP101089 and NCI T32 training program CA096520.
Footnotes
Supporting Information for this preprint is available from the Human Mutation editorial office upon request (humu@wiley.com)
REFERENCES
- Abkevich V, Zharkikh A, Deffenbaugh AM, Frank D, Chen Y, Shattuck D, Skolnick MH, Gutin A, Tavtigian SV. Analysis of missense variation in human BRCA1 in the context of interspecific sequence variation. J Med Genet. 2004;41:492–507. doi: 10.1136/jmg.2003.015867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramowitz M, Stegun IA. Handbook of Mathematical Functions with Formulas, Graphs, and Mathematical Tables. U.S. Government Printing Office; Washington, D.C.: 1972. p. 885. [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev S. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agresti A. Categorical Data Analysis. 2nd edition. John Wiley and Sons; Hoboken, New Jersey: 2002. [Google Scholar]
- Balasubramanian S, Xia Y, Freinkman E, Gerstein M. Sequence variation in G-protein-coupled receptors: analysis of single nucleotide polymorphisms. Nucl Acids Res. 2005;33:1710–1721. doi: 10.1093/nar/gki311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao L, Cui Y. Prediction of the phenotypic effects of non-synonymous single nucleotide polymorphisms using structural and evolutionary information. Bioinformatics. 2005;21:2185–2190. doi: 10.1093/bioinformatics/bti365. [DOI] [PubMed] [Google Scholar]
- Chan PA, Duraisamy S, Miller PJ, Newell JA, McBride C, Bond JP, Raevaara T, Ollila S, Nystrom M, Grimm AJ, et al. Interpreting missense variants: comparing computational methods in human disease genes CDKN2A, MLH1, MSH2, MECP2, and tyrosinase (TYR). Hum Mutat. 2007;28:683–693. doi: 10.1002/humu.20492. [DOI] [PubMed] [Google Scholar]
- Chao EC, Velasquez JL, Witherspoon MS, Rozek LS, Peel D, Ng P, Gruber SB, Watson P, Rennert G, Anton-Culver H, et al. Accurate classification of MLH1/MSH2 missense variants with multivariate analysis of protein polymorphisms-mismatch repair (MAPP-MMR). Hum Mutat. 2008;29:852–860. doi: 10.1002/humu.20735. [DOI] [PubMed] [Google Scholar]
- Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res. 2009;19:1553–1561. doi: 10.1101/gr.092619.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett T. An introduction to ROC analysis. Pattern Recognition Letters. 2006;27:861–874. [Google Scholar]
- Greenblatt MS, Beaudet JG, Gump JR, Godin KS, Trombley L, Koh J, Bond JP. Detailed computational study of p53 and p16: using evolutionary sequence analysis and disease-associated mutations to predict the functional consequences of allelic variants. Oncogene. 2003;22:1150–1163. doi: 10.1038/sj.onc.1206101. [DOI] [PubMed] [Google Scholar]
- Greenblatt MS, Brody LC, Foulkes WD, Genuardi M, Hofstra RM, Olivier M, Plon S, Sijmons RH, Sinilnikova O, Spurdle AB. Locus-specific databases and recommendations to strengthen their contributions to the classification of variants in cancer susceptibility genes. Hum Mutat. 2008;29:1273–1281. doi: 10.1002/humu.20889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldgar DE, Easton DF, Deffenbaugh AM, Monterio AN, Tavtigian SV, Couch FJ, Breast Cancer Information Core (BIC) Steering Committee Integrated evaluation of DNA sequence variants of unknown clinical significance: application to BRCA1 and BRCA2. Am J Hum Genet. 2004;75:535–544. doi: 10.1086/424388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanczar B, Hua J, Sima C, Weinstein J, Bittner M, Dougherty ER. Small-sample precision of ROC-related estimates. Bioinformatics. 2010;26:822–830. doi: 10.1093/bioinformatics/btq037. [DOI] [PubMed] [Google Scholar]
- Hogg RV, Tanis EA. Probability and Statistical Inference. 7th edition. Pearson Prentice Hall; Upper Saddle River, New Jersey: 2006. [Google Scholar]
- Jordan DM, Ramensky VE, Sunyaev SR. Human allelic variation: perspective from protein function, structure, and evolution. Curr Opin Struct Biol. 2010;20:342–350. doi: 10.1016/j.sbi.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karchin R, Mukesh A, Sali A, Couch F, Beattie MS. Classifying variants of undetermined significance in BRCA2 with protein likelihood ratios. Cancer Inform. 2008;6:203–216. doi: 10.4137/cin.s618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karchin R. Next generation tools for the annotation of human SNPs. Brief Bioinform. 2009;10:35–52. doi: 10.1093/bib/bbn047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryukov GV, Pennacchio LA, Sunyaev SR. Most rare missense alleles are deleterious in humans: implication for complex disease and association studies. Am J Hum Genet. 2007;80:727–739. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathe E, Olivier M, Kato S, Ishioka C, Hainaut P, Tavtigian SV. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucl Acids Res. 2006;34:1317–25. doi: 10.1093/nar/gkj518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002;12:436–446. doi: 10.1101/gr.212802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annu Rev Genomics Human Genet. 2006;7:61–80. doi: 10.1146/annurev.genom.7.080505.115630. [DOI] [PubMed] [Google Scholar]
- Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. The IARC TP53 Database: New Online Mutation Analysis and Recommendation to Users. Hum Mutat. 2002;19:607–614. doi: 10.1002/humu.10081. [DOI] [PubMed] [Google Scholar]
- Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucl Acids Res. 2002;30:3894–900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reva B, Antipin Y, Sander C. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol. 2007;8:R232. doi: 10.1186/gb-2007-8-11-r232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reva BA, Antipin YA, Sander C. Functional impact of protein mutations: evolutionary information score and application to cancer genomics. Nucl Acids Res. 2010 doi: 10.1093/nar/gkr407. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider TD, Stormo GD, Gold L, Ehrenfeucht A. Information content of binding sites on nucleotide sequences. J Mol Biol. 1986;188:415–431. doi: 10.1016/0022-2836(86)90165-8. [DOI] [PubMed] [Google Scholar]
- Sing T, Sander O, Beerenwinkel N, Lengauer T. ROCR: visualizing classifier performance in R. Bioinformatics. 2005;21:3940–3941. doi: 10.1093/bioinformatics/bti623. [DOI] [PubMed] [Google Scholar]
- Sunyaev S, Ramensky V, Koch I, Lathe W, Kondrashov AS, Bork P. Prediction of deleterious human alleles. Hum Mol Genet. 2001;10:591–597. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- Szabo C, Masiello A, Ryan JF, The BIC Consortium. Brody L. The breast cancer information core: database design, structure and scope. Hum Mutat. 2002;16:123–131. doi: 10.1002/1098-1004(200008)16:2<123::AID-HUMU4>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Tavtigian SV, Deffenbaugh AM, Yin L, Judkins T, Scholl T, Samollow PB, de Silva D, Zharkikh A, Thomas A. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J Med Genet. 2006;43:295–305. doi: 10.1136/jmg.2005.033878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavtigian SV, Greenblatt MS, Lesueur F, Byrnes GB, IARC Unclassified Genetic Variants Working Group In silico analysis of missense substitutions using sequence-alignment based methods. Hum Mutat. 2008;29:1327–1336. doi: 10.1002/humu.20892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thusberg J, Vihinen M. Pathogenic or not? And if so, then how? Studying the effects of missense mutations using bioinformatics methods. Hum Mutat. 2009;30:703–714. doi: 10.1002/humu.20938. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supp Table S1-S4