Interaction of SQSTM1 with the motor protein dynein – SQSTM1 is required for normal dynein function and trafficking (original) (raw)
ABSTRACT
The dynein motor protein complex is required for retrograde transport of vesicular cargo and for transport of aggregated proteins along microtubules for processing and degradation at perinuclear aggresomes. Disruption of this process leads to dysfunctional endosome accumulation and increased protein aggregation in the cell cytoplasm, both pathological features of neurodegenerative diseases. However, the exact mechanism of dynein functionality in these pathways is still being elucidated. Here, we show that the scaffolding protein SQSTM1 directly interacts with dynein through a previously unidentified dynein-binding site. This interaction is independent of HDAC6, a known interacting protein of both SQSTM1 and dynein. However, knockdown of HDAC6 increases the interaction of SQSTM1 with dynein, indicating a possible competitive interaction. Using different dynein cargoes, we show that SQSTM1 is required for proper dynein motility and trafficking along microtubules. Based on our results, we propose a new model of competitive interaction between SQSTM1 and HDAC6 with dynein. In this model, SQSTM1 would not only affect the association of polyubiquitylated protein aggregates and endosomes with dynein, but would also be required for normal dynein function.
KEY WORDS: Dynein intermediate chain, SQSTM1, Histone deacetylase 6, HDAC6, Late endosome, Aggresome
INTRODUCTION
Dynein is the major molecular motor for retrograde movement of cargoes, such as mitochondria, organelles and misfolded proteins, along the microtubule. Cytoplasmic dynein is a large protein complex containing heavy (DHC), intermediate (DIC) and light chains (DLC) (Paschal et al., 1987; King et al., 1998). Dynein binds to microtubules through dimerization of its two heavy chains, which also contain its ATP-binding activity. ATP is hydrolyzed in the globular heads of the intact motor to provide energy for dynein processivity. The tail domain of the heavy chains interacts with multiple intermediate and light chains, forming the cargo-binding complex. The association of dynein with many membrane cargos or target sites requires the accessory dynactin protein complex (Vallee et al., 2004). The dynactin complex has been shown to interact with dynein through DICs and the dynactin subunit p150glued (also known as DCTN1) in a manner that is crucial for dynein function. p150glued itself contains a microtubule-binding site, providing an additional microtubule contact for the motor protein complex. Dynactin has been reported to be required for cargo binding to dynein (Karki and Holzbaur, 1995; Schroer, 2004) and has also been reported to increase the processivity of the ATPase activity of dynein, allowing movement of cargoes along the microtubule (Vaughan and Vallee, 1995; Boylan et al., 2000).
Dynein is involved with a wide array of cellular processes, from regulating the spindle assembly checkpoint to localization of mRNAs (Vallee et al., 2004). Dynein is also associated with a number of disease processes, including viral transport, failure of neuron migration and axonemal disorders, such as ciliary dyskinesias (Dodding and May, 2011; Vallee et al., 2004; Hafezparast et al., 2003). However, the best-documented function of dynein is its role in retrograde transport of membranous organelles within neuronal cells (Schroer et al., 1989; Schnapp and Reese, 1989). Dynein is required in the maintenance of microtubules (Asthana et al., 2012) and is involved in lysosomal (Lin and Collins, 1992) and endosomal vesicular transport mechanisms (Aniento et al., 1993). Autophagosomal movement within the cell for efficient lysosomal encounters is also dependent on the dynein–dynactin motor complex (Kimura et al., 2008). Because lysosomes are located predominantly in the cytoplasm of the cell, a functional dynein transport system is essential for delivery of late endosomal cargo to the perinuclear region. This transport is required for fusion of endosomes with lysosomes for efficient degradation of internalized material (Cai et al., 2010). Recently, dynein has also been implicated in the clearance of misfolded and aggregated proteins where mutations, resulting in decreased dynein functionality, cause increased aggregate formation and impairment of the autophagic clearance of aggregated proteins (Ravikumar et al., 2005).
Increased protein aggregation is one of the most salient pathological hallmarks of neurodegenerative diseases. Disruption of motor-protein function and transport in the cell results in defective disposal of aggregation-prone proteins and increased aggregate formation, likely leading to neurodegeneration (Chevalier-Larsen and Holzbaur, 2006; De Vos et al., 2008). These cellular aggregates have been shown to consist of numerous ubiquitylated proteins that can no longer be efficiently disposed of by either the ubiquitin-proteasome system (UPS) or autophagic pathways (Johnston et al., 1998). Aggregated proteins can form inclusion bodies directly through self-assembly of non-native monomers. Alternatively, these oligomeric proteins seed throughout the cytoplasm and grow into small aggregates, which are subsequently delivered through dynein-mediated retrograde transport on microtubules to the microtubule-organizing center (MTOC). Once located at the MTOC, these juxtanuclear protein deposits are termed aggresomes (Kopito, 2000). It is generally accepted that aggresome formation is a protective cellular response for segregating misfolded proteins from the cytoplasmic environment and enhancing the degradation of these toxic protein aggregates (Kawaguchi et al., 2003; Hideshima et al., 2005). Disruption of the microtubule network or dynein motor function prevents aggresome formation, leading to the appearance of dispersed foci of misfolded protein aggregates throughout the cytoplasm (García-Mata et al., 1999). Disruption of either autophagy or an efficient dynein dependent retrograde transport system results in a buildup of aggregated proteins and dysfunctional organelles in the cell which is a driving feature of neurodegeneration (Hafezparast et al., 2003; Hara et al., 2006).
Owing to its ability to bind either directly to the proteasome, through its PB1 domain or to the autophagic precursor LC3, through its LIR domain, the multimeric protein SQSTM1 (also known as p62) has been implicated in protein degradation pathways. Recently, SQSTM1 has also been implicated in mitochondrial homeostasis (Seibenhener et al., 2013) and microtubule network equilibrium (Yan et al., 2013). Importantly, substantial evidence indicates that SQSTM1 levels are directly associated with protein aggregation in multiple neurodegenerative diseases (Kuusisto et al., 2001; Zatloukal et al., 2002; Nagaoka et al., 2004; Mizuno et al., 2006). Aggregates analyzed in plaques from brains of Alzheimer's disease patients or in Lewy bodies from Parkinson's disease samples contain SQSTM1 as a major constituent (Kuusisto et al., 2001; Nakaso et al., 2004). Removal of SQSTM1 results in increased aggregate formation in the cell, whereas overexpression of SQSTM1 results in increased aggregate clearance (Paine et al., 2005) leading to the proposal that SQSTM1 can function in garbage packing for the eventual disposal of toxic proteins (Moscat and Diaz-Meco, 2009). SQSTM1 is also involved in the elimination of defective mitochondria through modulation of Parkin- and PINK1-mediated mitochondrial clearance (Geisler et al., 2010). Taken together, it is clear that SQSTM1 is involved in multiple cellular events including cellular aggregation, dysfunctional organelle removal and neurodegenerative-disease-related cellular responses.
The potential for an overlap in function between SQSTM1 and dynein is intriguing. However, there has never been evidence of direct interaction between the two proteins reported despite the existence of circumstantial links. BAG3, a member of the Bcl-2-associated athanogen family, associates with the members of the HSP70 chaperone family to sequester misfolded proteins to autophagosomes in a SQSTM1-dependent manner that is reliant on dynein motor complexes for transport along microtubules (Behl, 2011). In addition, microtubule remodeling is associated with ATG12 colocalization with SQSTM1 in autolysosomes in a dynactin–dynein-dependent manner (Nunes et al., 2013). Although these studies place SQSTM1 and dynein in juxtaposition to each other during protein clearance, no study to date has addressed the potential for direct interaction between SQSTM1 and dynein.
With this in mind, we wished to investigate whether SQSTM1 and dynein interact themselves and whether SQSTM1 can somehow affect the normal function of dynein. Our results indicate that SQSTM1 is involved in, but is not solely responsible nor necessary for, formation of a complex between aggregated proteins and dynein. We report that SQSTM1 does, however, play a newly identified role in dynein function and motility of dynein cargoes.
RESULTS
SQSTM1 interacts with dynein
Aggresome formation is regulated by the motor protein dynein (Ouyang et al., 2012) with aggregated proteins being transported along microtubules to juxtanuclear positions for autophagic clearance. Two dynein subunits, intermediate chains 1 and 2 (DIC1 and DIC2, also known as DYNC1I1 and DYNC1I2) have been implicated in dynein binding to both aggregated proteins (Xu et al., 2013) and cellular organelles (Pfister et al., 2006; Mitchell et al., 2012) for retrograde transport to aggresomes. The adaptor protein SQSTM1 (Okatsu et al., 2010) has also been implicated in aggresome formation by contributing to the transport of both ubiquitylated (Behl, 2011) and non-ubiquitylated proteins (Watanabe and Tanaka, 2011). As these two proteins are both intimately involved in protein trafficking of ubiquitylated cargoes, we reasoned that some form of direct or indirect interaction was plausible.
To test for a general interaction, human embryonic kidney (HEK) cells were transfected with Myc-tagged SQSTM1, following which DICs were immunoprecipitated. The exogenous protein was found to associate with endogenous DICs in transfected cells (Fig. 1A). Wild-type (WT) and p62KO (i.e. SQSTM1-null) mouse embryonic fibroblast (MEF) cells were then immunoprecipitated for DICs and endogenous protein interactions examined. Misfolding protein stress was induced in the cell by MG132 treatment and detected by immunoblotting with antibody against K63-linked ubiquitin (K63Ub). SQSTM1 has been shown to provide a platform for predominantly K63-linked ubiquitylation to occur (Ramesh Babu et al., 2008). Endogenous SQSTM1 was seen to associate with DICs at basal levels. However, this association significantly increased when misfolding stress was applied to the cells (Fig. 1B). It should be noted that there was an increase in the amount of SQSTM1 protein in the cell after treatment with MG132; however, the association of this larger pool of protein with DICs was more than the overall increase observed in cell lysates, indicating a drive towards protein–protein interaction and not simply an increased basal amount of available protein. Levels of K63Ub-tagged proteins associated with DICs increased slightly in transgenic knockout cells, indicative of the increased stress induced by lack of SQSTM1 and that misfolded proteins can interact with the dynein motor in the absence of SQSTM1. In addition, the interaction between K63-ubiquitylated protein and DICs increased with MG132 treatment in a manner independent of SQSTM1. Both WT and p62KO cells showed a similar increase in K63Ub interaction with DICs upon the application of cellular stress.
Fig. 1.
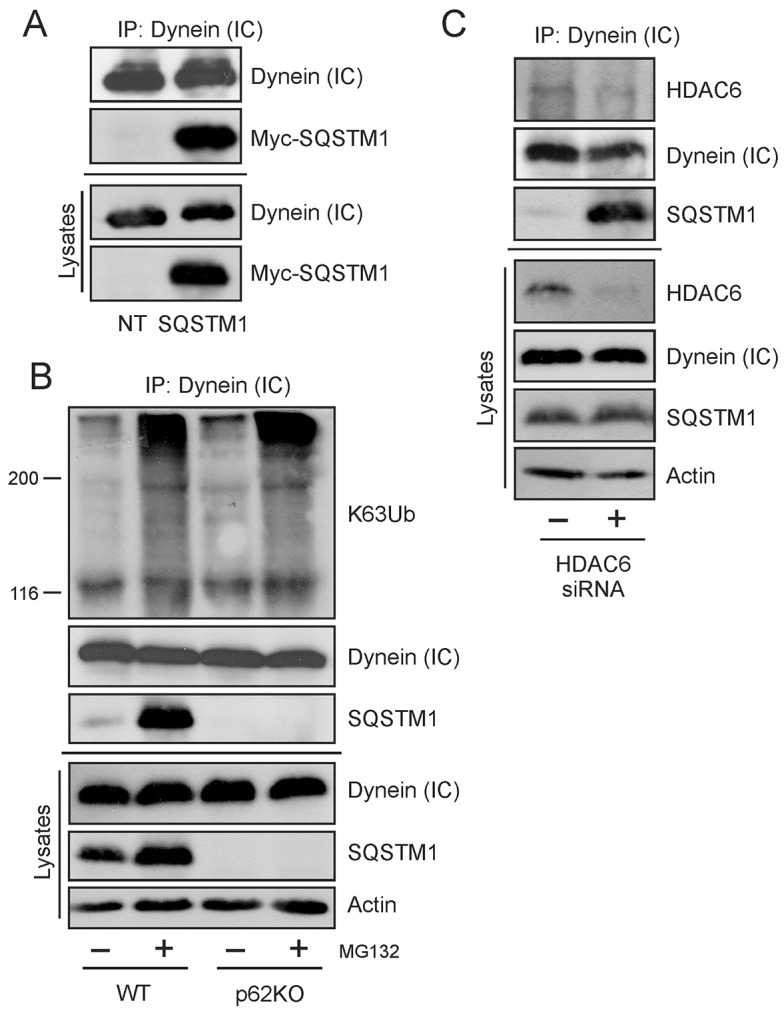
SQSTM1 interacts with dynein in a manner independent of HDAC6. (A) Exogenous Myc-tagged SQSTM1 was transfected into HEK cells and immunoprecipitated (IP) with endogenous DICs [dynein (IC)]. NT, non transfected cells. (B) Endogenous SQSTM1 immunoprecipitates with DICs in MEF cells but is not required for the interaction of K63Ub-tagged aggregates with the motor protein. The interaction of SQSTM1 with DICs increases under cellular misfolding stress induced by 5 µM MG132 for 6 h. (C) The endogenous interaction between SQSTM1 and DICs is not dependent on the presence of HDAC6. WT MEF cells had HDAC6 expression reduced by siRNA transfection resulting in increased SQSTM1 interaction with DICs.
SQSTM1 interaction with DICs is independent of HDAC6
It has been shown previously that the histone deacetylase HDAC6 directly interacts with dynein as an adaptor for loading of ubiquitylated cargos (Kawaguchi et al., 2003). We have recently demonstrated that HDAC6 also binds to SQSTM1 (Yan et al., 2013). We reasoned that because HDAC6 and SQSTM1 are both involved with dynein and ubiquitylated cargoes, that HDAC6 could be required for our observed SQSTM1–DIC association. If this were correct, we hypothesized that by removing HDAC6, we would see a corresponding decrease in SQSTM1 interaction with DICs. HDAC6 protein expression was knocked down in WT MEF cells using specific HDAC6 siRNA. However, our results were opposite of expectations. Knockdown of HDAC6 actually resulted in an increased association of SQSTM1 with dynein motor protein, even in the absence of induced cellular stress (Fig. 1C). These results provided strong evidence that when HDAC6 was reduced or absent, the interaction between SQSTM1 and DIC actually increased, suggesting that there is competition between the two proteins for binding to DICs (i.e. that they share an overlapping binding site on DICs). We concluded from these results that the SQSTM1–DIC interaction we observed was not dependent on HDAC6 and could thus be a direct interaction.
Identification of a specific binding domain in SQSTM1 that interacts with dynein
As SQSTM1 can interact with DIC in an HDAC6-independent fashion, we next hypothesized that this interaction could be by specific binding of the two proteins. To examine this possibility and to potentially map the location of any interaction site, SQSTM1 HA-tagged deletion constructs (Fig. 2A) were transfected into HEK cells and DIC was immunoprecipitated. Specific interaction of exogenous SQSTM1 constructs with DIC was observed only when the region between amino acids 163 and 229 of SQSTM1 was present in the construct (Fig. 2B). This is suggestive of a possible dynein-interacting domain located between the ZZ domain and the TRAF6-binding domain of SQSTM1.
Fig. 2.
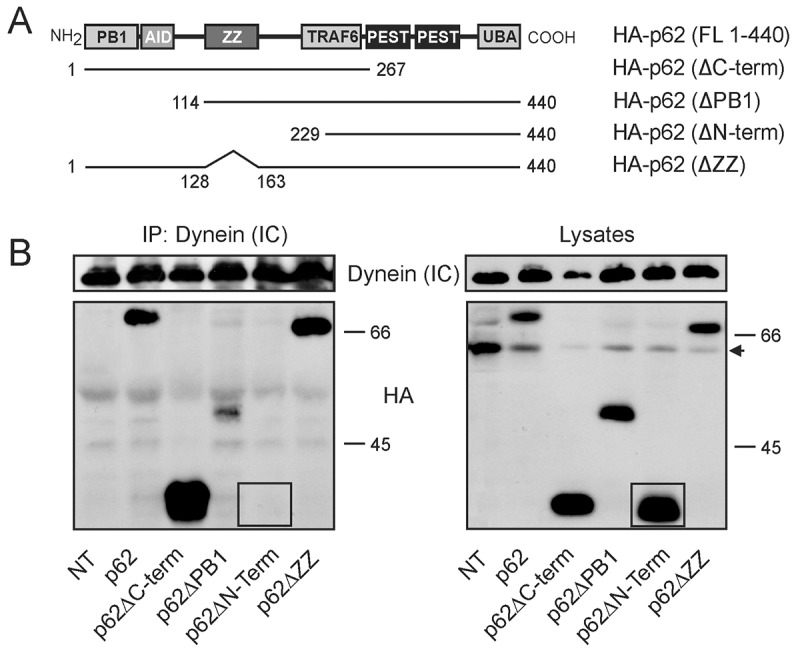
Mapping and identification of a domain in SQSTM1 required for dynein interaction. (A) HA-tagged expression constructs used for mapping the domains of SQSTM1 that interacted with DICs [dynein (IC)]. (B) Exogenous deletion constructs of SQSTM1 were transfected into HEK cells followed by immunoprecipitation with antibody to endogenous dynein motor protein. The amino acids between 163 and 229 of SQSTM1 are required for interaction of SQSTM1 with dynein. NT, non-transfected cells; the arrow points to a non-specific protein band in HEK lysates that interacts with the HA antibody and should not be considered in interpretation as this band does not show up in immunoprecipitates. Absence of p62ΔN-term interaction with DICs is indicated by comparing the corresponding regions (inside box) in each gel.
A random overlapping design was used to identify possible interaction sites within this defined dynein binding area in SQSTM1. Peptides of approximate 15-mer length were generated with a TAT-conjugated N-terminal sequence to aid in cell permeability (Nakielny and Dreyfuss, 1999; Pan et al., 2012) and used in an in vitro GST-pulldown assay to assess their ability to interfere with direct SQSTM1–DIC interaction (M.L.S., unpublished data). A potential binding domain was located in the peptide (E177GFSHSRWLRKVKHGHF194). Addition of this peptide in a pulldown assay prevented the interaction of agarose-bound GST-tagged SQSTM1 with DICs from HEK whole-cell lysates. Interestingly, dynein interaction with SQSTM1 was specifically inhibited, whereas the interaction with the motor protein kinesin was unaffected (Fig. 3A).
Fig. 3.

The specific interaction between SQSTM1 and dynein is inhibited by peptide to a newly identified interaction domain in SQSTM1. (A) The in vitro interaction between SQSTM1 and DICs [dynein (IC)] was examined by a GST-pulldown assay. Interaction between GST–SQSTM1 and DICs was specifically inhibited by a DIC peptide against a potential dynein-binding domain of SQSTM1 located at amino acids 163–229. Increasing peptide concentrations added to the pulldown assay were 0 µM, 50 µM and 100 µM. GST–SQSTM1 addition to the assay was assessed by using a GST-specific antibody, and equal lysate addition to the assay is indicated by actin staining. (B) The interaction between GST–SQSTM1 and purified DICs was examined by an in vitro assay. Purified DIC1 and DIC2 were incubated with GST–SQSTM1 in the absence or presence of either inhibitory peptide (50 µM, Dyn pep) or random peptide (50 µM, Ran pep) and binding assessed with the anti-DIC antibody. Inhibitory peptide prevented the interaction between DIC1 or DIC2 and GST–SQSTM1, whereas the random peptide had no effect. Specificity of DIC interaction with SQSTM1 is shown by the lack of binding of DIC to glutathione beads themselves. (C) In vivo interaction between SQSTM1 and dynein was examined by immunoprecipitation with the anti-DIC antibody. Addition of the cell-permeable DIC peptide against the dynein-binding domain of SQSTM1 to WT MEF cells in culture prior to immunoprecipitation resulted in inhibition of the interaction between SQSTM1 and dynein. Peptide concentrations were the same as indicated in A.
To confirm the specificity of the peptide to inhibit the dynein–SQSTM1 interaction, it was necessary to eliminate the availability of any co-binding factors present in the cell lysate. Agarose-bound bacterially expressed GST-tagged SQSTM1 was allowed to interact with commercially available purified DIC1 and DIC2 (Fig. 3B). GST–SQSTM1 bound both DIC1 and DIC2 in an in vitro assay. The binding domain inhibitory peptide was successful in preventing the binding of purified DICs with GST–SQSTM1, whereas a random peptide of equal length did not abrogate DIC association with GST–SQSTM1. It should also be noted that DIC2 appeared to show a greater level of interaction with GST–SQSTM1 compared to DIC1. Whether this indicates a preferred SQSTM1-binding site in DIC2 remains to be investigated. In summary, this is the first evidence for direct binding of DICs to SQSTM1.
The cell-permeable peptide was then added to WT MEF cells in culture to examine potential in vivo effects. Cells were treated with MG132 to initiate misfolding protein stress and proteins associated with DICs were immunoprecipitated. An increased concentration of competitive peptide decreased the amount of SQSTM1 that interacted with DICs (Fig. 3B) at the same concentration (100 µM) seen in the in vitro assay. Taken together, these data support the conclusion that SQSTM1 is capable of binding directly with DICs both in vitro and in vivo.
Dynein motor protein function is impaired in the absence of SQSTM1
Having established that SQSTM1 can bind directly with DIC, we next examined what effect, if any, this interaction might have on dynein motor transport function. K63-linked polyubiquitylation of misfolded proteins and their subsequent transport to aggresomes for degradation is a dynein-dependent process (Olzmann et al., 2007; Olzmann and Chin, 2008). Thus, we reasoned that we could examine the localization of K63-ubiquitylated substrates with dynein in the presence or absence of SQSTM1 to determine whether SQSTM1 was important for dynein functionality. WT and p62KO MEF cells were treated with MG132 for 6 h to allow misfolded proteins to accumulate, or with a combination of MG132 after prior treatment with erythro-9-[3-(2-hydroxynonyl)] adenine (EHNA) to disrupt ATPase-activity-dependent movement with dynein of K63-polyubiquitylated misfolded proteins. Cells were subsequently analyzed by immunofluorescence for colocalization of DICs and K63Ub-marked proteins (Fig. 4). By using WT and p62KO cells, we were able to specifically look at what role SQSTM1 plays in this process. MG132 treatment resulted in increased colocalization of DICs and K63Ub (seen as yellow cytoplasmic aggregates in merged images) in a SQSTM1-independent manner. Colocalized K63Ub–dynein aggregates (Fig. 4, yellow arrows indicating colocalized Oregon-Green-labeled dynein and Texas-Red-labeled K63Ub) could be seen to increase around the perinuclear region in WT cells upon misfolding stress with functional dynein trafficking. However, when dynein ATPase activity was disrupted by EHNA treatment, colocalized aggregates of dynein and K63Ub were more prevalently localized in the cytoplasm. Lack of SQSTM1 resulted in increased cytoplasmic colocalization of K63Ub-tagged proteins with dynein motor proteins above those basal levels seen in WT cells. Treatment with stress inducers showed no great difference between WT and p62KO cells. Importantly, we did observe that misfolding stress resulted in an increase in vesicular structures, exemplified by their ovoid morphology (Fig. 4, white arrows) throughout the treated cells. Importantly, these structures were obvious in untreated p62KO cells, which are already under cellular stress owing to the lack of SQSTM1 (Ramesh Babu et al., 2008; Rodriguez et al., 2006).
Fig. 4.

Dynein motor protein trafficking of aggregated proteins is impaired in the absence of SQSTM1. WT or p62KO MEF cells were immunostained for endogenous DICs [dynein (IC)] (Oregon Green) and K63-polyubiquitin (K63Ub, Texas Red) and imaged using confocal microscopy. Protein misfolding stress was induced with MG132 (5 µM, 6 h) and dynein ATPase activity inhibited with EHNA treatment (500 µM, 8 h). Colocalization is indicated by yellow arrows in inset images. Vesicle structures are indicated by white arrows. A total of 20 individual cells from each treatment condition were used to measure vesicle localization (Table 1). Scale bars: 10 µm.
We also noted that localization of these vesicles (Fig. 4, white arrows) was dependent not only on the presence of SQSTM1, but also on the functionality of dynein. In untreated WT MEF cells vesicles were scarce; however, an increase in cytoplasmic vesicles was seen in the absence of SQSTM1. When the distance of these structures from the nucleus of the cell was measured in p62KO cells, vesicles were localized significantly further from the nuclear envelope (5.21±0.43 µm) compared to in unstressed WT cells (3.45±0.39 µm; Student's _t_-test = 4.29, P<0.05, mean±s.e.m.) (Table 1). MG132 treatment resulted in perinuclear localization of WT vesicles (1.26±0.24 µm) consistent with the operation of a functional retrograde transport system, whereas the absence of SQSTM1 caused vesicles to remain in the cytoplasm of the cell at a distance substantially away from the nucleus (6.96±0.98 µm). Treatment with EHNA prior to MG132 resulted in blockage of vesicle movement to the perinuclear region in WT (4.63±0.41 µm), whereas there was no change from MG132 treatment alone in p62KO cells (5.48±0.58 µm) (Table 1).
Table 1. Vesicle distances measured from the nuclear membrane.
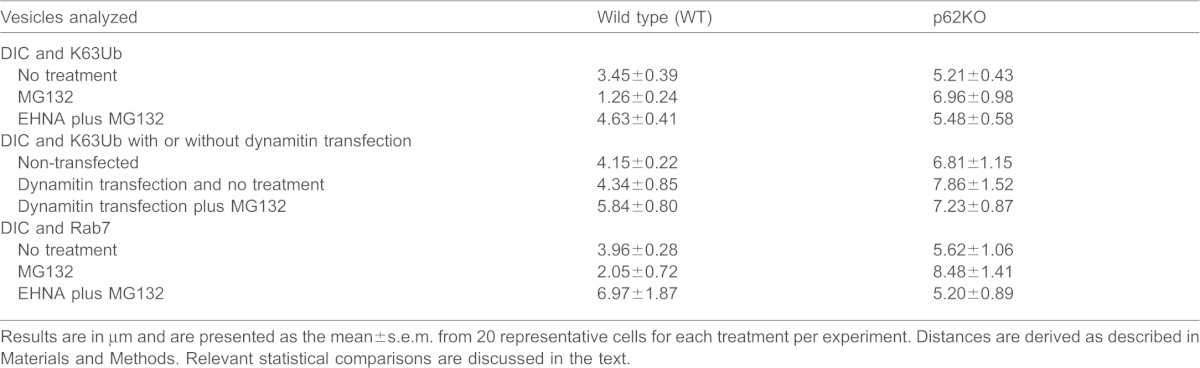
To confirm that the blockage of vesicle movement was indeed the result of dynein inhibition, WT or p62KO MEF cells were transfected with GFP–p50/dynamitin (also known as dynactin subunit 2, DCTN2) (Fig. 5). Overexpression of the dynactin subunit p50/dynamitin results in disruption of the dynactin complex thereby inhibiting dynein motility (Burkhardt et al., 1997; Engelke et al., 2011). When p50/dynamitin was expressed in WT cells, there was a marked visual increase in vesicle formation scattered throughout the cytoplasm over what was seen in non-transfected WT cells. The prevalence and distance of these vesicles from the nuclear envelope (4.34±0.85 µm) was consistent with that reported above in p62KO cells (6.81±1.15 µm). When treated with MG132, there was no substantial difference in number and localization (5.84±0.8 µm) compared to untreated cells. p62KO MEF cells overexpressing p50/dynamitin showed no change in vesicle localization (7.86±1.52 mm) compared to non-transfected p62KO cells (6.81±1.15 µm; Fig. 5, white arrows point to representative vesicles) (Table 1). Vesicles were still predominantly localized in the cytoplasmic compartment of the cell. Treatment of transfected cells with MG132, to allow accumulation of misfolded proteins, again showed no difference (7.23±0.87 µm) to cells overexpressing p50/dynamitin alone. Vesicles were still predominantly localized away from the perinuclear region and in the cytoplasm of the cells.
Fig. 5.

Dynein motor protein trafficking of aggregated proteins is inhibited by overexpression of GFP–p50/dynamitin. WT or p62KO MEF cells were transfected with GFP-tagged p50/dynamitin, and endogenous DICs [dynein (IC)] (Cascade Blue) and K63-ubiquitylated proteins (K63Ub, Texas Red) localization were visualized by confocal microscopy. Protein misfolding stress was induced with MG132 (5 µM, 6 h). Vesicle structures are indicated by white arrows. A total of 20 individual cells from each treatment condition were used to measure vesicle localization (Table 1). Scale bars: 10 µm.
In summary, K63-ubiquitylated proteins localized with dynein effectively in the presence or absence of SQSTM1, indicating that SQSTM1 is not required for loading misfolded cargoes to dynein motor proteins. However, localization of vesicle structures away from perinuclear aggresomes in the absence of SQSTM1 clearly indicates a requirement for SQSTM1 in dynein motility.
Dynein transport of late endosomes along degradative pathways is impaired in the absence of SQSTM1
DICs have been implicated in the retrograde transport of Trk-containing signaling endosomes (Mitchell et al., 2012). The basic endosomal maturation program begins with endocytosis of cargo from the plasma membrane into endocytic vesicles, which then fuse into early endosomes (Huotari and Helenius, 2011). These are now recognized as the main sorting facility of the endocytic pathway, and individually have a complex tubular and vacuolar morphology (Zerial and MacBride, 2001). Late endosomes are derived from the vacuolar regions of early endosomes, through a not completely defined molecular sorting process involving domain switching and under the direction of Rab GTPases (Rink et al., 2005; Vonderheit and Helenius, 2005). Prior to dissociating from the maturing endosome, the early endosome marker Rab5 is thought to recruit Rab7. Once the Rab7 domain is generated and Rab5 is lost, the mature late endosome is trafficked to the perinuclear region where it fuses with lysosomes in a degradative pathway (Eskelinin, 2008; Kinchen and Ravichandran, 2008; Kerr and Teasdale, 2009).
Mature late endosomes are typically round or oval in morphology (Huotari and Helenius, 2011). Based on the round and oval morphology of the vesicles observed in our MEF cells and the dependence on functional dynein for perinuclear localization, we reasoned that they could be mature late endosomes. Using immunofluorescence, we sought to determine whether the absence of SQSTM1 affected the retrograde transport of late endosomes by assessing whether the late endosome marker Rab7 colocalized with DICs. WT and p62KO MEF cells were treated with MG132, with or without preceding EHNA treatment as described previously, and cells were analyzed by confocal microscopy (Fig. 6). Untreated WT cells showed very little colocalization of Rab7 with DIC and, as seen previously with K63Ub and dynein staining, a marked lack of vesicle formation. Upon MG132 treatment, vesicles (Fig. 6, white arrows) were noticeably present in the perinuclear region (2.05±0.72 µm) as was colocalization of Rab7 with DIC (yellow in the images), indicating that these vesicles are indeed late endosomes. When the ATPase-dependent dynein function was inhibited by EHNA treatment, colocalization still occurred, however, perinuclear localization was abrogated (6.97±1.87 µm), in agreement with our observed results with dynein and K63Ub staining. When SQSTM1 was absent, localization of Rab7 and dynein still occurred. This indicates that cargo loading, in this case late endosomes, is still functional and, thus, SQSTM1 is not required for dynein–cargo association. Late endosomes were prevalent in knockout cells, however, they remained mostly cytoplasmic in their localization with or without cellular misfolding stress being applied (no treatment, 5.62±1.06 µm; MG132, 8.48±1.41 µm; EHNA and MG132, 5.2±0.89 µm) (Table 1). Thus, as seen with aggregated proteins, late endosome trafficking to the perinuclear region for fusion with lysosomes in the endocytic maturation pathway relies on the presence of SQSTM1.
Fig. 6.

Dynein motor protein trafficking of Rab7-positive late endosomes is impaired in the absence of SQSTM1. WT or p62KO MEF cells were immunostained for endogenous DICs [dynein (IC)] (Oregon Green) and Rab7 (Texas Red) and imaged using confocal microscopy. Protein misfolding stress was induced with MG132 (5 µM, 6 h) and dynein function inhibited with EHNA treatment (500 µM, 8 h). Colocalization at vesicular structures is indicated by white arrows. A total of 20 individual cells from each treatment condition were used to measure vesicle localization (Table 1). Scale bars: 10 µm.
Expression of SQSTM1 in the null background restores dynein retrograde transport
We have shown that the interaction of dynein with both K63Ub (Figs 4 and 5) and Rab7 at the late endosome (Fig. 6) is unaffected by the absence of SQSTM1, whereas dynein motor function related to transport of both protein aggregates and late endosomes is inhibited. To further investigate this observation, we used the SOD1 A4V mutant to analyze dynein motility. Wild-type SOD1 does not interact with dynein, whereas the A4V-mutant–dynein interaction contributes to motor neuron cell death associated with amyotrophic lateral sclerosis (ALS) (Ström et al., 2008; Tang et al., 2012). We have shown that removal of SQSTM1 from the system results in disruption of the retrograde function of the dynein motor protein. To further support this hypothesis, we wished to return SQSTM1 to the knockout background and show restoration of dynein motility. Thus, GFP-tagged SQSTM1 was introduced into WT and p62KO cells along with the FLAG-tagged SOD1 A4V aggregation mutant to see whether expression of SQSTM1 could restore the retrograde function of dynein for known aggregation-prone cargoes.
In untreated WT or p62KO MEF cells, colocalization of DICs with the SOD1 A4V mutant was minimal. However, the vesicles we have identified above as late endosomes could still be seen in the cytoplasm of the knockout cells (Fig. 7, yellow arrows). When SQSTM1 was overexpressed in WT cells, colocalization of dynein–SQSTM1–SOD1-A4V complexes was observed throughout the cell, but notably, large aggregated complexes were found at perinuclear locations (Fig. 7, white arrows). Induction of misfolded protein stress by MG132 treatment increased the amount of the complex, which was mainly located in regions around the nucleus of the cell (Fig. 7). It is also worth noting that perinuclear vesicles were again seen with misfolding stress and were localized perinuclearly. Overexpression of SQSTM1 resulted in large protein aggregates throughout the cell, presumably because there was excess SQSTM1 available to contribute to the formation of cellular misfolded protein aggregates. In p62KO cells, where SQSTM1 was restored to the null-background, localization of dynein–SQSTM1–SOD1-A4V complexes was readily seen at perinuclear locations (Fig. 7, white arrows). Importantly, the presence of late endosome vesicles was markedly reduced compared to their presence in previous experiments (Figs 5 and 6) and those vesicles still present (Fig. 7, yellow arrows) were all now located in the perinuclear region. These results indicate that dynein motility is restored when SQSTM1 is expressed. MG132 treatment of p62KO cells expressing GFP–SQSTM1 caused the same large protein aggregate formation as seen in overexpressing WT cells, as well as perinuclear localization of late endosome vesicles. Restoring expression of SQSTM1 to the null background rescued the trafficking-deficient phenotype of p62KO cells resulting in a phenotype that could not be visually differentiated from WT overexpressing cells.
Fig. 7.

Introduction of SQSTM1 into a null background restores the colocalization of mutant SOD1 A4V and dynein and facilitates aggregate and endosomal trafficking to the perinuclear region. WT and p62KO cells transfected with GFP–SQSTM1 and SOD1 A4V (SODmut) prior to immunostaining for DICs [dynein (IC)] (Cascade Blue) and exogenously expressed SOD1 A4V mutant (Texas Red). Protein misfolding stress was induced with MG132 (5 µM, 6 h). Colocalization of dynein–SQSTM1–SOD1-A4V complexes (white in the merged image) can be seen localized around the nucleus of the cell (indicated by white arrows). Endosomal vesicles are indicated by yellow arrows and are predominantly perinuclear in WT and p62KO MEF cells. Scale bars: 10 µm.
Taken together, these results indicate that SQSTM1 and DIC functionally associate in MEF cells. By examining dynein cargo transport paradigms, we have shown that SQSTM1 plays an as yet undefined role in the functional working of the dynein motor protein. Although cargo loading does not appear to be significantly inhibited by the loss of SQSTM1, transport of three different dynein cargos to the perinuclear region of the cell along degradative pathways is inhibited by the absence of SQSTM1. Highlighted here is that restoration of SQSTM1 expression to a null background restores dynein functionality and defines a new role for SQSTM1 in the cell.
DISCUSSION
Evidence implicating dynein dysfunction in multiple neurodegenerative diseases is accumulating. However, in most cases, this evidence is largely indirect (Eschbach and Dupuis, 2011). The accumulation of aggregated, ubiquitylated and misfolded proteins is a hallmark of cellular pathology associated with neurodegenerative disease and represents a general response of eukaryotic cells to the presence of misfolded and non-degraded proteins. Dynein dysfunction leads to defective retrograde transport resulting in decreased mitochondrial transport along axons back to the cell body (Hollenbeck and Saxton, 2005) and decreased signaling endosome transport along microtubules (Rishal and Fainzilber, 2010). Decreased dynein functionality also leads to the accumulation of amyloid precursor protein (APP), as well as amyloid β (Aβ) peptide, which is associated with Alzheimer's disease (Cataldo et al., 2000; Driskell et al., 2007). Thus dynein is involved in multiple pathways that lead to abnormal neuronal physiology as a result of incomplete processing of misfolded proteins and defective organelles.
Late endosome trafficking along microtubules, which is mediated by dynein motors, from distal processes to the soma of neurons is required for their maintenance and survival (Cai et al., 2010), and dynein mutation and dysfunction leads to prominent neurodegeneration associated with lack of endosome maturation (Hafezparast et al., 2003; Komatsu et al., 2006). Previous studies strongly support the hypothesis that SQSTM1 is intimately linked to and is a key modulator of neurodegeneration (Bjørkøy et al., 2009; Du et al., 2009). Maturation and clearance of late endosomes depends on proper dynein function. Inhibiting the ATPase activity of dynein leads to a buildup of late endosomes in the cytoplasm of MEFs. As we report here, absence of SQSTM1 leads to an identical phenotypic buildup of late endosomes in the cytoplasm of the cell. Reintroduction of SQSTM1 into knockout cells rescues the defective phenotype, indicating a crucial role for SQSTM1 in completing the maturing and degradative endocytic pathway. Thus, SQSTM1 is integral to dynein functionality. Based on the collective results of our study, we propose a model where SQSTM1 directly interacts with dynein, not affecting its cargo-binding capability, but instead modulating the ATPase-dependent trafficking of the motor protein along microtubules to the degradative centers around the nucleus.
The ability of dynein ability to transport substrates is driven by hydrolysis of ATP in the globular heads of dynein heavy chains (Kon et al., 2005). We have shown here, with multiple cargoes, that inhibition of the ATPase activity of dynein results in abrogated aggregate and vesicle trafficking. It is thus reasonable that dynein motor complex movement is affected in the absence of SQSTM1 because loss of SQSTM1 leads to decreased ATP levels in MEF cells (Seibenhener et al., 2013). Therefore, dynein motor complex processivity could be disrupted by the decreased levels of ATP available in the cell. Under this hypothesis, SQSTM1 could not only play a direct role in transport by interacting with dynein and dynein cargoes, but also an important indirect role because of its documented influence on ATP production by mitochondria (Seibenhener et al., 2013).
HDAC6 and SQSTM1 have both been identified as components of functional aggresomes. HDAC6 binds ubiquitin chains, with a preference for K63-polyubiquitin, through its C-terminal BUZ domain. Either polyubiquitylated substrates or free ubiquitin chains can interact with HDAC6 (Ouyang et al., 2012). However, in either case, once associated with HDAC6, the polyubiquitylated proteins are relocated to aggresomes through the interaction of HDAC6 with the motor protein dynein. Loss of HDAC6 has been reported to result in the failure to colocalize aggregated proteins to the aggresome for degradation leading to aggregate buildup, neurodegeneration (Ju et al., 2008; Lee et al., 2010) and inclusion body formation (Guthrie and Kraemer, 2011; Richter-Landsberg and Leyk, 2013). SQSTM1 also binds ubiquitylated substrates, again with a preference for K63-polyubiquitin, through its C-terminal UBA domain. SQSTM1 interacts with the autophagy protein LC3 and is required for autophagosome formation (Pankiv et al., 2007). In the absence of SQSTM1, autophagic clearance of ubiquitylated substrates is impaired and inclusion bodies are formed throughout the cytoplasm (Wooten et al., 2008). Lack of SQSTM1 also leads to abrogation of polyubiquitylated protein trafficking to the proteasome for degradation (Seibenhener et al., 2004). Thus, both proteins appear to be involved in the accumulation and efficient disposal of polyubiquitylated protein aggregates.
In a recent manuscript (Yan et al., 2013), we reported the existence of a direct interaction between SQSTM1 and HDAC6. Loss of SQSTM1 results in hyperactivation of HDAC6 and deacetylation of α-tubulin and cortactin (Yan et al., 2013), an integral component of F-actin-remodeling assemblies in quality control autophagy (Lee et al., 2010). Tubulin acetylation levels are becoming recognized as regulators of microtubule function affecting processes from cell motility to intracellular trafficking (Perdiz et al., 2011). Here, we expand on this previous work and identify SQSTM1 as not only a regulator of HDAC6-associated microtubule acetylation, but also an dynein-interacting protein involved in trafficking of misfolded proteins and late endosomes to perinuclear degradation centers. An intriguing question is what is the precise arrangement of this dynein–SQSTM1–HDAC6 complex? We outline two potential interaction models in Fig. 8.
Fig. 8.
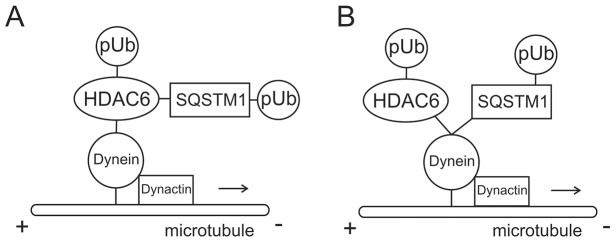
Proposed models for SQSTM1 interaction with both HDAC6 and dynein. (A) Model for SQSTM1 interaction with HDAC6 and polyubiquitylated cargoes (pUb) with dynein motor complex. HDAC6 is the linker between SQSTM1 and the dynein motor protein. Both HDAC6 and SQSTM1 attach polyubiquitylated cargo to dynein for trafficking. (B) Proposed and favored model for SQSTM1 and HDAC6 competitive direct interaction with dynein motor complex. Both SQSTM1 and HDAC6 associate with polyubiquitylated aggregated proteins (or organelles) and compete for a binding site(s) on with DICs. Dynein function is dependent on the presence of SQSTM1.
Reciprocal interaction domains found in both SQSTM1 and HDAC6 indicate obvious interaction between these two proteins. Based on current literature (Kawaguchi et al., 2003; Watabe and Nakaki, 2011), it was natural to conclude that HDAC6 was the dynein-interacting protein of the complex because a dynein-binding domain has previously been identified (Kawaguchi et al., 2003). Under this model SQSTM1 binds directly to HDAC6 with both being capable of interactions with aggregated polyubiquitylated proteins or other degradative cargoes such as late endosomes (Fig. 8A). HDAC6 could then bind with dynein, leading to retrograde transport of cargoes to perinuclear degradation centers for disposal. This is consistent with our observation of reduced microtubule transport efficiency. It should also be noted that the absence of SQSTM1 resulted in an increase in the deacetylation activity of HDAC6 (Yan et al., 2013). The dynamic interplay between acetylation and deacetylation of tubulin subunits of the microtubule has been suggested as a mechanism for motor protein movement along the microtubule (Reed et al., 2006; Dompierre et al., 2007). It has also been argued that the recruitment of motor proteins to microtubules, leading to vesicular transport, is enhanced by tubulin acetylation (Reed et al., 2006). This suggests that inhibiting HDAC6 deacetylase activity restores the MT-dependent transport mechanisms in neurons by increasing the recruitment of kinesin and dynein and/or dynactin to the more acetylated microtubules (Dompierre et al., 2007). Therefore, dynein transport could be disrupted in the absence of SQSTM1 because HDAC6 activity is increased, resulting in reduced acetylation of the microtubule network and preventing the recruitment of the motor protein complex. This would suggest that SQSTM1 could play a role in motor protein processivity by preventing the deacetylation of microtubules by HDAC6. Absence of SQSTM1 results in hypo-acetylation of α-tubulin as HDAC6 activity is unregulated and could result in the motor protein mobility along the microtubule being impaired.
Although a mechanism including aggregated proteins and SQSTM1 binding to HDAC6 and then that complex interacting with dynein for transport is plausible, our data here support an alternative model (Fig. 8B). Based on our results, we favor a model where SQSTM1 can itself interact with the dynein motor protein, not as just a linker to polyubiquitylated cargos and cellular vesicles, but as an associating protein. Our results identify for the first time a dynein-binding domain in SQSTM1 and show specific interaction between these two proteins. Furthermore, it appears that SQSTM1 plays a key role in controlling dynein specific motility. When SQSTM1 is absent from embryonic fibroblasts, endogenous transport of late endosomes and aggregates to aggresomes localized around the juxtanuclear MTOC is impaired. In further support of this, the A4V SOD1 mutant specifically interacts with the dynein motor protein complex for transport to the aggresome, where it colocalizes with γ-tubulin, an MTOC marker (Ström et al., 2008). In p62KO MEF cells, interaction of this mutant protein with dynein was evident (Fig. 7). However, transport to the perinuclear MTOC region was impaired in the absence of SQSTM1, further strengthening the hypothesis that SQSTM1 is important for dynein functionality. As we have described above, this lack of dynein function could be attributed to decreased ATP levels present in the cell. However, we cannot discount other possibilities for SQSTM1 to control dynein function. What we can support, is that when SQSTM1 is introduced into the knockout null background, mutant SOD1 protein is again trafficked and localized with dynein to the perinuclear MTOC region (Fig. 7). We can also identify ultrastructure late-endosome-like vesicles localized to this same region when SQSTM1 expression is restored. We conclude from these results that SQSTM1 is an important interaction protein for the dynein motor complex and is required for dynein function.
Phosphorylation of dynein subunits has also been implicated in dynein regulation (Dillman and Pfister, 1994; Lin et al., 1994; Vaughan et al., 2001), with some examples showing that phosphorylation can inhibit or enhance dynein binding to organelles or some cargoes (Mitchell et al., 2012). Atypical PKC (aPKC) can phosphorylate DICs, regulating their binding with paxillin, thus controlling the focal adhesion disassembly (Rosse et al., 2012). Given that SQSTM1 is an interacting partner of aPKC, providing a scaffold for phosphorylation to occur (Puls et al., 1997; Jiang et al., 2009), it is possible that, in the absence of SQSTM1, dynein motor function is disrupted owing to a lack of phosphorylation. However, this is speculative at this point as dynein motility has not been shown to require PKC phosphorylation. It does, however, open the door to interesting possibilities of motor protein complex functionality.
In reviewing all of the results from this study, we now propose a new model that defines a multifaceted association between SQSTM1, HDAC6 and dynein. It is clear that SQSTM1 and HDAC6 can both directly interact with the motor protein dynein (Fig. 7B). Interestingly, our data suggest that the interaction sites possibly overlap because knockdown of HDAC6 by siRNA resulted in increased SQSTM1 binding to dynein. Whereas SQSTM1 might indeed play a role in the recruitment of polyubiquitylated aggregates to the dynein motor complex, we have shown here that it is also required for dynein motility. Without SQSTM1, although dynein is still capable of interacting with cargoes for degradation, these cargoes are unable to traffic to degradation centers localized at the MTOC. Whether this is due to changes in the activity of HDAC6 or in the ability of the dynein motor complex itself to be active and move along the microtubule is the subject of further research. However, our findings provide insight into the mechanisms underlying dynein motility and suggest that SQSTM1 plays a newly defined role in clearance of misfolded proteins and cellular organelles.
MATERIALS AND METHODS
Reagents and Antibodies
Lipofectamine 2000 was purchased from Life Technologies (Carlsbald, CA). The Mammalian Transfection kit was from EMD-Millipore, Billerica, MA. INTERFERin reagent was from Polyplus (Berkley, CA). Antibodies were against the following proteins: DIC1/2 (i.e. against both the DICs), Rab7, Myc, HA, GFP and GST (Santa Cruz Biotechnology, Santa Cruz, CA); K63-ubiquitin and FLAG (Cell Signaling, Danvers, MA USA); actin (Sigma-Aldrich, St Louis, MO); SQSTM1 and HDAC6 (Abcam, Cambridge, MA); and kinesin (Chemicon, Temecula, CA). HDAC6 siRNA was from Santa Cruz Biotechnology. MG132 was from ENZO Lifesciences (Farmingdale, NY USA) and EHNA was from EMD-Millipore (San Diego, CA USA). Fluorescent secondary antibodies (conjugated to Cascade Blue, Texas Red and Oregon Green) were from Life Technologies (Carlsbad, CA). Synthetic peptide to the SQSTM1 sequence 177–194 was generated by Alpha Diagnostic International (San Antonio, TX). The GFP–p50/dynamitin construct was a generous gift of Richard Vallee, Columbia University, NY.
Cell culture and transfection
Human embryonic kidney (HEK) 293 cells from the American Type Culture Collection were grown as described previously (Wooten et al., 2005). Transfection was achieved using the Mammalian Cell Transfection Kit following the manufacturer's protocol. Wild type (WT) and p62-knockout (p62KO) mouse embryonic fibroblasts (MEFs) were derived from E13.5 mouse embryos (Durán et al., 2004) and grown in DMEM supplemented with 10% fetal calf serum and antibiotics at 37°C under high humidity and 5% CO2. Transfection of MEF cells was carried out using Lipofectamine 2000 (Life Technologies, Grand Island, NY). Where indicated, MEF cells were transfected with HDAC6 siRNA using the INTERFERin (Polyplus, New York, NY) reagent following the ‘insert’ instructions.
Immunoprecipitation and western blot analysis
Either HEK or MEF cells were lysed on tissue culture plates on ice with Triton lysis buffer (TLB; 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 10 mM NaF, 1 mM Na3VO4, 0.5% Triton X-100, 10 µg/ml leupeptin, 10 µg/ml aprotinin and 1 mM PMSF). Protein amounts were determined by using the Bradford assay. Equal amounts of whole-cell lysate (500 µg) were incubated with 3 µg of appropriate primary antibody for 3 h at 4°C. IgG conjugated to agarose beads was then added. Immunoprecipitation reactions were allowed to rotate overnight at 4°C. The immunoprecipitates were then washed five times with TLB. Proteins were released from agarose beads by boiling for 2 min in SDS-PAGE sample buffer and were then separated by 12% SDS-PAGE followed by transfer onto nitrocellulose membrane and western blot analysis with the corresponding antibodies. ECL solution (GE Healthcare, Piscataway, NJ) was used to visualize proteins.
GST–SQSTM1 pulldown assay
JM109 E. coli cells transformed with vector to GST-tagged full-length SQSTM1 were induced with isopropyl 1-thio-β-D-galactopyranoside and lysed on ice in NETN buffer (20 mM Tris-HCl pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.1% Nonidet P-40, 2 µg/ml aprotinin, 2 µg/ml leupeptin and 1 mM phenylmethylsulfonyl fluoride). The GST-tagged SQSTM1 was bound to glutathione–agarose beads overnight at 4°C followed by washing five times with NETN buffer. Captured protein on agarose beads was washed three times with NETN buffer prior to use. For pulldown assays, 3.5 µg of GST–SQSTM1 was added to 500 µg of MEF cell lysates and rotated for 2 h. Beads were subsequently washed three times with binding buffer and SDS-PAGE sample buffer was added. Proteins were separated by 10% SDS-PAGE and analyzed by western blotting as described above. For purified dynein interaction experiments, 3.5 µg of GST–SQSTM1 was rotated for 1 h at 4°C with 50 µM inhibitory or random peptide (LEQALSMGPSDEGFWLS) in ten volumes of TEN buffer (20 mM Tris-HCl pH 8.0, 100 mM NaCl and 1 mM EDTA). Purified dynein intermediate chain 1 or 2 (0.3 µg; Abcam, Cambridge, MA) was added and incubation continued as above for 4 h. Glutathione beads were used to confirm the specificity of DIC interaction with SQSTM1. Bound chains were washed three times with TEN prior to the addition of SDS-PAGE sample buffer and proteins separated by 10% SDS-PAGE. Bound DIC was identified by western blotting with anti-DIC1/2 antibody (Santa Cruz Biotechnology, Santa Cruz, CA), whereas GST–SQSTM1 was recognized using anti-SQSTM1 antibody (Abcam, Cambridge, MA). Equal loading of beads was confirmed by silver staining (Pierce Biotechnology, Rockford, IL).
Immunofluorescence
Wild type (WT) and p62KO MEF cells were grown on coverslips coated with poly-D-lysine and rat tail collagen (3∶1) in 24-well plates and treated with 5 µM MG132 for 6 h and 500 µM EHNA for 8 h as described in the figure legends. MEF cells were washed with phosphate-buffered saline (PBS), followed by fixation with freshly made warm 4% paraformaldehyde in PBS. Permeabilization was in 0.1% Triton X-100 in PBS for 15 min prior to blocking with 3% milk in PBS for 1 h at room temperature. Endogenous proteins were detected with specific antibody in blocking solution overnight at 4°C. Following removal of primary antibody by washing three times with PBS, fluorescently tagged secondary antibody was added in blocking solution for 1 h at room temperature. Cells were washed a total of five times with PBS prior to mounting on slides. Where indicated, cells were transfected with construct for specific protein expression as described above. Fluorescently tagged proteins were analyzed with a 60× oil immersion lens on a Nikon A1/T1 confocal microscope and images were processed employing NIS Elements Software (Nikon, Melville, NY).
For analysis of vesicle distance from the perinuclear region, 20 individual cells, identified as the most representative samples, from each cell line and treatment panel were blinded to the individual doing the analysis and used to determine distance of vesicles from the perinuclear region using the Nikon Elements Software AR package 3.22.13 (Build 730). Briefly, a simple line measurement was taken from the nuclear envelope to the approximate center of each easily identifiable vesicle in the panel. Each panel was automatically calibrated by the microscope software as the cell was being imaged. Data was exported into Microsoft EXCEL 2010 for analysis of mean±s.e.m. Comparison of measurements for each figure is presented in Table 1.
Acknowledgments
We would like to thank Richard Vallee for his generous gift of GFP-tagged p50/dynamitin.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
L.C.B., M.L.S., M.W.W. and M.C.W. conceived of and designed experiments. L.C.B., M.L.S. and Y.D. performed experiments. M.T.D-M. and J.M. contributed materials and reagents used in experiments. L.C.B., M.L.S. and M.C.W. analyzed the data. L.C.B., M.L.S., Y.D., J.Y., M.W.W. and M.C.W. contributed intellectual discussions. L.C.B., M.L.S. and M.C.W. wrote the manuscript.
Funding
This work was supported by the National Institutes of Health [grant number 2RO1NS033661 to M.C.W.]; and by the Alabama Agricultural Experiment Station [grant number HATCH ALA021-1-09017 to M.C.W.]. Deposited in PMC for release after 12 months.
References
- Aniento F., Emans N., Griffiths G., Gruenberg J. (1993). Cytoplasmic dynein-dependent vesicular transport from early to late endosomes. J. Cell Biol. 123, 1373–1387 10.1083/jcb.123.6.1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asthana J., Kuchibhatla A., Jana S. C., Ray K., Panda D. (2012). Dynein light chain 1 (LC8) association enhances microtubule stability and promotes microtubule bundling. J. Biol. Chem. 287, 40793–40805 10.1074/jbc.M112.394353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behl C. (2011). BAG3 and friends: co-chaperones in selective autophagy during aging and disease. Autophagy 7, 795–798 10.4161/auto.7.7.15844 [DOI] [PubMed] [Google Scholar]
- Bjørkøy G., Lamark T., Pankiv S., Øvervatn A., Brech A., Johansen T. (2009). Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 452, 181–197 10.1016/S0076-6879(08)03612-4 [DOI] [PubMed] [Google Scholar]
- Boylan K., Serr M., Hays T. (2000). A molecular genetic analysis of the interaction between the cytoplasmic dynein intermediate chain and the glued (dynactin) complex. Mol. Biol. Cell. 11, 3791–3803 10.1091/mbc.11.11.3791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt J. K., Echeverri C. J., Nilsson T., Vallee R. B. (1997). Overexpression of the dynamitin (p50) subunit of the dynactin complex disrupts dynein-dependent maintenance of membrane organelle distribution. J. Cell Biol. 139, 469–484 10.1083/jcb.139.2.469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q., Lu L., Tian J. H., Zhu Y. B., Qiao H., Sheng Z. H. (2010). Snapin-regulated late endosomal transport is critical for efficient autophagy-lysosomal function in neurons. Neuron 68, 73–86 10.1016/j.neuron.2010.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo A. M., Peterhoff C. M., Troncoso J. C., Gomez-Isla T., Hyman B. T., Nixon R. A. (2000). Endocytic pathway abnormalities precede amyloid β deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 157, 277–286 10.1016/S0002-9440(10)64538-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier-Larsen E., Holzbaur E. L. F. (2006). Axonal transport and neurodegenerative disease. Biochim. Biophys. Acta 1762, 1094–1108 10.1016/j.bbadis.2006.04.002 [DOI] [PubMed] [Google Scholar]
- De Vos K. J., Grierson A. J., Ackerley S., Miller C. C. J. (2008). Role of axonal transport in neurodegenerative diseases. Annu. Rev. Neurosci. 31, 151–173 10.1146/annurev.neuro.31.061307.090711 [DOI] [PubMed] [Google Scholar]
- Dillman J. F., 3rd, Pfister K. K. (1994). Differential phosphorylation in vivo of cytoplasmic dynein associated with anterogradely moving organelles. J. Cell Biol. 127, 1671–1681 10.1083/jcb.127.6.1671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodding M. P., Way M. (2011). Coupling viruses to dynein and kinesin-1. EMBO J. 30, 3527–3539 10.1038/emboj.2011.283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dompierre J. P., Godin J. D., Charrin B. C., Cordelières F. P., King S. J., Humbert S., Saudou F. (2007). Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J. Neurosci. 27, 3571–3583 10.1523/JNEUROSCI.0037-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driskell O. J., Mironov A., Allan V. J., Woodman P. G. (2007). Dynein is required for receptor sorting and the morphogenesis of early endosomes. Nat. Cell Biol. 9, 113–120 10.1038/ncb1525 [DOI] [PubMed] [Google Scholar]
- Du Y., Wooten M. C., Wooten M. W. (2009). Oxidative damage to the promoter region of SQSTM1/p62 is common to neurodegenerative disease. Neurobiol. Dis. 35, 302–310 10.1016/j.nbd.2009.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durán A., Serrano M., Leitges M., Flores J. M., Picard S., Brown J. P., Moscat J., Diaz-Meco M. T. (2004). The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis. Dev. Cell 6, 303–309 10.1016/S1534-5807(03)00403-9 [DOI] [PubMed] [Google Scholar]
- Engelke M. F., Burckhardt C. J., Morf M. K., Greber U. F. (2011). The dynactin complex enhances the speed of microtubule-dependent motions of adenovirus both towards and away from the nucleus. Viruses 3, 233–253 10.3390/v3030233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eschbach J., Dupuis L. (2011). Cytoplasmic dynein in neurodegeneration. Pharmacol. Ther. 130, 348–363 10.1016/j.pharmthera.2011.03.004 [DOI] [PubMed] [Google Scholar]
- Eskelinen E. L. (2008). New insights into the mechanisms of macroautophagy in mammalian cells. Int. Rev. Cell Mol. Biol. 266, 207–247 10.1016/S1937-6448(07)66005-5 [DOI] [PubMed] [Google Scholar]
- García-Mata R., Bebök Z., Sorscher E. J., Sztul E. S. (1999). Characterization and dynamics of aggresome formation by a cytosolic GFP-chimera. J. Cell Biol. 146, 1239–1254 10.1083/jcb.146.6.1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S., Holmström K. M., Skujat D., Fiesel F. C., Rothfuss O. C., Kahle P. J., Springer W. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131 10.1038/ncb2012 [DOI] [PubMed] [Google Scholar]
- Guthrie C. R., Kraemer B. C. (2011). Proteasome inhibition drives HDAC6-dependent recruitment of tau to aggresomes. J. Mol. Neurosci. 45, 32–41 10.1007/s12031-011-9502-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafezparast M., Klocke R., Ruhrberg C., Marquardt A., Ahmad-Annuar A., Bowen S., Lalli G., Witherden A. S., Hummerich H., Nicholson S. et al. (2003). Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science 300, 808–812 10.1126/science.1083129 [DOI] [PubMed] [Google Scholar]
- Hara T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R., Yokoyama M., Mishima K., Saito I., Okano H. et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889 10.1038/nature04724 [DOI] [PubMed] [Google Scholar]
- Hideshima T., Bradner J. E., Chauhan D., Anderson K. C. (2005). Intracellular protein degradation and its therapeutic implications. Clin. Cancer Res. 11, 8530–8533 10.1158/1078-0432.CCR-05-2305 [DOI] [PubMed] [Google Scholar]
- Hollenbeck P. J., Saxton W. M. (2005). The axonal transport of mitochondria. J. Cell Sci. 118, 5411–5419 10.1242/jcs.02745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huotari J., Helenius A. (2011). Endosome maturation. EMBO J. 30, 3481–3500 10.1038/emboj.2011.286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J., Parameshwaran K., Seibenhener M. L., Kang M. G., Suppiramaniam V., Huganir R. L., Diaz-Meco M. T., Wooten M. W. (2009). AMPA receptor trafficking and synaptic plasticity require SQSTM1/p62. Hippocampus 19, 392–406 10.1002/hipo.20528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston J. A., Ward C. L., Kopito R. R. (1998). Aggresomes: a cellular response to misfolded proteins. J. Cell Biol. 143, 1883–1898 10.1083/jcb.143.7.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju J. S., Miller S. E., Hanson P. I., Weihl C. C. (2008). Impaired protein aggregate handling and clearance underlie the pathogenesis of p97/VCP-associated disease. J. Biol. Chem. 283, 30289–30299 10.1074/jbc.M805517200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki S., Holzbaur E. L. F. (1995). Affinity chromatography demonstrates a direct binding between cytoplasmic dynein and the dynactin complex. J. Biol. Chem. 270, 28806–28811 10.1074/jbc.270.48.28806 [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y., Kovacs J. J., McLaurin A., Vance J. M., Ito A., Yao T. P. (2003). The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115, 727–738 10.1016/S0092-8674(03)00939-5 [DOI] [PubMed] [Google Scholar]
- Kerr M. C., Teasdale R. D. (2009). Defining macropinocytosis. Traffic 10, 364–371 10.1111/j.1600-0854.2009.00878.x [DOI] [PubMed] [Google Scholar]
- Kimura S., Noda T., Yoshimori T. (2008). Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct. Funct. 33, 109–122 10.1247/csf.08005 [DOI] [PubMed] [Google Scholar]
- Kinchen J. M., Ravichandran K. S. (2008). Phagocytic signaling: you can touch, but you can't eat. Curr. Biol. 18, R521–R524 10.1016/j.cub.2008.04.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King S. M., Barbarese E., Dillman J. F., 3rd, Benashski S. E., Do K. T., Patel-King R. S., Pfister K. K. (1998). Cytoplasmic dynein contains a family of differentially expressed light chains. Biochemistry 37, 15033–15041 10.1021/bi9810813 [DOI] [PubMed] [Google Scholar]
- Komatsu M., Waguri S., Chiba T., Murata S., Iwata J., Tanida I., Ueno T., Koike M., Uchiyama Y., Kominami E. et al. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884 10.1038/nature04723 [DOI] [PubMed] [Google Scholar]
- Kon T., Mogami T., Ohkura R., Nishiura M., Sutoh K. (2005). ATP hydrolysis cycle-dependent tail motions in cytoplasmic dynein. Nat. Struct. Mol. Biol. 12, 513–519 10.1038/nsmb930 [DOI] [PubMed] [Google Scholar]
- Kopito R. R. (2000). Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 10, 524–530 10.1016/S0962-8924(00)01852-3 [DOI] [PubMed] [Google Scholar]
- Kuusisto E., Salminen A., Alafuzoff I. (2001). Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport 12, 2085–2090 10.1097/00001756-200107200-00009 [DOI] [PubMed] [Google Scholar]
- Lee J. Y., Koga H., Kawaguchi Y., Tang W., Wong E., Gao Y. S., Pandey U. B., Kaushik S., Tresse E., Lu J. et al. (2010). HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 29, 969–980 10.1038/emboj.2009.405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S. X., Collins C. A. (1992). Immunolocalization of cytoplasmic dynein to lysosomes in cultured cells. J. Cell Sci. 101, 125–137 [DOI] [PubMed] [Google Scholar]
- Lin S. X., Ferro K. L., Collins C. A. (1994). Cytoplasmic dynein undergoes intracellular redistribution concomitant with phosphorylation of the heavy chain in response to serum starvation and okadaic acid. J. Cell Biol. 127, 1009–1019 10.1083/jcb.127.4.1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell D. J., Blasier K. R., Jeffery E. D., Ross M. W., Pullikuth A. K., Suo D., Park J., Smiley W. R., Lo K. W., Shabanowitz J. et al. (2012). Trk activation of the ERK1/2 kinase pathway stimulates intermediate chain phosphorylation and recruits cytoplasmic dynein to signaling endosomes for retrograde axonal transport. J. Neurosci. 32, 15495–15510 10.1523/JNEUROSCI.5599-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno Y., Amari M., Takatama M., Aizawa H., Mihara B., Okamoto K. (2006). Immunoreactivities of p62, an ubiqutin-binding protein, in the spinal anterior horn cells of patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 249, 13–18 10.1016/j.jns.2006.05.060 [DOI] [PubMed] [Google Scholar]
- Moscat J., Diaz-Meco M. T. (2009). To aggregate or not to aggregate? A new role for p62. EMBO Rep. 10, 804 10.1038/embor.2009.172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaoka U., Kim K., Jana N. R., Doi H., Maruyama M., Mitsui K., Oyama F., Nukina N. (2004). Increased expression of p62 in expanded polyglutamine-expressing cells and its association with polyglutamine inclusions. J. Neurochem. 91, 57–68 10.1111/j.1471-4159.2004.02692.x [DOI] [PubMed] [Google Scholar]
- Nakaso K., Yoshimoto Y., Nakano T., Takeshima T., Fukuhara Y., Yasui K., Araga S., Yanagawa T., Ishii T., Nakashima K. (2004). Transcriptional activation of p62/A170/ZIP during the formation of the aggregates: possible mechanisms and the role in Lewy body formation in Parkinson's disease. Brain Res. 1012, 42–51 10.1016/j.brainres.2004.03.029 [DOI] [PubMed] [Google Scholar]
- Nakielny S., Dreyfuss G. (1999). Transport of proteins and RNAs in and out of the nucleus. Cell 99, 677–690 10.1016/S0092-8674(00)81666-9 [DOI] [PubMed] [Google Scholar]
- Nunes P., Ernandez T., Roth I., Qiao X., Strebel D., Bouley R., Charollais A., Ramadori P., Foti M., Meda P. et al. (2013). Hypertonic stress promotes autophagy and microtubule-dependent autophagosomal clusters. Autophagy 9, 550–567 10.4161/auto.23662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okatsu K., Saisho K., Shimanuki M., Nakada K., Shitara H., Sou Y. S., Kimura M., Sato S., Hattori N., Komatsu M. et al. (2010). p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 15, 887–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann J. A., Chin L. S. (2008). Parkin-mediated K63-linked polyubiquitination: a signal for targeting misfolded proteins to the aggresome-autophagy pathway. Autophagy 4, 85–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann J. A., Li L., Chudaev M. V., Chen J., Perez F. A., Palmiter R. D., Chin L. S. (2007). Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol. 178, 1025–1038 10.1083/jcb.200611128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang H., Ali Y. O., Ravichandran M., Dong A., Qiu W., MacKenzie F., Dhe-Paganon S., Arrowsmith C. H., Zhai R. G. (2012). Protein aggregates are recruited to aggresome by histone deacetylase 6 via unanchored ubiquitin C termini. J. Biol. Chem. 287, 2317–2327 10.1074/jbc.M111.273730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paine M. G., Babu J. R., Seibenhener M. L., Wooten M. W. (2005). Evidence for p62 aggregate formation: role in cell survival. FEBS Lett. 579, 5029–5034 10.1016/j.febslet.2005.08.010 [DOI] [PubMed] [Google Scholar]
- Pan L., He Q., Liu J., Chen Y., Ma M., Zhang L., Shi J. (2012). Nuclear-targeted drug delivery of TAT peptide-conjugated monodisperse mesoporous silica nanoparticles. J. Am. Chem. Soc. 134, 5722–5725 10.1021/ja211035w [DOI] [PubMed] [Google Scholar]
- Pankiv S., Clausen T. H., Lamark T., Brech A., Bruun J. A., Outzen H., Øvervatn A., Bjørkøy G., Johansen T. (2007). p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145 10.1074/jbc.M702824200 [DOI] [PubMed] [Google Scholar]
- Paschal B. M., Shpetner H. S., Vallee R. B. (1987). MAP 1C is a microtubule-activated ATPase which translocates microtubules in vitro and has dynein-like properties. J. Cell Biol. 105, 1273–1282 10.1083/jcb.105.3.1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdiz D., Mackeh R., Poüs C., Baillet A. (2011). The ins and outs of tubulin acetylation: more than just a post-translational modification? Cell. Signal. 23, 763–771 10.1016/j.cellsig.2010.10.014 [DOI] [PubMed] [Google Scholar]
- Pfister K. K., Shah P. R., Hummerich H., Russ A., Cotton J., Annuar A. A., King S. M., Fisher E. M. (2006). Genetic analysis of the cytoplasmic dynein subunit families. PLoS Genet. 2, e1 10.1371/journal.pgen.0020001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puls A., Schmidt S., Grawe F., Stabel S. (1997). Interaction of protein kinase C ζ with ZIP, a novel protein kinase C-binding protein. Proc. Natl. Acad. Sci. USA 94, 6191–6196 10.1073/pnas.94.12.6191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh Babu J., Lamar Seibenhener M., Peng J., Strom A. L., Kemppainen R., Cox N., Zhu H., Wooten M. C., Diaz-Meco M. T., Moscat J. et al. (2008). Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J. Neurochem. 106, 107–120 10.1111/j.1471-4159.2008.05340.x [DOI] [PubMed] [Google Scholar]
- Ravikumar B., Acevedo-Arozena A., Imarisio S., Berger Z., Vacher C., O'Kane C. J., Brown S. D., Rubinsztein D. C. (2005). Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat. Genet. 37, 771–776 10.1038/ng1591 [DOI] [PubMed] [Google Scholar]
- Reed N. A., Cai D., Blasius T. L., Jih G. T., Meyhofer E., Gaertig J., Verhey K. J. (2006). Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 16, 2166–2172 10.1016/j.cub.2006.09.014 [DOI] [PubMed] [Google Scholar]
- Richter-Landsberg C., Leyk J. (2013). Inclusion body formation, macroautophagy, and the role of HDAC6 in neurodegeneration. Acta Neuropathol. 126, 793–807 10.1007/s00401-013-1158-x [DOI] [PubMed] [Google Scholar]
- Rink J., Ghigo E., Kalaidzidis Y., Zerial M. (2005). Rab conversion as a mechanism of progression from early to late endosomes. Cell 122, 735–749 10.1016/j.cell.2005.06.043 [DOI] [PubMed] [Google Scholar]
- Rishal I., Fainzilber M. (2010). Retrograde signaling in axonal regeneration. Exp. Neurol. 223, 5–10 10.1016/j.expneurol.2009.08.010 [DOI] [PubMed] [Google Scholar]
- Rodriguez A., Durán A., Selloum M., Champy M. F., Diez-Guerra F. J., Flores J. M., Serrano M., Auwerx J., Diaz-Meco M. T., Moscat J. (2006). Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab. 3, 211–222 10.1016/j.cmet.2006.01.011 [DOI] [PubMed] [Google Scholar]
- Rosse C., Boeckeler K., Linch M., Radtke S., Frith D., Barnouin K., Morsi A. S., Hafezparast M., Howell M., Parker P. J. (2012). Binding of dynein intermediate chain 2 to paxillin controls focal adhesion dynamics and migration. J. Cell Sci. 125, 3733–3738 10.1242/jcs.089557 [DOI] [PubMed] [Google Scholar]
- Schnapp B. J., Reese T. S. (1989). Dynein is the motor for retrograde axonal transport of organelles. Proc. Natl. Acad. Sci. USA 86, 1548–1552 10.1073/pnas.86.5.1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroer T. A. (2004). Dynactin. Annu. Rev. Cell Dev. Biol. 20, 759–779 10.1146/annurev.cellbio.20.012103.094623 [DOI] [PubMed] [Google Scholar]
- Schroer T. A., Steuer E. R., Sheetz M. P. (1989). Cytoplasmic dynein is a minus end-directed motor for membranous organelles. Cell 56, 937–946 10.1016/0092-8674(89)90627-2 [DOI] [PubMed] [Google Scholar]
- Seibenhener M. L., Babu J. R., Geetha T., Wong H. C., Krishna N. R., Wooten M. W. (2004). Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol. Cell. Biol. 24, 8055–8068 10.1128/MCB.24.18.8055-8068.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibenhener M. L., Du Y., Diaz-Meco M. T., Moscat J., Wooten M. C., Wooten M. W. (2013). A role for sequestosome 1/p62 in mitochondrial dynamics, import and genome integrity. Biochim. Biophys. Acta 1833, 452–459 10.1016/j.bbamcr.2012.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ström A. L., Shi P., Zhang F., Gal J., Kilty R., Hayward L. J., Zhu H. (2008). Interaction of amyotrophic lateral sclerosis (ALS)-related mutant copper-zinc superoxide dismutase with the dynein-dynactin complex contributes to inclusion formation. J. Biol. Chem. 283, 22795–22805 10.1074/jbc.M800276200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X., Seyb K. I., Huang M., Schuman E. R., Shi P., Zhu H., Glicksman M. A. (2012). A high-throughput screening method for small-molecule inhibitors of the aberrant mutant SOD1 and dynein complex interaction. J. Biomol. Screen. 17, 314–326 10.1177/1087057111429595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallee R. B., Williams J. C., Varma D., Barnhart L. E. (2004). Dynein: An ancient motor protein involved in multiple modes of transport. J. Neurobiol. 58, 189–200 10.1002/neu.10314 [DOI] [PubMed] [Google Scholar]
- Vaughan K. T., Vallee R. B. (1995). Cytoplasmic dynein binds dynactin through a direct interaction between the intermediate chains and p150Glued. J. Cell Biol. 131, 1507–1516 10.1083/jcb.131.6.1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan P. S., Leszyk J. D., Vaughan K. T. (2001). Cytoplasmic dynein intermediate chain phosphorylation regulates binding to dynactin. J. Biol. Chem. 276, 26171–26179 10.1074/jbc.M102649200 [DOI] [PubMed] [Google Scholar]
- Vonderheit A., Helenius A. (2005). Rab7 associates with early endosomes to mediate sorting and transport of Semliki forest virus to late endosomes. PLoS Biol. 3, e233 10.1371/journal.pbio.0030233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe M., Nakaki T. (2011). Protein kinase CK2 regulates the formation and clearance of aggresomes in response to stress. J. Cell Sci. 124, 1519–1532 10.1242/jcs.081778 [DOI] [PubMed] [Google Scholar]
- Watanabe Y., Tanaka M. (2011). p62/SQSTM1 in autophagic clearance of a non-ubiquitylated substrate. J. Cell Sci. 124, 2692–2701 10.1242/jcs.081232 [DOI] [PubMed] [Google Scholar]
- Wooten M. W., Geetha T., Seibenhener M. L., Babu J. R., Diaz-Meco M. T., Moscat J. (2005). The p62 scaffold regulates nerve growth factor-induced NF-kappaB activation by influencing TRAF6 polyubiquitination. J. Biol. Chem. 280, 35625–35629 10.1074/jbc.C500237200 [DOI] [PubMed] [Google Scholar]
- Wooten M. W., Geetha T., Babu J. R., Seibenhener M. L., Peng J., Cox N., Diaz-Meco M. T., Moscat J. (2008). Essential role of SQSTM1/p62 in accumulation of K63-ubiquitinated proteins. J. Biol. Chem. 283, 6783–6789 10.1074/jbc.M709496200 [DOI] [PubMed] [Google Scholar]
- Xu Z., Graham K., Foote M., Liang F., Rizkallah R., Hurt M., Wang Y., Wu Y., Zhou Y. (2013). 14-3-3 protein targets misfolded chaperone-associated proteins to aggresomes. J. Cell Sci. 126, 4173–4186 10.1242/jcs.126102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J., Seibenhener M. L., Calderilla-Barbosa L., Diaz-Meco M. T., Moscat J., Jiang J., Wooten M. W., Wooten M. C. (2013). SQSTM1/p62 interacts with HDAC6 and regulates deacetylase activity. PLoS ONE 8, e76016 10.1371/journal.pone.0076016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatloukal K., Stumptner C., Fuchsbichler A., Heid H., Schnoelzer M., Kenner L., Kleinert R., Prinz M., Aguzzi A., Denk H. (2002). p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am. J. Pathol. 160, 255–263 10.1016/S0002-9440(10)64369-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerial M., McBride H. (2001). Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2, 107–117 10.1038/35052055 [DOI] [PubMed] [Google Scholar]