Quantitative Analyses of SMN1 and SMN2 Based on Real-Time LightCycler PCR: Fast and Highly Reliable Carrier Testing and Prediction of Severity of Spinal Muscular Atrophy (original) (raw)
Abstract
Spinal muscular atrophy (SMA) is a common autosomal recessive disorder in humans, caused by homozygous absence of the survival motor neuron gene 1 (SMN1). SMN2, a copy gene, influences the severity of SMA and may be used in somatic gene therapy of patients with SMA in the future. We present a new, fast, and highly reliable quantitative test, based on real-time LightCycler PCR that amplifies either SMN1 or SMN2. The SMN1 copies were determined and validated in 329 carriers and controls. The specificity of the test is 100%, whereas the sensitivity is 96.2%. The quantitative analysis of SMN2 copies in 375 patients with type I, type II, or type III SMA showed a significant correlation between SMN2 copy number and type of SMA as well as duration of survival. Thus, 80% of patients with type I SMA carry one or two SMN2 copies, and 82% of patients with type II SMA carry three SMN2 copies, whereas 96% of patients with type III SMA carry three or four SMN2 copies. Among 113 patients with type I SMA, 9 with one SMN2 copy lived <11 mo, 88/94 with two SMN2 copies lived <21 mo, and 8/10 with three SMN2 copies lived 33–66 mo. On the basis of SMN2 copy number, we calculated the posterior probability that a child with homozygous absence of SMN1 will develop type I, type II, or type III SMA.
Introduction
With an incidence of 1/6,000 to 1/10,000 and a carrier frequency of 1/40 to 1/50, spinal muscular atrophy (SMA) is the second-most-frequent autosomal recessive disease in Europeans (Pearn 1980). It is a neuromuscular disorder caused by the degeneration of α-motor neurons of the spinal cord anterior horns, leading to progressive atrophy of proximal muscles, paralysis, respiratory failure, and infant death. Patients with SMA have been classified into three types, on the basis of age at onset and clinical severity (International SMA Consortium 1992; Zerres and Rudnik-Schöneborn 1995): type I (MIM 253300) is the most severe form, type II (MIM 253550) is the intermediate form, and type III (MIM 253400) is the mildest form.
The SMA-determining gene called the “survival motor neuron” gene (SMN) is present on 5q13 in two copies, SMN1 and SMN2, which differ by only five nucleotide exchanges within their 3′ ends (Lefebvre et al. 1995; Bürglen et al. 1996). Two of these base-pair exchanges, located in exons 7 and 8, allow SMN1 to be distinguished from SMN2 and currently are used for direct diagnosis of SMA (for review, see Scheffer et al. 2001). In a summary of 1,122 patients with type I, type II, or type III SMA, it has been shown that 94% revealed homozygous absence of SMN1 (for review, see Wirth 2000). Approximately half of the remaining patients were identified as compound heterozygotes (with deletions and intragenic SMN1 mutations), whereas the other half, without SMN1 mutations, were considered as distinct genetic entities (Wirth et al. 1999).
Each patient affected with SMA retains at least one SMN2 copy. An inverse correlation between SMN2 protein level and SMA severity has been reported in humans and transgenic SMA mice (Coovert et al. 1997; Lefebvre et al. 1997; Hsieh-Li et al. 2000; Monani et al. 2000). However, SMN2 fails to provide complete protection from SMA due to a single, translationally silent nucleotide difference in exon 7 that disrupts an exonic splicing enhancer and causes exon 7 skipping (Lorson et al. 1999; Monani et al. 1999). It has been shown that SMN2 full-length mRNA can be restored by the splicing factor Htra2-β1 and the nontoxic drug sodium butyrate (Hofmann et al. 2000; Chang et al. 2001). Therefore, prevention of exon 7 skipping by up-regulating the full-length SMN2 mRNA represents an exciting goal for SMA therapy.
Since homozygous absence of exon 7 of SMN1 is a common variant, SMA carrier testing based on direct quantitative SMN1 analysis is a major issue. So far, several different SMN quantitative PCR tests have been developed that include multiple steps and complicated procedures that are laborious and subject to errors (Velasco et al. 1996; McAndrew et al. 1997; Taylor et al. 1998; Wirth et al. 1999; Scheffer et al. 2000).
We therefore developed a fast and highly reliable quantitative test for either SMN1 or SMN2, on the basis of real-time PCR on a LightCycler instrument (Roche Diagnostics). This test allowed us for the first time to analyze a large number of patients affected with SMA for their SMN2 copy number and to correlate the SMN2 copy number with type of SMA and duration of survival. An important aim of the study was to examine whether the identification of SMN2 copy number in a patient with SMA could serve as a prognostic tool.
Subjects, Material, and Methods
Carriers and Noncarriers
One hundred twenty-four carriers (parents of multiplex families with SMA) and 65 unaffected sibs of patients with SMA (noncarriers), as determined by haplotype analyses, were used to validate the SMN1 copy number by real-time PCR. Furthermore, we used 140 DNA samples from control individuals (38 healthy spouses of SMA carriers and 102 unrelated unaffected individuals) to determine the rate of SMN1 duplication on one chromosome and the SMA carrier frequency.
Patient Samples
The quantification of the SMN2 copy number was performed on DNA samples from 375 patients with SMA who previously have been classified as follows: 188 have type I SMA, 110 have type II SMA, and 77 have type III SMA (Wirth et al. 1997, 1999). The age of survival was available from 113 patients with type I SMA. All patients fulfilled the diagnostic criteria defined by the International SMA Consortium (1992) and by Zerres and Rudnik-Schöneborn (1995) for proximal spinal muscular atrophy. Informed consent was obtained from all subjects.
DNA Isolation and Quantification and Haplotype Analysis
Genomic DNA was isolated from blood samples by the salting-out method (Miller et al. 1988). DNA was dissolved in TE−4 buffer (10 mM Tris/0.1 mM EDTA pH 8) and was stored for at least 5 d before measurement of DNA concentration. Photometric measuring of DNA was performed on a Gene Quant II (Pharmacia). Only samples that had a 260 nm:280 nm ratio in the range 1.75–1.85 were used. DNA was diluted first to 20 ng/μl and then to a final concentration of 5 ng/μl. The exact measurement of the DNA concentration (with maximum deviation of ±3%) is the most important prerequisite for reliable determination of the gene copy number. The multicopy polymorphic markers Ag1-CA and C212 were used for haplotype analysis, as described elsewhere (Wirth et al. 1995).
Quantitative Real-Time PCR of SMN1 and SMN2 Copy Numbers
For determination of SMN1 and SMN2 gene dosages, we established a new quantification assay on the basis of real-time PCR. The quantification was performed with a LightCycler instrument (Roche Diagnostics) by use of the fluorescence-resonance energy-transfer technique. Online measurements of PCR products were based on the use of either SYBR Green I or specific hybridization probes labeled with 3′-fluorescein and 5′-LightCycler Red 640. The primers were designed to distinguish between SMN1 and SMN2, ending on or very close to the nucleotide differences between SMN1 and SMN2 (italicized letters) in exon 7 at position 6 and intron 7 at position +214. SMN1 was amplified by use of the forward primer telSMNex7forw (5′-TTTATTTTCCTTACAGGGTTT_C_-3′) and the reverse primer telSMNint7rev (5′-GTGAAAGTATGTTTCTTCCACg_T_A-3′). SMN2 was amplified by using the forward primer cenSMNex7forw (5′-TTTATTTTCCTTACAGGGTTT_T_A-3′) and the reverse primer cenSMNint7rev (5′-GTGAAAGTATGTTTCTTCCACg_C_A-3′). The hybridization probe detects both SMN1 and SMN2 and consists of a primer pair with a 3′-fluorescein (SMN FL: 5′-AATGCTGGCAGACTTACTCCTTAATTTA FL-3′) and a 5′-LightCycler Red 640 (SMN LC: 5′-LCR640 AATGTGAGCACCTTCCTTCTTTTTG-3′). Lowercase letters indicate mutations introduced to obtain adequate gene-specific PCR.
The PCR was performed in a total volume of 10 μl, containing 7.5 ng (for SMN1) or 6 ng (for SMN2) of genomic DNA, 10 pmol of each primer, 1 μl of Faststart DNA Kit SYBR Green I (Roche Molecular Biochemicals), 4 mM MgCl2 or Faststart Kit hybridization probes (Roche Molecular Biochemicals), 4 mM MgCl2 and 2 pmol of each hybridization probe. The PCR conditions for SMN1 or SMN2 with SYBR Green I were: 95°C for 10 min followed by 35 cycles with 95°C for 15 s, 58°C for 5 s, 72°C for 25 s, and a fluorescent detection step at 76°C for 1 s. During the entire program, we used the maximum temperature transition rate of 20°C/s. This quantification program was followed by a melting program to detect the melting points for every PCR product for each sample. The analysis was performed with the second derivative maximum method of the LightCycler software.
The PCR conditions for SMN1 or SMN2 with hybridization probes were: 95°C for 10 min, followed by 35 cycles of 95°C for 10 s, 58°C for 10 s (in this step, the measurement takes place), and 72°C for 20 s. This PCR program is followed by a melting curve program consisting of 15 s of denaturation at 95°C, 30 s of annealing at 55°C, and a melting and continuous measuring step at 0.1°C/s up to 85°C. The analysis was performed with the second derivative maximum method of the LightCycler software.
During the log-linear phase, amplification can be described as _N_=_N_0(1+E const)n where N is the number of amplified molecules; _N_0 is the initial number of molecules; E is the amplification efficiency; and n is the number of cycles. Since the amplification efficiency during the log-phase is constant, the initial concentration of the sample was calculated on the basis of the above formula, by use of human genomic DNA (external standard).
For SMN1, the DNA from a healthy individual with homozygous absence of SMN2 and two Ag1-CA and two C212 alleles was used as an external standard. This individual can be considered to possess two SMN1 copies. We used 1.5 μl of genomic DNA in concentrations of 1.25, 2.5, 5, and 7.5 ng/μl, respectively, corresponding to 0.5, 1, 2, and 3 fictive copies of SMN1.
For SMN2, the DNA of a patient with SMA who had homozygous absence of SMN1 and four Ag1-CA and C212 alleles was used as an external standard. This patient can be considered to possess four SMN2 copies. Genomic DNA (1.2 μl) was used in concentrations of 1.25, 2.5, 5, and 10 ng/μl, respectively, corresponding to one, two, four, and eight copies of SMN2. In addition, for a proper SMN2 standard, five further patients with SMA with three or four SMN2 copies were chosen and were compared to one another by defining each of the DNA samples as standards and the other DNAs as unknown. By means of this procedure, we were able to make sure that the initially postulated copy number was correct. All individuals used as standards and controls have also been tested by competitive multiplex quantitative PCR (Wirth et al. 1999). The standards were used to calculate the regression curve. In each experiment, the reproducibility of the calibration curve was analyzed by evaluation of the slope and the correlation coefficient of the curve. Sample concentrations were inferred on the basis of the regression curve in each experiment. All samples were measured at least twice, by use of independent DNA dilutions of 5 ng/μl.
Sequencing of Genomic PCR Products
Mutations within the annealing region of the hybridization probe were detected by direct sequencing of genomic PCR products of SMN2 exon 7 of patients 4929 and 4437, amplified by the primers cenSMNex7forw and cenSMNex7rev. Sequencing was performed on an ABI Prism 377, according to the manufacturer’s instructions.
Statistical Analysis
A special database in Access (Microsoft) was programmed for collection of all data for patients with SMA and their families (personal data, clinical features, and molecular genetic results, as well as data for direct calculation of mean ± SD LightCycler SMN1 and SMN2 values and coefficient of variation (CV). The SPSS program package was used to construct the Kaplan-Meier curves.
Prior probabilities for type I, type II, and type III SMA were based on cumulative frequencies found in cohorts of patients with SMA (Hausmanova-Petrusewicz et al. 1985). Odds ratios (ORs) were calculated using the SAS/STAT software, specifically the procedures FREQ and LOGISTIC in release 6.11 (SAS Institute 1996), according to the method of Hosmer and Lemesshow (2000). The ORs and their 95% Wald confidence limits were calculated for type I, type II, and type III SMA and for one to four copies of SMN2. The OR for one copy was used as a reference and was fixed to unity. ORs and prior probabilities were used in a Bayesian fashion to calculate posterior probabilities for type I, type II, and type III SMA, depending on the SMN2 copy number.
Results
Establishment of a Real-Time PCR for SMN1 or SMN2 on a LightCycler Instrument
On the basis of real-time LightCycler PCR, we developed a quantitative test that enables a fast and highly reliable differentiation between either one to four SMN1 copies or one to four SMN2 copies. To specifically detect either SMN1 or SMN2, primers were designed that end on or very close to the nucleotide differences between SMN1 and SMN2 in exon 7 at position 6 and intron 7 at position +214. To analyze the gene-specific amplification, DNA from individuals who carry SMN1 only or from patients with SMA who carry SMN2 only were used. The production of gene-specific amplicons (fig. 1a and 1_b_) allowed us to use both systems for online fluorescence resonance energy-transfer measurement: SYBR Green I and LightCycler hybridization probes labeled with 3′-fluorescein and 5′-LightCycler red 640. Since the data for both systems were almost identical and the SYBR Green I format is less expensive, we will present only data based on this PCR format.
Figure 1.

Fluorescence versus cycle number, melting peaks, and sequence of new point mutation in SMN2. _a,_SYBR Green I fluorescence plot versus cycle number resulting from amplification of genomic DNA (external standard; st 0.5, st 1, st 2, st 3) of an unaffected individual carrying two SMN1 copies and homozygous absence of SMN2. In addition, four negative controls (three patients with SMA with homozygous absence of SMN1 and one water control) were tested. Slope = −4.112, error = 0.0279, _r_=-1.00. b, SYBR Green I fluorescence plot versus cycle number resulting from amplification of genomic DNA (external standard; st 1, st 2, st 4, st 8) of a patient with SMA with four SMN2 copies and homozygous absence of SMN1. In addition, four negative controls (three normal individuals with homozygous absence of SMN2 and one water control) were analyzed. Slope = −4.057, error = 0.0469, _r_=-1.00. c, Normalized SYBR Green I fluorescence plot of the external standard (st) for SMN1 and samples of carriers (C) and controls (ctr) with one, two, or three SMN1 copies. Each copy number was detected in three different DNA samples. Slope = −3.860, error = 0.0213, _r_=-1.00. d, Normalized SYBR Green I fluorescence plot of the external standard (st) for SMN2 and of patients with type I, type II, and type III SMA with one, two, three, or four SMN2 copies. Slope = − 3,819, error = 0.0181, _r_=-1.00. e, Melting-curve analysis of SMN2, with hybridization probe indicating the mutation 892G→C. The DNA of the patient with SMA with the homozygous 892G→C mutation in SMN2 shows a lower melting temperature (m/m, T _m_=58.0°C) than an SMA patient with the wild-type SMN2 sequence (w/w, T _m_=64.5°C). Patients with SMA with heterozygous genotype (w/m) display two peaks. f, New intragenic SMN2 mutation 892G→C in patients with SMA, found by sequencing the DNA samples with melting peaks at 58.0°C. Reverse nucleotide sequences obtained from direct sequencing of genomic PCR products from patients 4437 (heterozygous) and 4929 (homozygous ) for the mutation 892G→C; arrows indicate the positions of the mutation.
The reproducibility of the method was determined in 10 independent PCR reactions on 10 different days. For SMN1, we used DNA from carriers with one copy or noncarriers with two or three copies, whereas, for SMN2, we used DNA from patients with type I, type II, and type III SMA with one to four SMN2 copies. The number of SMN1 or SMN2 copies in these individuals were also determined by the previously reported competitive multiplex quantitative PCR (Wirth et al. 1999), by haplotype analysis of the Ag1-CA and C212 marker alleles, and by quantitative analysis of the SMN genes in further relatives in three generations. The newly developed method allows a clear differentiation between one to four SMN1 or SMN2 copies, without any region of overlap (table 1).
Table 1.
Reproducibility of Real-Time LightCycler Quantitative Analyses of SMN1 and SMN2, on the Basis of 10 Independent PCRs
| Test and Subjects | SMN1/SMN2 | No. ofAg1-CA/C212Marker Alleles | No. ofSMN Copies | Minimum/MaximumAverage Values | Mean ± SDValues | Coefficient ofVariation(%) |
|---|---|---|---|---|---|---|
| SMN1 quantitative test: | ||||||
| Carrier | +/+ | 3/3 | 1 | .989/1.141 | 1.072 ± .044 | 4.0 |
| Carrier | +/+ | 3/3 | 1 | .975/1.150 | 1.060 ± .056 | 5.2 |
| Control | +/+ | 3/3 | 2 | 1.912/2.276 | 2.043 ± .105 | 5.1 |
| Control | +/+ | 4/4 | 3 | 2.974/3.307 | 3.112 ± .119 | 3.8 |
| SMN2 quantitative test: | ||||||
| Type I SMA | del/+ | 1/1 | 1 | .902/1.132 | 1.004 ± .083 | 8.2 |
| Type I SMA | del/+ | 2/2 | 2 | 1.768/2.136 | 1.918 ± .104 | 5.4 |
| Type I SMA | del/+ | 3/3 | 3 | 2.784/3.200 | 2.998 ± .136 | 4.5 |
| Type III SMA | del/+ | 4/4 | 4 | 3.694/4.233 | 3.927 ± .172 | 4.3 |
Validity of the Heterozygosity SMN1 Test
This test represents a fast molecular genetic tool for carrier screening both in families with SMA and in the general population, as well as for identification of compound heterozygous patients with SMA. A representative quantitative SMN1 PCR of the external standard and several unknown samples (carriers and controls) is shown in figure 1c. To validate the test, we analyzed 124 carriers (parents from multiplex families with SMA) and 65 unaffected sibs of patients with SMA (noncarriers, according to haplotype analysis). The mean value obtained for each person is plotted in figure 2. Maximum and minimum average values, mean values ± SD, and CVs are given in table 2. All but 5 of 124 carriers showed one SMN1 copy, as expected. The remaining five carriers presented two SMN1 copies. Among the unaffected sibs, 61 of 65 presented two SMN1 copies, whereas the remaining 4 carried three SMN1 copies. These data suggest that, in 9 of 254 normal chromosomes (124 from carriers and 130 from unaffected sibs), two SMN1 copies are present on one chromosome. To determine the frequency of two SMN1 copies per chromosome in a larger population, we analyzed 140 additional individuals from the general population (38 spouses of SMA carriers and 102 control individuals without muscle weakness; see fig. 2). Of 140 controls, 4 (2.85%) showed only one SMN1 copy and therefore were identified as SMA carriers. This corresponds to a heterozygosity frequency of 1 in 35. Additionally, we identified, in 3 of 140 controls, three SMN1 copies and, in 1 of 140 controls, four SMN1 copies. In summary, 14 (2.6%) of 530 normal chromosomes carry two SMN1 copies per chromosome (table 3).
Figure 2.

Plot of the average SMN1 values for carriers (_N_=124), noncarriers (unaffected sibs of patients with SMA) (_N_=65), and controls (_N_=140).
Table 2.
LightCycler Mean Values ± SD and CVs of Both SMN1, Tested in 329 Carriers and Controls, and SMN2, Tested in 375 Patients with Type I, Type II, and Type III SMA
| Test andNo. ofCopies | Na | Minimum/MaximumAverage Values | Mean ± SDValues | CV(%) |
|---|---|---|---|---|
| SMN1: | ||||
| 1 | 123 | .599/1.176 | .971±.077 | 7.9 |
| 2 | 198 | 1.683/2.436 | 2.053±.146 | 7.1 |
| 3 | 7 | 2.775/3.410 | 3.027±.200 | 6.6 |
| 4 | 1 | 4.035 | 4.035 | … |
| SMN2: | ||||
| 1 | 13 | .860/1.390 | 1.146±.162 | 14.0 |
| 2 | 153 | 1.555/2.466 | 1.900±.174 | 9.2 |
| 3 | 166 | 2.518/3.411 | 2.866±.203 | 7.1 |
| 4 | 43 | 3.524/4.245 | 3.863±.183 | 5.0 |
Table 3.
Distribution of Both SMN1 Copies in Carriers, Noncarriers (Unaffected Sibs of Patients with SMA), and Controls and SMN2 Copies in Patients with SMA
| Distribution, for SMN Copy Number | |||||
|---|---|---|---|---|---|
| Test andPatient Status | 1 | 2 | 3 | 4 | Total |
| SMN1: | |||||
| Carrier | 119 (96.0%) | 5 (4.0%) | 0 (0.0%) | 0 (0.0%) | 124 |
| Noncarrier | 0 (0.0%) | 61 (93.8%) | 4 (6.2%) | 0 (0.0%) | 65 |
| Controls | 4 (2.9%) | 132 (94.3%) | 3 (2.1%) | 1 (0.7%) | 140 |
| Total | 329 | ||||
| SMN2: | |||||
| Type I SMA | 13 (6.9%) | 138 (73.4%) | 37 (19.7%) | 0 (0.0%) | 188 |
| Type II SMA | 0 (0.0%) | 12 (10.9%) | 90 (81.8%) | 8 (7.3%) | 110 |
| Type III SMA | 0 (0.0%) | 3 (3.9%) | 39 (50.6%) | 35 (45.5%) | 77 |
| Total | 375 |
Quantitative Analyses of SMN2 Copy Number in Patients with SMA
This fast and highly reliable test allowed us to analyze a large number of patients with SMA for their SMN2 copy number and to correlate these with the type of SMA and duration of survival. We analyzed the SMN2 copy number in 188 patients with type I SMA, 110 patients with type II SMA, and 77 patients with type III SMA with both SYBR Green I and the hybridization probe. In four patients, a discrepancy between the SMN2 copies determined by the two detection systems was observed. In addition, in four patients (three heterozygous and one homozygous), the PCR products detected with hybridization probes showed a different melting curve (fig. 1_e_). Direct sequencing of the SMN2 PCR products in two patients revealed a point mutation at nucleotide position 892, G→C, that causes an amino acid substitution from glycine to arginine at codon 287 (G287R) (fig. 1_f_). The nucleotide exchange is localized within the annealing region of the hybridization probe, thereby explaining the altered melting curves.
The quantitative analysis of the SMN2 copy number in 375 patients with SMA are given in tables 2 and 3. In 80% of patients with type I SMA, one or two SMN2 copies were found, 82% of patients with type II SMA carried three SMN2 copies, and 96% of patients with type III SMA carried three or four SMN2 copies. However, ∼20% of patients with type I SMA showed three SMN2 copies, and none showed four SMN2 copies. Similarly, among patients with type II SMA, ∼11% showed two SMN2 copies, and ∼7% showed four SMN2 copies, whereas, among patients with type III SMA, none showed one SMN2 copy, and ∼4% revealed two SMN2 copies (fig. 3).
Figure 3.
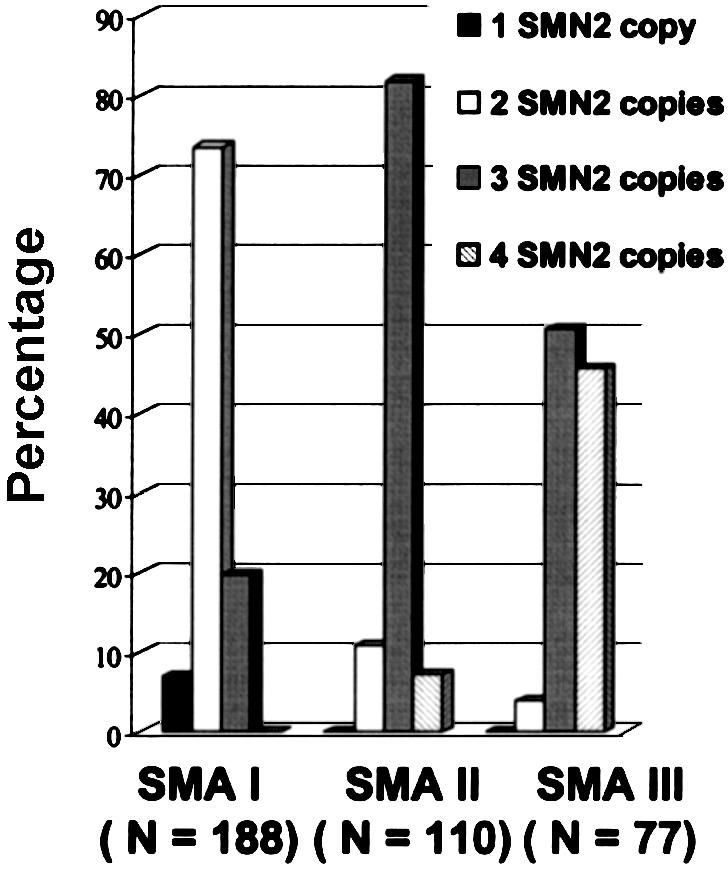
Diagram of the frequency of patients with type I, type II, and type III SMA versus SMN2 copy number.
A strong correlation between SMN2 copy number and SMA phenotype was observed; however, this correlation is not absolute. This may be because of (1) true variation caused by modifying genes or other external factors, (2) intragenic mutations within SMN2, or (3) SMN2 genes partially deleted or duplicated as a result of deletions or gene conversions involving either the 5′ end or the 3′ end of the SMN genes. To verify the first hypothesis, we analyzed 13 families with SMA that had siblings carrying homozygous mutations of SMN1 but discordant phenotypes (Helmken et al. 2000_a,_ 2000_b_). In each case, identical numbers of SMN2 copies were determined in haploidentical siblings. Although, in all these families, the unaffected or milder affected siblings were females, there is no overall significant difference in the distribution of SMN2 copies between sexes (table 4). For the second hypothesis, we already presented in this article an intragenic SMN2 mutation in exon 7 (G278R), revealing that intragenic mutations may indeed be a reason that can disturb the correlation between SMN2 copy number and SMA phenotype. To verify the third hypothesis, we analyzed, in our patients with SMA Ag1-CA and C212—two highly polymorphic markers localized at the 5′ end of the SMN genes (Wirth et al. 1995)—and correlated the total number of marker alleles in each individual with the SMN2 copy number (table 5). In 80% of patients with type I SMA, 77% of patients with type II SMA, and 64% of patients with type III SMA, identical numbers of Ag1-CA alleles and SMN2 copies were found, whereas C212 alleles were identical with SMN2 copies in 75% of patients with type I SMA, 62% of patients with type II SMA, and 63% of patients with type III SMA, respectively. As expected, Ag1-CA showed a slightly higher correlation, on the basis of its closer proximity to the 5′ end of the SMN gene, than C212. In some cases, false interpretation of the autoradiographies (duplicated alleles are sometimes not easy to recognize) may be the cause of the discordant data. However, these data also suggest that, at least in part, incomplete deletions or duplications of SMN2 copies may be responsible for the discrepancies seen in SMN2 copy number versus Ag1-CA/C212 alleles.
Table 4.
Distribution of SMN2 Copy Number in Females and Males of Patients with Type I, Type II, and Type III SMA
| Distribution forSMN2 Copy Number | |||||
|---|---|---|---|---|---|
| Sex and Disease | 1 | 2 | 3 | 4 | Total |
| Female: | |||||
| Type I SMA | 7 | 66 | 13 | 0 | 86 |
| Type II SMA | 0 | 11a | 38 | 1 | 50 |
| Type III SMA | 0 | 1 | 15 | 18 | 34 |
| Male: | |||||
| Type I SMA | 6 | 72 | 24 | 0 | 102 |
| Type II SMA | 0 | 1a | 52 | 7 | 60 |
| Type III SMA | 0 | 2 | 24 | 17 | 43 |
Table 5.
Correlation between SMN2 Copy Number and Ag1-CA and C212 Marker Alleles
| No. of Marker Allelesa | ||||||
|---|---|---|---|---|---|---|
| PolymorphicMarker,Type of SMA,and SMN2Copy Number | 1 | 2 | 3 | 4 | 5 | % ofPatientsb |
| Ag1-CA: | ||||||
| Type I SMA: | ||||||
| 1 | 1 | 10 | 2 | |||
| 2 | 3 | 114 | 9 | 5 | 81 | |
| 3 | 1 | 9 | 27 | |||
| Type II SMA: | ||||||
| 2 | 7 | 4 | 1 | |||
| 3 | 11 | 77 | 1 | 1 | 77 | |
| 4 | 1 | 6 | 1 | |||
| Type III SMA: | ||||||
| 2 | 2 | 1 | ||||
| 3 | 3 | 28 | 7 | 64 | ||
| 4 | 5 | 11 | 19 | |||
| C212: | ||||||
| Type I SMA: | ||||||
| 1 | 1 | 10 | 2 | |||
| 2 | 8 | 108 | 11 | 4 | 75 | |
| 3 | 1 | 8 | 27 | 1 | ||
| Type II SMA: | ||||||
| 2 | 1 | 6 | 4 | 1 | ||
| 3 | 26 | 60 | 2 | 62 | ||
| 4 | 2 | 5 | 1 | |||
| Type III SMA: | ||||||
| 2 | 2 | 1 | ||||
| 3 | 6 | 28 | 4 | 63 | ||
| 4 | 4 | 13 | 18 |
Furthermore, we checked for a correlation between number of SMN2 copies and age of survival in 113 patients with type I SMA (fig. 4_a_). In 9 of 113 patients with type I SMA, one SMN2 copy was found with a median age of survival of 7 mo; none survived longer than 11 mo. In 94 of 113 patients with type I SMA, two SMN2 copies were found, with a median age of survival of 8 mo. All but six patients with two SMN2 copies died before age 21 mo. In 10 of 113 patients, three SMN2 copies were determined, with a median age of survival of 37.5 mo; 8 of these 10 patients lived 33–66 mo, and 6 of those 8 also revealed three Ag1-CA alleles, indicating that these patients with type I SMA owe their increased survival to the increased number of SMN2 copies. On the basis of Kaplan-Meier survival curves, we calculated the probability of survival for patients with type I SMA in accordance with their SMN2 copy number (fig. 4_b_).
Figure 4.

a, Age of survival of children with type I SMA, correlated with the average SMN2 value. Circles correspond to one SMN2 copy, squares to two SMN2 copies, and triangles to three SMN2 copies. The line represents a trendline for the age of survival versus the SMN2 copy number. b, Kaplan-Meier curves show the probabilities of survival for patients with type I SMA who carry one, two, or three SMN2 copies.
Prediction of SMA Severity
Finally, we calculated the posterior probability of a child with homozygous absence of SMN1 developing type I, type II, or type III SMA, conditional on the number of SMN2 copies (table 6). A child with one SMN2 copy has a risk of >99%—and, with two SMN2 copies, a risk of 97%—of developing type I SMA. A child with three SMN2 copies has a risk of 82.8% of developing type II SMA, and a child with four SMN2 copies has a risk of 83.6% of developing type III SMA. These data demonstrate nicely that SMN2 copy number can be used as a prognostic tool.
Table 6.
Probabilities That an Unaffected Child Who Has Been Tested after Birth and Has Been Found to Carry a Homozygous Absence of SMN1 Will Develop Type I, II, or III SMA, on the Basis of Number of SMN2 Copies
| Probability ofDeveloping SMA | |||
|---|---|---|---|
| Probability Type andNo. of SMN2 Copies | Type I | Type II | Type III |
| Prior probabilities | .51 | .32 | .17 |
| Conditional probabilities (OR):a | |||
| One copy | 1.00 | <.001 | <.001 |
| Two copies | 1.00 | .044 | .0145 |
| Three copies | 1.00 | 18.4 | 4.19 |
| Four copies | 1.00 | 14.7 | 156.7 |
| Joint probabilities: | |||
| One copy | .51 | <.00032 | <.00017 |
| Two copies | .51 | .0141 | .000255 |
| Three copies | .51 | 5.89 | .712 |
| Four copies | .51 | 4.70 | 26.6 |
| Posterior probabilities (%): | |||
| One copy | >99.9 | <.1 | <.1 |
| Two copies | 97.26 | 2.7 | .04 |
| Three copies | 7.2 | 82.8 | 10.0 |
| Four copies | 1.6 | 14.8 | 83.6 |
Discussion
Here, we present the first quantitative test of SMN1 or SMN2 based on real-time PCR by use of the LightCycler Instrument. The possibility of direct measurement of the PCR product accumulated after each cycle, without any intermediate steps, ensures the high specificity of real-time PCR assays. We succeeded in developing the test so that gene-specific primers amplify only SMN1 but not SMN2 (or vice versa).
SMN1 dosage analysis is frequently requested in the context of genetic counseling, either as carrier testing or as a means of identifying patients with compound mutations (deletions and intragenic SMN1 mutation). Unfortunately, the presence of two SMN1 copies per chromosome or intragenic mutations in some of the SMA chromosomes slightly diminishes the sensitivity of the test (McAndrew et al. 1997; Wirth et al. 1999; Scheffer et al. 2000; present study). On the basis of all reported data, including this one, in 20/834 (2.4%) healthy chromosomes, two SMN1 copies were found. Consequently, 4.8% of normal individuals would be misinterpreted as noncarriers on the basis of the direct SMN1 test. The sensitivity of the test is 95.2%. In addition, 1.7% of SMA carriers reveal intragenic SMN1 mutations—given that 3.4% of patients with SMA show compound SMN1 mutations, deletion plus intragenic mutation (Wirth et al. 1999)—and would not be recognized by the SMN1 quantitative test. This reduces the sensitivity of the test to 93.5% for a person from the general population.
The quantification of SMN2 copies performed in this paper in 375 patients with type I, type II, and type III SMA demonstrates a significant correlation between SMN2 copy number and type of SMA, as well as duration of survival. Approximately 80% of patients with type I SMA showed only one or two SMN2 copies, suggesting that these patients mainly carry genuine deletions of SMN1, whereas SMN2 is present either on one chromosome (genotype 0:1 or 0:2) or on both chromosomes (genotype 1:1). Furthermore, for patients with type I SMA, we were able to show an inverse correlation between SMN2 copy number and duration of survival; patients with one SMN2 copy had a median survival of 7 mo, whereas those with two or three SMN2 copies survived 8 and 37.5 mo, respectively. Although, in the first months, the survival curves are relatively similar, the probability of survival for type I patients with one SMN2 copy is significantly lower than that for patients with two SMN2 copies (fig. 4_b_). The median age of survival for patients with one SMN2 copy might even be biased, given the inclusion criteria for the quantitative analysis. We considered only those patients from whom DNA was isolated by the same method. Patients who died soon after birth had to be excluded sometimes, since only small amounts of DNA or DNA from muscle biopsies were available.
Our results of SMN2 copy number also fit well with our previously reported SMA model (Wirth et al. 1995), where we hypothesized that patients with type I SMA carry two “severe” SMA chromosomes and that patients with type II SMA carry one “severe” and one “mild” SMA chromosome, whereas patients with type III SMA carry two “mild” SMA chromosomes. In ∼80% of patients with type I SMA, we identified one or two SMN2 copies, corresponding to deletions of SMN1 on both chromosomes. About 80% of patients with type II SMA carry three SMN2 copies, corresponding to a deletion of SMN1 on one chromosome and a gene conversion of SMN1 in SMN2 on the second chromosome, whereas ∼45% of patients with type III SMA showed four SMN2 copies, corresponding to gene conversion on both chromosomes.
In previous work, Burghes (1997) reported a quantitative analysis of SMN2 copies in 21 patients with type I SMA, 38.1%, 42.9%, and 19% of whom had SMN2 copy numbers of one, two, and three, respectively, and in 58 patients with type II and type III SMA, 12%, 48.3%, 34.5%, and 5.2% of whom had SMN2 copy numbers of one, two, three, and four, respectively. Since the number of patients with type I SMA is small and the numbers of patients with type II and type III SMA are not listed separately, it is difficult to compare the data. Similarly, Taylor et al. (1998) performed a quantitative analysis of SMN2 in 26 patients with type I SMA and 23 patients with type II or type III SMA and showed a significant correlation between lower ratios for SMN2 versus MPZ (control gene) in type I and between higher ratios in type II and III. However, the authors stated that they were unable to assess exact copy number from those ratios.
The impressive progress achieved in the last few years in the understanding of the molecular basis of SMA and the recent identification of drugs that increase full-length SMN2 mRNA open the possibility of a therapy for patients with SMA in the near future. Quantification of the SMN2 copies in patients with SMA will be an essential prerequisite of therapy. Clinicians could start therapy and adapt drug treatment depending on the SMN2 copies before motor neurons are affected and children show first symptoms.
Our data show that, although various factors—such as modifying genes, external factors, intragenic SMN2 mutations, and incomplete SMN2 copies—can slightly influence the SMA phenotype, the risk calculation for a child with homozygous absence of SMN1 to develop a type I, II or III SMA can be reliably predicted. This study will open new diagnostic possibilities for routine laboratories and will be essential in clinical trials in patients with SMA.
Acknowledgments
We are grateful to all families with SMA and clinicians, especially Dr. K. Zerres and Dr. S. Rudnik-Schöneborn. We thank S. Raeder and H. Raschke for excellent technical assistance and Dr. Olfert Landt (TIB MOLBIOL) for contributing to the PCR primer design. We are grateful to Drs. W. Friedl, P. Propping, and Y. Hofmann for critical reading of the manuscript. This work was supported by the Deutsche Forschungsgemeinschaft (SFB400 A6 and Z2, and Graduiertenkolleg 246 T6), Families of SMA, and BONFOR.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for type I SMA [Werdnig-Hoffmann disease; MIM 253300], type II SMA [MIM 253550], and type III SMA [Kugelberg-Welander disease; MIM 253400])
References
- Burghes AHM (1997) When is a deletion not a deletion? When it is converted. Am J Hum Genet 61:9–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bürglen L, Lefebvre S, Clermont O, Burlet P, Viollet L, Cruaud C, Munnich A, et al (1996_a_) Structure and organization of the human survival motor neuron (SMN) gene. Genomics 32:479–482 [DOI] [PubMed] [Google Scholar]
- Chang JG, Hsieh-Li HM, Jong YJ, Wang NM, Tsai CH, Li H (2001) Treatment of spinal muscular atrophy by sodium butyrate. Proc Natl Acad Sci USA 98:9808–9813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coovert DD, Le TT, McAndrew PE, Strasswimmer J, Crawford TO, Mendell JR, Coulson SE, Androphy EJ, Prior TW, Burghes AH (1997) The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet 6:1205–1214 [DOI] [PubMed] [Google Scholar]
- Hausmanowa-Petrusewicz I, Zaremba J, Borkowska J (1985) Chronic proximal spinal muscular atrophy of childhood and adolescence: problems of classification and genetic counseling. J Med Genet 22:350–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmken C, Wetter A, Rudnik-Schöneborn S, Liehr T, Zerres K, Wirth B (2000_a_) An essential SMN interacting protein (SIP1) is not involved in the phenotypic variability of spinal muscular atrophy (SMA). Eur J Hum Genet 8:493–499 [DOI] [PubMed] [Google Scholar]
- Helmken C, Wirth B (2000_b_) Exclusion of Htra2-b1, an up-regulator of full-length SMN2 transcript, as a modifying gene for spinal muscular atrophy. Hum Genet 107:554–558 [DOI] [PubMed] [Google Scholar]
- Hofmann Y, Lorson CL, Stamm S, Androphy EJ, Wirth B (2000) Htra2-β 1 stimulates an exonic splicing enhancer and can restore full-length SMN expression to survival motor neuron 2 (SMN2). Proc Natl Acad Sci USA 97:9618–9623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosmer DW, Lemeshow S (2000) Applied logistic regression. John Wiley & Sons, New York [Google Scholar]
- Hsieh-Li HM, Chang JG, Jong YJ, Wu MH, Wang NM, Tsai CH, Li H (2000) A mouse model for spinal muscular atrophy. Nat Genet 24:66–70 [DOI] [PubMed] [Google Scholar]
- International SMA Consortium (1992) Meeting report: International SMA Consortium Meeting. Neuromusc Disord 2:423–428 [DOI] [PubMed] [Google Scholar]
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, et al (1995) Identification and characterization of the spinal muscular atrophy determining gene. Cell 80:155–165 [DOI] [PubMed] [Google Scholar]
- Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, Dreyfuss G, Melki J (1997) Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 16:265–269 [DOI] [PubMed] [Google Scholar]
- Lorson CL, Hahnen E, Androphy EJ, Wirth B (1999) A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA 96:6307–6311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAndrew PE, Parsons DW, Simard LR, Rochette C, Ray PN, Mendell J, Prior TW, et al (1997) Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNt and SMNc gene copy number. Am J Hum Genet 60:1411–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AH, McPherson JD (1999) A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet 8:1177–1183 [DOI] [PubMed] [Google Scholar]
- Monani UR, Sendtner M, Coovert DD, Parsons DW, Andreassi C, Le TT, Jablonka S, et al (2000) The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(−/−) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet 9:333–339 [DOI] [PubMed] [Google Scholar]
- Pearn J (1980) Classification of spinal muscular atrophies. Lancet 1:919–922 [DOI] [PubMed] [Google Scholar]
- SAS Institute (1996) SAS/STAT software: changes and enhancements through release 6.11. SAS Institute, Cary, NC [Google Scholar]
- Scheffer H, Cobben JM, Matthijes G, Wirth B (2001) Best practice guidelines for molecular analysis in spinal muscular atophy. Eur J Hum Genet 9:484–491 [DOI] [PubMed] [Google Scholar]
- Scheffer H, Cobben JM, Mensink RG, Stulp RP, van der Steege G, Buys CH (2000) SMA carrier testing--validation of hemizygous SMN exon 7 deletion test for the identification of proximal spinal muscular atrophy carriers and patients with a single allele deletion. Eur J Hum Genet 8:79–86 [DOI] [PubMed] [Google Scholar]
- Taylor JE, Thomas NH, Lewis CM, Abbs SJ, Rodrigues NR, Davies KE, Mathew CG (1998) Correlation of SMNt and SMNc gene copy number with age of onset and survival in spinal muscular atrophy. Eur J Hum Genet 6:467–474 [DOI] [PubMed] [Google Scholar]
- Velasco E, Valero C, Valero A, Moreno F, Hernandez-Chico C (1996) Molecular analysis of the SMN and NAIP genes in Spanish spinal muscular atrophy (SMA) families and correlation between number of copies of cBCD541 and SMA phenotype. Hum Mol Genet 5:257–263 [DOI] [PubMed] [Google Scholar]
- Wirth B (2000) An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum Mutat 15:228–223 [DOI] [PubMed] [Google Scholar]
- Wirth B, Hahnen E, Morgan K, DiDonato CJ, Dadze A, Rudnik-Schoneborn S, Simard LR, Zerres K, Burghes AH (1995) Allelic association and deletions in autosomal recessive proximal spinal muscular atrophy: association of marker genotype with disease severity and candidate cDNAs. Hum Mol Genet 4:1273–1284 [DOI] [PubMed] [Google Scholar]
- Wirth B, Herz M, Wetter A, Moskau S, Hahnen E, Rudnik-Schöneborn S, Wienker T, Zerres K (1999) Quantitative analysis of survival motor neuron copies: identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implication for genetic counseling. Am J Hum Genet 64:1340–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth B, Schmidt T, Hahnen E, Rudnik-Schöneborn S, Krawczak M, Müller-Myhsok B, Schönling J, et al (1997) De novo rearrangements found in 2% index patients with spinal muscular atrophy (SMA): mutational mechanisms, parental origin, mutation rate and implications for prenatal diagnosis. Am J Hum Genet 61:1102–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerres K, Rudnik-Schöneborn S (1995) Natural history in proximal spinal muscular atrophy (SMA): clinical analysis of 445 patients and suggestions for a modification of existing classifications. Arch Neurol 52:518–523 [DOI] [PubMed] [Google Scholar]