Modulation of mitochondrial function and autophagy mediates carnosine neuroprotection against ischemic brain damage (original) (raw)
. Author manuscript; available in PMC: 2015 Aug 1.
Abstract
Background and Purpose
Despite the rapidly increasing global burden of ischemic stroke, no therapeutic options for neuroprotection against stroke currently exist. Recent studies have shown that autophagy plays a key role in ischemic neuronal death and treatments that target autophagy may represent a novel strategy in neuroprotection. We investigated whether autophagy is regulated by carnosine, an endogenous pleiotropic dipeptide which has robust neuroprotective activity against ischemic brain damage.
Methods
We examined the effect of carnosine on mitochondrial dysfunction and autophagic processes in rat focal ischemia and in neuronal cultures.
Results
Autophagic pathways such as reduction of phosphorylated mTOR/p70S6K and the conversion of LC3-I to LC3-II were enhanced in the ischemic brain. However, treatment with carnosine significantly attenuated autophagic signaling in the ischemic brain, with improvement of brain mitochondrial function and mitophagy signaling. The protective effect of carnosine against autophagy was also confirmed in primary cortical neurons.
Conclusion
Taken together, our data suggest that the neuroprotective effect of carnosine is at least partially mediated by mitochondrial protection, and attenuation of deleterious autophagic processes. Our findings shed new light on the mechanistic pathways that this exciting neuroprotective agent influences.
Keywords: carnosine, neuroprotection, ischemic stroke, autophagy, mitochondria
Introduction
Despite the high prevalence and the increasing global burden of ischemic stroke, there are no approved neuroprotective agents in clinical use. The only approved therapy is thrombolysis with tissue plasminogen activator (tPA), which has a narrow therapeutic window and hemorrhagic side effects that limit clinical use. There have been extensive efforts to develop novel therapeutic candidates for ischemic stroke.1,2 However, numerous promising candidates have failed in clinical trials due to a number of factors which include poor preclinical study design, illogical clinical translation of preclinical data, poor efficacy and serious side effects.3,4 Moreover, understanding the precise mechanisms through which candidate agents exert their protective effects is an important and critical part of therapy development. Agents that influence multiple deleterious pathways are more likely to be efficacious clinically.5,6
There is increasing evidence that autophagy, a highly regulated cellular process that involves degradation of cellular proteins and organelles, can contribute to neuronal death during brain ischemia. Enhancement of autophagic processes was observed in brain after hypoxic-ischemia,7 and the occurrence of autophagy measured by conversion of LC3-I to LC3-II during brain ischemia has been confirmed by in vivo imaging.8 Although controversy exists whether autophagy contributes to cell death or cell survival,9-11 recent observations using inhibitors or modulators of autophagy revealed that autophagy mediates neuronal cell death during ischemia.12,13 Wen et al14 observed autophagy in focal cerebral ischemia, and demonstrated that treatment with inhibitors of autophagy significantly reduced brain damage. Data also exist showing that neuronal death during ischemia is mediated by oxidative stress generated from autophagosomes and mitochondria that are participating in the autophagic process.15
Activation of autophagic pathways is associated with perturbations in mitochondrial function.16 Mitochondrial damage is known to result in activation of mitophagy, a specific type of autophagy that eliminates dysfunctional mitochondria,17,18 under normal as well as pathological conditions including cerebral ischemia.19 Despite the increasing attention on autophagy as a novel target for stroke therapy development, studies on agents that modulate autophagy and that could be used clinically are still limited.
Carnosine, an endogenous dipeptide, is a pleotropic agent that exhibits diverse activities including anti-oxidant, anti-matrix metalloproteinase, heavy metal chelating and anti-excitotoxic properties.20,21 We recently showed that carnosine robustly reduced brain damage after ischemic stroke.22-25 Post-treatment with carnosine protected against histological brain damage both in permanent- and transient-ischemic rat models with a wide clinically relevant therapeutic window of 9 hr and 6 hr, respectively, along with improvements in functional outcomes.23 Carnosine did not exhibit any side effects or organ toxicity.23,25 Along with our observation, others have also reported the robust neuroprotective activity of carnosine.26-28 However, it is not known whether carnosine can influence autophagy in the ischemic brain.
In the current study, we have investigated whether carnosine has the ability to modulate autophagic processes in the ischemic brain using both in vitro and in vivo approaches. We extended our studies to mitochondria and showed that carnosine has a significant and profound effect on autophagy and associated mitochondrial perturbations that occur during ischemia. Our findings support the pleiotropic multimodal action of carnosine and provide, for the first time, proof of its influence on autophagy.
Materials and Methods
More details are provided in the online supplemental material.
Animals
All animal experiments were conducted using adult male Sprague-Dawley rats weighing 250 to 300 g (Harlan, Koatech, Korea) and performed in accordance with the NIH Policy and Animal Welfare Act under the approval by Institutional Animal Care and Use Committee (IACUC) at Hanyang University.
Blinding and Randomization
Treatment groups were allocated in a randomized fashion. Investigators were blind to the allocation of treatment during surgeries and outcome evaluations.
Treatments
Carnosine was obtained from Sigma and dissolved in saline. Carnosine (1,000 mg/kg) was administered into the lateral tail vein at 6 hr after ischemic onset both in permanent and transient models. The choice of this dose and time window is based on previous dose finding studies.22-25
Ischemic stroke in rats
Permanent or transient focal cerebral ischemia was induced by intraluminal middle cerebral artery occlusion (MCAO).23 Ischemia was initiated by a silicone-coated 4-0 monofilament nylon suture (Doccol Co.) as described previously.23,29
Calculation of infarct volume
At 24 hr after onset of ischemia, rats were euthanized by isoflurane overdose, and the isolated brains were cut into 2 mm sections. The infarct volume for each section was calculated by 2% triphenyltetrazolium chloride (TTC).30
Assessment of neurological function
Deficit in neurological function was evaluated by behavioral tests including the adhesive tape removal test and a Rota Rod test, at 24 hr after tMCAO (6 hr ischemia).23,31 All rats were trained to the tests for 5 consecutive days before focal ischemia.
Brain homogenization and mitochondria isolation
Brain samples between bregma levels +2 and -4 mm, which include ischemic core and penumbra, were rapidly isolated at 24 hrs after MCAO, and brain homogenates were obtained by homogenization in isolation buffer. Brain mitochondria was further isolated using Percoll gradient centrifugation.32
Western Blot of brain homogenate or isolated brain mitochondria
Processed brain homogenates or brain mitochondria were examined in western blot using Tris-HCl SDS-PAGE.23,32 Detailed information on primary antibodies is described in the online supplemental material.
Complex I activity
Complex I activity in isolated brain mitochondria was measured using colorimetric method as previously described with 2,6 dichloroindophenol (DCIP).33
In vitro culture of primary cortical neurons
Primary cortical neuronal cultures were established as described previously.22 Neuronal cultures were maintained in a CO2 incubator at 37°C, and used between days in vitro (DIV) 7 and 11.
NMDA-induced excitotoxicity
Ischemic neuronal damage was examined by N-methyl-daspartate (NMDA)-induced excitotoxicity.34 NMDA-induced cytotoxicity was measured at 24 hr after NMDA exposure by leakage of lactate dehydrogenase (LDH). Alterations in cellular proteins were assessed by western blot as described earlier, with cell lysates extracted from neuronal cells using RIPA buffer (Thermo Scientific). To examine carnosine protection, cells were pretreated with carnosine for 30 min prior to NMDA stimulation.
Statistics
We calculated the means and standard errors of means (SEM) for all treatment groups. Differences in values were analyzed using Student t-test or analysis of variance (ANOVA), as appropriate, using SPSS software (Chicago, IL). Multiple comparisons were made using one-way ANOVA followed by Tukey test. Two-tailed Student’s t-test analysis was used for comparing values between two groups. In all cases, a p value of < 0.05 was considered significant.
Results
Carnosine protects the ischemic brain in focal stroke
First, we examined the neuroprotective effect of carnosine in rat focal ischemia. All physiological variables including body temperature and cerebral blood flow (CBF) were maintained in the reference range. Induction of focal ischemia was attained by middle cerebral artery occlusion (MCAO) and verified by monitoring of CBF. Post-treatment with carnosine (1000 mg/kg) at 6 hr significantly reduced brain infarct volume (Fig. 1A), measured by TTC-staining. Similarly, we found that carnosine improved functional outcomes following 6 hr transient MCAO, using a variety of tests which included the latency for removal of adhesive tape placed on forelimbs and the latencies to fall off from the accelerating Rota Rod, respectively.23,31 (Fig. 1B and 1C).
Figure 1. Protective effect of carnosine against brain damage during ischemic stroke.

Ischemic stroke was achieved by middle cerebral artery occlusion (MCAO) in rats. (A) Carnosine (1000 mg/kg) was administered 6 hr after onset of ischemia. Infarct volume was determined by 2,3,5-triphenyltetrazolium chloride staining at 24 hr after MCAO. The representative photos are shown. N=13-15. *p<0.05 vs. saline-treated rats. (B and C) Carnosine (1000 mg/kg) was administered to rats at 6 hr after ischemic onset during transient MCAO (6 hr ischemia/18 hr reperfusion). Behavioral tests were performed at 24 hr before and after ischemia. (B) Somatosensory deficit was determined using the Adhesive Tape tests, where required time to remove adhesives on fore limbs were measured. (C) In the RotaRod test, motor-ambultatory function was determined. Latencies to fall off from the rotarod with accelerated speeds were measured. B: N=13-15, C: N=15-16. **p<0.01, #p<0.05 vs. the corresponding group. Data were expressed as mean ± SEM and analyzed by Student's t-test.
Carnosine reduced autophagy in brain homogenates
To investigate whether autophagic processes are involved in carnosine mediated protection, we examined the extent of conversion of LC3-I to LC3-II, an important marker of autophagy that is responsible for formation of autophagosome.35 A significant increase in LC3-II formation was observed in the ipsilateral hemisphere following ischemia. However, this increase in LC3-II formation was attenuated by treatment with carnosine (Fig. 2A). It is also well established that inhibition of the mTOR pathway plays a key role in autophagy.36 To investigate the effect of carnosine on the autophagic signaling pathway, we measured the levels of phospho-mTOR (p-mTOR) and phospho-p70S6K (p-p70S6K), a representative downstream target of mTOR,37 in brain homogenates after ischemia. Carnosine did not affect the basal activity of mTOR; similar levels of p-mTOR were observed in hemispheres contralateral to the ischemia in both saline- and carnosine-treated rats (Figure 2B). Ischemia inhibited the phosphorylated levels of mTOR, but this inhibition was blocked by carnosine. Similarly, reductions in the levels of p-p70S6K in ischemic brain were also reversed by carnosine (Fig. 2B). Taken together, these findings support the modulating role of carnosine on autophagy in the ischemic brain. While mTOR-autophagy pathways were significantly influenced by ischemia and reversed by carnosine, the level of phosphorylated ERK 1/2 was not changed either by ischemia or carnosine treatment (Fig. 2B), showing that the modulation of autophagic proteins by carnosine is not a non-specific epi-phenomenon.
Figure 2. Inhibitory effect of carnosine on autophagy in ischemic brain.
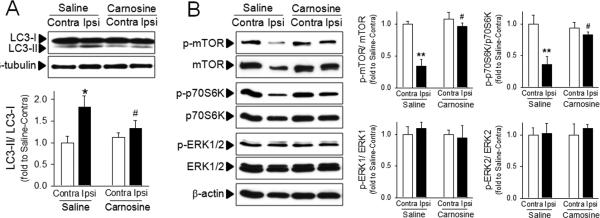
Brain homogenates were isolated from contralateral (Contra) or ipsilateral (Ipsi) hemispheres from saline- or carnosine (1000 mg/kg; 6 hr post treatment)-administered rats following pMCAO. (A) The extent of autophagy was examined using the conversion of LC3-II from LC3-I. (B) Autophagic signaling was examined by phosphorylation of mTOR, p70S6K and ERK. The representative bands are shown. Relative density of each band was analyzed by ImageJ. N=4. *p<0.05, **p<0.01 vs. contralateral hemisphere from saline-treated rats. #p<0.05 vs. ipsilateral hemisphere from saline-treated rats. Data were expressed as mean ± SEM and analyzed by Student's t-test.
Carnosine attenuates ischemic injury to mitochondria
We have previously reported that carnosine reversed the impairment of mitochondrial permeability transition in primary neurons and astrocytes. Since it is well established that mitochondrial dysfunction contributes to autophagy induction,16,18 we examined whether carnosine protected against mitochondrial damage and mitophagy. Ischemia resulted in decreased activity of complex I in isolated brain mitochondria suggesting impairment in mitochondrial respiratory function. Ischemic mitochondrial dysfunction was significantly reversed in mitochondria isolated from carnosine-treated rats (Fig. 3A). To determine if there is a link between mitochondrial dysfunction and autophagy, we examined the levels of p-Drp1 and Parkin which play key roles in mitochondrial fragmentation and mitophagy during cell death, respectively.38-40 The mitochondrial levels of p-Drp1 and Parkin were significantly increased by ischemia, but the increase of p-Drp1 and Parkin were attenuated by carnosine treatment (Fig. 3B).
Figure 3. Protective effect of carnosine on mitochondrial damage in ischemic brain.
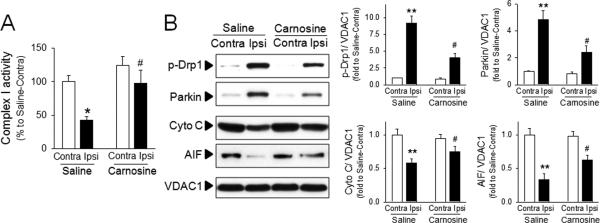
Brain mitochondria were isolated from contralateral (Contra) or ipsilateral (Ipsi) hemispheres from saline- or carnosine (1000 mg/kg; 6 hr post treatment)-administered rats following pMCAO. (A) Complex I activity was measured using colorimetric method. (B) The extent of mitochondrial fragmentation and mitophagy was examined using the level of p-Drp 1 and Parkin. Mitochondrial levels of apoptosis inducing factor (AIF) and cytochrome C were measured. Relative density of each band was analyzed by ImageJ. N=4. *p<0.05, **p<0.01 vs. contralateral hemisphere from saline-treated rats. #p<0.05 vs. ipsilateral hemisphere from saline-treated rats. Data were expressed as mean ± SEM and analyzed by Student's t-test.
While the levels of p-Drp1 and Parkin were increased by ischemia, the levels of cytochrome C and apoptosis-inducing factor (AIF) were significantly decreased in brain mitochondria following ischemic insult. Since cytochrome C and AIF are released from mitochondria to the cytosol during mitochondrial damage,32,41 these results were consistent with mitochondrial dysfunction. Carnosine potently inhibited the release of AIF and cytochrome C, demonstrating its protective activity on mitochondrial damage (Fig. 3B).
Carnosine protects against neuronal autophagy in culture
Primary cortical neurons were transiently exposed to toxic levels NMDA, and cytotoxicity and autophagic signaling pathways were examined. As shown in Figure 4A, NMDA induced significant cytotoxicity in primary cortical neurons, and NMDA-cytotoxicity was reduced by carnosine treatment. Interestingly, autophagic signaling pathways including LC3-II formation and mTOR de-phosphorylation were significantly enhanced by NMDA exposure, and carnosine reversed these changes (Fig. 4B), confirming the protective effect of carnosine against ischemia-induced neuronal autophagy.
Figure 4. Inhibitory effect of carnosine on neuronal autophagy following NMDA stimulation.
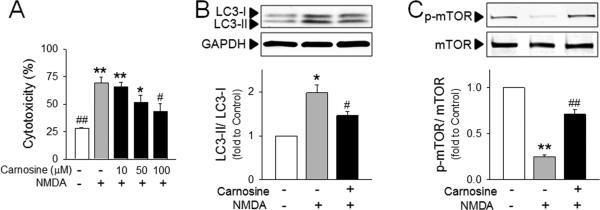
Primary cortical neurons were pre-treated with carnosine 30 min prior to exposure to NMDA (N-methyl-d-aspartate; 25 μM). (A) Neuronal cell death was determined by extent of lactate dehydrogenase leakage at 24 hr after NMDA stimulation. N=5. (B and C) The conversion of LC3-II from LC3-I (B) and the phosphorylation of mTOR (C) in NMDA (25 μM)-stimulated primary neurons with or without carnosine (100 μM) pretreatment. The representative bands are shown. Relative density of each band was analyzed by ImageJ. N=3. *p<0.05, **p<0.01 vs. control group. #p<0.05 vs. NMDA-treated group. Data were expressed as mean ± SEM and analyzed by one way ANOVA followed by Tukey test (A) or by Student's t-test (B and C).
Discussion
Stroke involves a cascade activation of multiple deleterious pathways,2,42,43 and therefore a drug candidate that specifically modulates a single pathway is not likely to show clinical efficacy against ischemic brain damage. Many therapeutic candidates including neuroprotectants which had strong protective activity pre-clinically have failed in clinical trials.1,4 One major reason for this is that past strategies have focused on targeting one pathway. We have shown that carnosine is an exciting candidate for development as a stroke therapy.23,25 It is safe and efficacious with a large clinically relevant therapeutic time window. Moreover, it is a pleiotropic agent that favorably modulates several deleterious pathways that contribute to cell injury and cell death during and after ischemia.21,44 We show here, using in vitro and in vivo approaches that carnosine has a profound and significant effect on autophagy, a recently identified noxious pathway in ischemic stroke. We believe that the current study underlines the translational importance of carnosine as a therapeutic candidate against ischemic stroke where multiple deleterious pathways aggravate neuronal damage.
Autophagy is the cellular process that mediates degradation of cellular proteins and organelles and maintains homeostasis.45 Despite its essential role in normal cellular physiology, excessive activation of autophagic pathways is also reported to be highly associated with many disease states including brain damage.46,47 Autophagic cell death has been referred to as type II cell death, which is one of the major types of cell death along with apoptotic (type I) and necrotic (type III) cell death.48,49 While necrotic and apoptotic cell deaths have long been considered as the main pathological events in ischemic stroke,50,51 autophagy has been recently recognized as a possible deleterious event also. Activation of autophagic signaling was observed in ischemic brain,52 mediating ischemic neuronal death.10 Notably, autophagic cell death was found to be the most important contributing pathway in neonatal cerebral ischemia relative to apoptosis and necrosis.53 Autophagy-inhibitors such as 3-MA significantly reverse ischemic brain damage14 and inhibition of autophagy was suggested to be the main mechanism of ischemic post-conditioning neuroprotection.54 Conversely, it has also been reported that autophagy may play a dual role in neuronal survival and death during ischemia,10 and further studies on the exact molecular targets which switch beneficial autophagy to detrimental autophagy would give valuable insights for development of treatments that modulate autophagy.
The role of mitochondrial dysfunction has been proposed as a contributor to autophagy.16 We and others have previously shown that ischemic insults to the brain induced mitochondrial permeability transition (MPT) resulting in damage to mitochondrial function in neurons.23,41 Onset of mitochondrial dysfunction is closely linked to initiation of autophagy in I/R injured myocytes,46 in rat hepatocytes,55 and in neurons.15 Damaged mitochondria releases cytochrome C (cyt C), AIF, and reactive oxygen species,17 which promote mitophagy, a form of autophagy that is involved in the removal of dysfunctional mitochondria. Recent data suggests that Parkin, an ubiquitin ligase that mediates mitophagy,40 is recruited to the damaged mitochondria.36,56 In this report, we observed the increased recruitment of Parkin to the mitochondria, and loss of AIF and cyt C from mitochondria in ischemic brain, which were significantly attenuated by carnosine, demonstrating its protective effect against mitophagy and ultimately autophagic neuronal death. Similarly, Mehta et al57 showed that selenium conserved mitochondrial function and stimulated mitochondria biogenesis, along with reduced autophagy in glutamate-induced neuronal toxicity.
Interest in the development of carnosine as an endogenous pleiotropic molecule for therapeutic use clinically has been increasing.20,44,58-60 Here we focused on the potential of carnosine against ischemic stroke. Several previous reports showed that carnosine also had beneficial activities in neurodegenerative diseases including Alzheimer diseases,61 and dementia.62 Of note, dysregulation of autophagic processes have been recently recognized to contribute to the progress of these neurodegenerative diseases.63,64 Further elucidation of carnosine's effects on autophagy in these neurodegenerative diseases is needed.
In summary, we have demonstrated that carnosine inhibits ischemia-induced autophagy and mitochondrial damage. This novel action of carnosine adds to the other body of compelling data that supports the development of carnosine as a therapeutic agent against ischemic stroke.
Supplementary Material
Online supplement
Acknowledgments
Source of Funding: This study was supported by the NIH and American Heart Association grants to Arshad Majid. This work was also supported by NRF-2012M3A9C6049935 and the DGIST Convergence Science Center Program (14-BD-04) to Seong Woon Yu, and by NRF-2012R1A1A3013240 to Ok-Nam Bae, funded by the Ministry of Science, ICT and Future Planning of Korea.
Footnotes
Conflict of interests; None
Disclosure: None
References
- 1.Fisher M. New approaches to neuroprotective drug development. Stroke. 2011;42:S24–27. doi: 10.1161/STROKEAHA.110.592394. [DOI] [PubMed] [Google Scholar]
- 2.Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dirnagl U, Macleod MR. Stroke research at a road block: the streets from adversity should be paved with meta-analysis and good laboratory practice. Br J Pharmacol. 2009;157:1154–1156. doi: 10.1111/j.1476-5381.2009.00211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Green AR. Pharmacological approaches to acute ischaemic stroke: reperfusion certainly, neuroprotection possibly. Br J Pharmacol. 2008;153(Suppl 1):S325–338. doi: 10.1038/sj.bjp.0707594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lapchak PA. Emerging Therapies: Pleiotropic Multi-target Drugs to Treat Stroke Victims. Transl Stroke Res. 2011;2:129–135. doi: 10.1007/s12975-011-0074-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xin H, Li Y, Cui Y, Yang JJ, Zhang ZG, Chopp M. Systemic administration of exosomes released from mesenchymal stromal cells promote functional recovery and neurovascular plasticity after stroke in rats. J Cereb Blood Flow Metab. 2013;33:1711–1715. doi: 10.1038/jcbfm.2013.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ginet V, Puyal J, Clarke PG, Truttmann AC. Enhancement of autophagic flux after neonatal cerebral hypoxia-ischemia and its region-specific relationship to apoptotic mechanisms. Am J Pathol. 2009;175:1962–1974. doi: 10.2353/ajpath.2009.090463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pamenter ME, Perkins GA, McGinness AK, Gu XQ, Ellisman MH, Haddad GG. Autophagy and apoptosis are differentially induced in neurons and astrocytes treated with an in vitro mimic of the ischemic penumbra. PLoS One. 2012;7:e51469. doi: 10.1371/journal.pone.0051469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu CL, Chen S, Dietrich D, Hu BR. Changes in autophagy after traumatic brain injury. J Cereb Blood Flow Metab. 2008;28:674–683. doi: 10.1038/sj.jcbfm.9600587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rami A, Kogel D. Apoptosis meets autophagy-like cell death in the ischemic penumbra: Two sides of the same coin? Autophagy. 2008;4:422–426. doi: 10.4161/auto.5778. [DOI] [PubMed] [Google Scholar]
- 11.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 13.Shi R, Weng J, Zhao L, Li XM, Gao TM, Kong J. Excessive autophagy contributes to neuron death in cerebral ischemia. CNS Neurosci Ther. 2012;18:250–260. doi: 10.1111/j.1755-5949.2012.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wen YD, Sheng R, Zhang LS, Han R, Zhang X, Zhang XD, et al. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy. 2008;4:762–769. doi: 10.4161/auto.6412. [DOI] [PubMed] [Google Scholar]
- 15.Kubota C, Torii S, Hou N, Saito N, Yoshimoto Y, Imai H, et al. Constitutive reactive oxygen species generation from autophagosome/lysosome in neuronal oxidative toxicity. J Biol Chem. 2010;285:667–674. doi: 10.1074/jbc.M109.053058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vosler PS, Graham SH, Wechsler LR, Chen J. Mitochondrial targets for stroke: focusing basic science research toward development of clinically translatable therapeutics. Stroke. 2009;40:3149–3155. doi: 10.1161/STROKEAHA.108.543769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gottlieb RA, Carreira RS. Autophagy in health and disease. 5. Mitophagy as a way of life. Am J Physiol Cell Physiol. 2010;299:C203–210. doi: 10.1152/ajpcell.00097.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kubli DA, Gustafsson AB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res. 2012;111:1208–1221. doi: 10.1161/CIRCRESAHA.112.265819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gabryel B, Kost A, Kasprowska D. Neuronal autophagy in cerebral ischemia--a potential target for neuroprotective strategies? Pharmacol Rep. 2012;64:1–15. doi: 10.1016/s1734-1140(12)70725-9. [DOI] [PubMed] [Google Scholar]
- 20.Vistoli G, Carini M, Aldini G. Transforming dietary peptides in promising lead compounds: the case of bioavailable carnosine analogs. Amino Acids. 2012;43:111–126. doi: 10.1007/s00726-012-1224-z. [DOI] [PubMed] [Google Scholar]
- 21.Horning MS, Blakemore LJ, Trombley PQ. Endogenous mechanisms of neuroprotection: role of zinc, copper, and carnosine. Brain Res. 2000;852:56–61. doi: 10.1016/s0006-8993(99)02215-5. [DOI] [PubMed] [Google Scholar]
- 22.Bae ON, Majid A. Role of histidine/histamine in carnosine-induced neuroprotection during ischemic brain damage. Brain Res. 2013;1527:246–254. doi: 10.1016/j.brainres.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 23.Bae ON, Serfozo K, Baek SH, Lee KY, Dorrance A, Rumbeiha W, et al. Safety and efficacy evaluation of carnosine, an endogenous neuroprotective agent for ischemic stroke. Stroke. 2013;44:205–212. doi: 10.1161/STROKEAHA.112.673954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Min J, Senut MC, Rajanikant K, Greenberg E, Bandagi R, Zemke D, et al. Differential neuroprotective effects of carnosine, anserine, and N-acetyl carnosine against permanent focal ischemia. J Neurosci Res. 2008;86:2984–2991. doi: 10.1002/jnr.21744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rajanikant GK, Zemke D, Senut MC, Frenkel MB, Chen AF, Gupta R, et al. Carnosine is neuroprotective against permanent focal cerebral ischemia in mice. Stroke. 2007;38:3023–3031. doi: 10.1161/STROKEAHA.107.488502. [DOI] [PubMed] [Google Scholar]
- 26.Dobrota D, Fedorova T, Stvolinsky S, Babusikova E, Likavcanova K, Drgova A, et al. Carnosine protects the brain of rats and Mongolian gerbils against ischemic injury: after-stroke-effect. Neurochem Res. 2005;30:1283–1288. doi: 10.1007/s11064-005-8799-7. [DOI] [PubMed] [Google Scholar]
- 27.Gallant S, Kukley M, Stvolinsky S, Bulygina E, Boldyrev A. Effect of carnosine on rats under experimental brain ischemia. Tohoku J Exp Med. 2000;191:85–99. doi: 10.1620/tjem.191.85. [DOI] [PubMed] [Google Scholar]
- 28.Zhang X, Song L, Cheng X, Yang Y, Luan B, Jia L, et al. Carnosine pretreatment protects against hypoxia-ischemia brain damage in the neonatal rat model. Eur J Pharmacol. 2011;667:202–207. doi: 10.1016/j.ejphar.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 29.Lee KY, Bae ON, Serfozo K, Hejabian S, Moussa A, Reeves M, et al. Asiatic acid attenuates infarct volume, mitochondrial dysfunction, and matrix metalloproteinase-9 induction after focal cerebral ischemia. Stroke. 2012;43:1632–1638. doi: 10.1161/STROKEAHA.111.639427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang G, Park D, Lee SH, Bae DK, Yang YH, Kyung J, et al. Neuroprotective Effects of a Butanol Fraction of Rosa hybrida Petals in a Middle Cerebral Artery Occlusion Model. Biomol Ther (Seoul) 2013;21:454–461. doi: 10.4062/biomolther.2013.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Fan X, Yu Z, Liao Z, Zhao J, Mandeville E, et al. Effects of tissue plasminogen activator and annexin A2 combination therapy on long-term neurological outcomes of rat focal embolic stroke. Stroke. 2014;45:619–622. doi: 10.1161/STROKEAHA.113.003823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baek SH, Bae ON, Kim EK, Yu SW. Induction of mitochondrial dysfunction by poly(ADP-ribose) polymer: implication for neuronal cell death. Mol Cells. 2013;36:258–266. doi: 10.1007/s10059-013-0172-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Janssen AJ, Trijbels FJ, Sengers RC, Smeitink JA, van den Heuvel LP, Wintjes LT, et al. Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin Chem. 2007;53:729–734. doi: 10.1373/clinchem.2006.078873. [DOI] [PubMed] [Google Scholar]
- 34.Panickar KS, Nonner D, White MG, Barrett JN. Overexpression of Cdk5 or non-phosphorylatable retinoblastoma protein protects septal neurons from oxygen-glucose deprivation. Neurochem Res. 2008;33:1852–1858. doi: 10.1007/s11064-008-9647-3. [DOI] [PubMed] [Google Scholar]
- 35.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 36.Yu J, Parkhitko AA, Henske EP. Mammalian target of rapamycin signaling and autophagy: roles in lymphangioleiomyomatosis therapy. Proc Am Thorac Soc. 2010;7:48–53. doi: 10.1513/pats.200909-104JS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koh PO, Cho JH, Won CK, Lee HJ, Sung JH, Kim MO. Estradiol attenuates the focal cerebral ischemic injury through mTOR/p70S6 kinase signaling pathway. Neurosci Lett. 2008;436:62–66. doi: 10.1016/j.neulet.2008.02.061. [DOI] [PubMed] [Google Scholar]
- 38.Jagasia R, Grote P, Westermann B, Conradt B. DRP-1-mediated mitochondrial fragmentation during EGL-1-induced cell death in C. elegans. Nature. 2005;433:754–760. doi: 10.1038/nature03316. [DOI] [PubMed] [Google Scholar]
- 39.Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012–2022. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]
- 40.Springer W, Kahle PJ. Regulation of PINK1-Parkin-mediated mitophagy. Autophagy. 2011;7:266–278. doi: 10.4161/auto.7.3.14348. [DOI] [PubMed] [Google Scholar]
- 41.Perez-Pinzon MA, Stetler RA, Fiskum G. Novel mitochondrial targets for neuroprotection. J Cereb Blood Flow Metab. 2012;32:1362–1376. doi: 10.1038/jcbfm.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Majid A. Neuroprotection in Stroke: Past, Present, and Future. ISRN Neurology. 2014;2014:515716. doi: 10.1155/2014/515716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saleem S, Zhuang H, Biswal S, Christen Y, Dore S. Ginkgo biloba extract neuroprotective action is dependent on heme oxygenase 1 in ischemic reperfusion brain injury. Stroke. 2008;39:3389–3396. doi: 10.1161/STROKEAHA.108.523480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bellia F, Vecchio G, Cuzzocrea S, Calabrese V, Rizzarelli E. Neuroprotective features of carnosine in oxidative driven diseases. Mol Aspects Med. 2011;32:258–266. doi: 10.1016/j.mam.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 45.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gustafsson AB, Gottlieb RA. Autophagy in ischemic heart disease. Circ Res. 2009;104:150–158. doi: 10.1161/CIRCRESAHA.108.187427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He Y, Wan S, Hua Y, Keep RF, Xi G. Autophagy after experimental intracerebral hemorrhage. J Cereb Blood Flow Metab. 2008;28:897–905. doi: 10.1038/sj.jcbfm.9600578. [DOI] [PubMed] [Google Scholar]
- 48.Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krysko DV, Vanden Berghe T, D'Herde K, Vandenabeele P. Apoptosis and necrosis: detection, discrimination and phagocytosis. Methods. 2008;44:205–221. doi: 10.1016/j.ymeth.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 50.Harraz MM, Dawson TM, Dawson VL. Advances in neuronal cell death 2007. Stroke. 2008;39:286–288. doi: 10.1161/STROKEAHA.107.511857. [DOI] [PubMed] [Google Scholar]
- 51.Lo EH. A new penumbra: transitioning from injury into repair after stroke. Nat Med. 2008;14:497–500. doi: 10.1038/nm1735. [DOI] [PubMed] [Google Scholar]
- 52.Tian F, Deguchi K, Yamashita T, Ohta Y, Morimoto N, Shang J, et al. In vivo imaging of autophagy in a mouse stroke model. Autophagy. 2010;6:1107–1114. doi: 10.4161/auto.6.8.13427. [DOI] [PubMed] [Google Scholar]
- 53.Puyal J, Vaslin A, Mottier V, Clarke PG. Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann Neurol. 2009;66:378–389. doi: 10.1002/ana.21714. [DOI] [PubMed] [Google Scholar]
- 54.Gao L, Jiang T, Guo J, Liu Y, Cui G, Gu L, et al. Inhibition of autophagy contributes to ischemic postconditioning-induced neuroprotection against focal cerebral ischemia in rats. PLoS One. 2012;7:e46092. doi: 10.1371/journal.pone.0046092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001;15:2286–2287. doi: 10.1096/fj.01-0206fje. [DOI] [PubMed] [Google Scholar]
- 56.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mehta SL, Kumari S, Mendelev N, Li PA. Selenium preserves mitochondrial function, stimulates mitochondrial biogenesis, and reduces infarct volume after focal cerebral ischemia. BMC Neurosci. 2012;13:79. doi: 10.1186/1471-2202-13-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barski OA, Xie Z, Baba SP, Sithu SD, Agarwal A, Cai J, et al. Dietary carnosine prevents early atherosclerotic lesion formation in apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2013;33:1162–1170. doi: 10.1161/ATVBAHA.112.300572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hipkiss AR. Carnosine and its possible roles in nutrition and health. Adv Food Nutr Res. 2009;57:87–154. doi: 10.1016/S1043-4526(09)57003-9. [DOI] [PubMed] [Google Scholar]
- 60.Hipkiss AR, Cartwright SP, Bromley C, Gross SR, Bill RM. Carnosine: can understanding its actions on energy metabolism and protein homeostasis inform its therapeutic potential? Chem Cent J. 2013;7:38. doi: 10.1186/1752-153X-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Corona C, Frazzini V, Silvestri E, Lattanzio R, La Sorda R, Piantelli M, et al. Effects of dietary supplementation of carnosine on mitochondrial dysfunction, amyloid pathology, and cognitive deficits in 3×Tg-AD mice. PLoS One. 2011;6:e17971. doi: 10.1371/journal.pone.0017971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ma J, Xiong JY, Hou WW, Yan HJ, Sun Y, Huang SW, et al. Protective effect of carnosine on subcortical ischemic vascular dementia in mice. CNS Neurosci Ther. 2012;18:745–753. doi: 10.1111/j.1755-5949.2012.00362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:5. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 64.Martinet W, Agostinis P, Vanhoecke B, Dewaele M, De Meyer GR. Autophagy in disease: a double-edged sword with therapeutic potential. Clin Sci (Lond) 2009;116:697–712. doi: 10.1042/CS20080508. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Online supplement