Transnitrosylation from DJ-1 to PTEN Attenuates Neuronal Cell Death in Parkinson's Disease Models (original) (raw)
Abstract
Emerging evidence suggests that oxidative/nitrosative stress, as occurs during aging, contributes to the pathogenesis of Parkinson's disease (PD). In contrast, detoxification of reactive oxygen species and reactive nitrogen species can protect neurons. DJ-1 has been identified as one of several recessively inherited genes whose mutation can cause familial PD, and inactivation of DJ-1 renders neurons more susceptible to oxidative stress and cell death. DJ-1 is also known to regulate the activity of the phosphatase and tensin homolog (PTEN), which plays a critical role in neuronal cell death in response to various insults. However, mechanistic details delineating how DJ-1 regulates PTEN activity remain unknown. Here, we report that PTEN phosphatase activity is inhibited via a transnitrosylation reaction [i.e., transfer of a nitric oxide (NO) group from the cysteine residue of one protein to another]. Specifically, we show that DJ-1 is _S_-nitrosylated (forming SNO-DJ-1); subsequently, the NO group is transferred from DJ-1 to PTEN by transnitrosylation. Moreover, we detect SNO-PTEN in human brains with sporadic PD. Using x-ray crystallography and site-directed mutagenesis, we find that Cys106 is the site of _S_-nitrosylation on DJ-1 and that mutation of this site inhibits transnitrosylation to PTEN. Importantly, _S_-nitrosylation of PTEN decreases its phosphatase activity, thus promoting cell survival. These findings provide mechanistic insight into the neuroprotective role of SNO-DJ-1 by elucidating how DJ-1 detoxifies NO via transnitrosylation to PTEN. Dysfunctional DJ-1, which lacks this transnitrosylation activity due to mutation or prior oxidation (e.g., sulfonation) of the critical cysteine thiol, could thus contribute to neurodegenerative disorders like PD.
Keywords: DJ-1, nitric oxide, Parkinson's disease, PTEN, _S_-nitrosylation, transnitrosylation
Introduction
Overproduction of nitric oxide (NO) is known to cause deleterious consequence to neuronal function or survival, contributing to neurodegenerative disorders such as Parkinson's disease (PD; Nakamura and Lipton, 2013). Conversely, physiological levels of NO play an important role as a regulator of neuronal signaling pathways, homeostasis, and activity. A major signaling pathway that can mediate the biological functions of NO is protein _S_-nitrosylation, a post-translational redox modification of a cysteine thiol group by NO-related species (Nakamura et al., 2013). Emerging evidence suggests that protein–protein transnitrosylation (or transfer of an NO group from one _S_-nitrosothiol (SNO) protein to the free thiol of another protein) represents an important mechanism whereby proteins can be _S_-nitrosylated or denitrosylated (Nakamura and Lipton, 2013).
DJ-1 is a redox-active protein known to be involved in some cases of PD (Canet-Avilés et al., 2004). For example, loss-of-function mutations in the DJ-1 gene cause early-onset autosomal-recessive PD (Bonifati et al., 2003). Structurally, DJ-1 possesses three potentially redox-active cysteine residues (Cys46, Cys53, and Cys106). Among these cysteine residues, Cys106 is the most oxidation sensitive, forming a cysteine sulfinic acid (−SO2H) in the high-resolution crystal structure (Wilson et al., 2003; Canet-Avilés et al., 2004; Premkumar et al., 2011). Moreover, oxidation of Cys106 is critical for the neuroprotective activity of DJ-1 (Blackinton et al., 2009; Kim et al., 2009; Wilson, 2011). Providing neuroprotection, DJ-1 scavenges reactive oxygen species (ROS) via oxidation of Cys106; Cys106 oxidation enhances mitochondrial localization and neuroprotection of DJ-1 (Canet-Avilés et al., 2004; Andres-Mateos et al., 2007). In addition to sulfination or sulfonation (−SO3H) of DJ-1, NO has been reported to _S_-nitrosylate cysteine residues of DJ-1 in vitro (Ito et al., 2006), although the specific cysteines involved in this reaction remain contentious.
DJ-1 acts as a neuroprotective protein via modulation of multiple cellular survival signaling pathways, including the phosphatase and tensin homolog (PTEN)–phosphatidylinositol 3-kinase (PI3K)–Akt cascade (Kim et al., 2005; Yang et al., 2005; Aleyasin et al., 2010). PTEN phosphatase activity antagonizes PI3K and hence, downstream, neuroprotective Akt activity. Inactivation of PTEN in dopaminergic neurons provides protection against neurotoxic insults, pointing to a critical role of the PTEN–PI3K–Akt pathway in PD pathogenesis (Diaz-Ruiz et al., 2009). Prior studies using Drosophila and mouse models showed that DJ-1 functions as a negative regulator of PTEN, thus offering neuroprotection from oxidative stress (Kim et al., 2005; Aleyasin et al., 2010). Moreover, direct interaction between DJ-1 and PTEN appeared to inhibit PTEN phosphatase activity (Kim et al., 2009).
Recently, our group reported another mechanism of PTEN inhibition whereby NO _S_-nitrosylates PTEN to inhibit its phosphatase activity (Kwak et al., 2010; Numajiri et al., 2011). Here, linking these prior observations, we report that DJ-1 inhibits PTEN activity via transnitrosylation, thus suppressing PTEN-dependent cell death. Our findings therefore show that under conditions of mild nitrosative stress, formation of SNO-DJ-1 leads to transfer of the NO group to PTEN, thus inhibiting PTEN activity and providing neuroprotection.
Materials and Methods
Reagents, antibodies, and cell cultures.
The calcium ionophore A23187 was from Life Technologies, and NG-nitro-l-arginine was from Enzo Life Sciences. Anti-PTEN (138G6), myc (9B11), and Akt (catalog #9272) antibodies were from Cell Signaling Technology; mouse monoclonal DJ-1 antibody (3E8) was from Enzo Life Sciences; rabbit polyclonal DJ-1 antibody (NB300-270) was from Novus Biologicals; anti-V5 antibody (R960-25) was from Life Technologies; and anti-actin antibody (C4) was from Millipore. _S_-Nitroso cysteine (SNOC) was prepared as described previously (Lei et al., 1992). Lipofectamine 2000, Lipofectamine LTX, and Plus Reagent were from Life Technologies. Human embryonic kidney (HEK) cells stably expressing neuronal NO synthase (nNOS; a gift from Drs. Solomon Snyder and David Bredt, Johns Hopkins University School of Medicine, Baltimore, MD) and neuroblastoma SH-SY5Y cells (from ATCC) were grown in DMEM (Life Technologies) containing 10% fetal bovine serum at 37°C under 5% CO2/95% air. A plasmid encoding HA-tagged PTEN was obtained from Addgene, and myc- or V5-tagged DJ-1 plasmids were provided by Drs. M. Cookson (National Institutes of Health, Bethesda) and C. Haass (German Center for Neurodegenerative Diseases, Munich) (Canet-Avilés et al., 2004; Görner et al., 2007). Silencer select siRNA for PARK7/DJ-1 (siRNA ID: s230250) was from Life Technologies.
Biotin-switch assay.
Cell or brain lysates were prepared in HENTS buffer (100 mm HEPES, 1 mm EDTA, 0.1 mm neocuproine, 1% Triton X-100, 0.1% SDS, pH 7.4) at a concentration of 1 μg/μl. The biotin-switch assay was performed as described previously (Jaffrey et al., 2001) with minor modifications. In brief, free thiols in the lysate were blocked by incubation in 10 mm methyl methanethiosulfonate (MMTS) at 50°C for 20 min with continuous mixing. The MMTS was then removed by acetone precipitation, and the pellet was resuspended in HENS buffer (100 mm HEPES, 1 mm EDTA, 0.1 mm neocuproine, plus 1% SDS). _S_-Nitrosothiols were selectively reduced by 20 mm ascorbate, and those reformed free thiols were labeled by 1 mm _N_-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio)-propionamide (biotin-HPDP) for 1 h at room temperature (RT). The biotinylated proteins were then collected on NeutrAvidin agarose beads (Thermo Scientific), which were then washed three times by Neutralization buffer (20 mm HEPES-NaOH pH 7.4, 100 mm NaCl, 1 mm EDTA, 0.5% Triton X-100). Proteins were eluted from the beads by SDS-PAGE loading buffer and subjected to immunoblot analysis.
Preparation of recombinant proteins for PTEN assay and transnitrosylation reactions.
Escherichia coli BL21 (DE3) cells transformed with pGEX-KG-PTEN, pGEX-2T-PTEN, pGEX-5X-1-DJ-1, or pET28a-DJ-1 plasmid were cultured at 37°C. Protein production was induced by 0.5 mm isopropyl β-d-thiogalactopyranoside, and GST fusion proteins were affinity purified using glutathione 4B Sepharose (GE Healthcare Life Sciences) or TALON metal affinity resin (Clontech). Thrombin or FactorXa protease was used for cleavage of the GST-tagged fusion proteins. Alternatively, GST-tagged proteins were recovered using elution buffer (10 mm reduced glutathione in 50 mm Tris-HCl, pH 7.3). Free reduced glutathione in eluted protein fractions was removed using a PD-10 desalting column (GE Healthcare Life Sciences).
For the transnitrosylation assays, recombinant PTEN (0.2 μm) or DJ-1 (2 μm), prepared from bacteria, was incubated with 25 μm SNOC for 45 min at RT in the dark. Subsequently, each of the resulting _S_-nitrosylated proteins was used as a potential NO donor to a non-nitrosylated version of the other protein in a 45 min incubation at RT in the dark to test for potential transnitrosylation. Resulting samples were subjected to the biotin-switch assay to monitor the _S_-nitrosylation status of each protein.
Mass spectrometry.
Tandem mass spectrometry (MS/MS) spectra to detect SNO-Cys were obtained from ArgC-digested human PTEN protein after performing the biotin-switch protocol on in vitro transnitrosylated PTEN. The protein digest was analyzed on an Orbitrap Velos mass spectrometer (Thermo Fisher Scientific). Protein identification was performed with Integrated Proteomics Pipeline-IP2 (Integrated Proteomics Applications, http://www.integratedproteomics.com/) using ProLuCID (Xu et al., 2006). MS/MS spectra were extracted using RawXtract (version 1.9.9; McDonald et al., 2004) and searched with ProLuCID against a Uniprot human protein database (2013_02_09) concatenated to a decoy database in which the sequence for each entry in the original database was reversed. The changes in mass of 45.988 (free thiols blocked by MMTS) and 428.192 (biotinylated cysteine after biotin switch) were set as differential modifications. Collision-induced dissociation-mediated fragmentation of the PTEN peptide resulted in formation of “b” and “y” ion series corresponding to the N-terminal and C-terminal fragments, respectively.
PTEN phosphatase activity assay.
Phosphatase activity of purified PTEN was measured by a Malachite Green Phosphatase Assay Kit (Echelon Biosciences), using PtdIns(3,4,5)P3 (Echelon Biosciences) as a substrate, according to the manufacturer's protocol. In brief, purified PTEN (25 μm) was exposed to SNOC (200 μm) or old SNOC (from which NO had been dissipated) for 20 min. Subsequently, 1 μm SNOC- or old SNOC-exposed PTEN (25 μl) was incubated with PtdIns(3,4,5)P3 and Malachite Green Solution (Echelon Biosciences) to quantify the released phosphate by reading the absorbance at 640 nm.
For cellular PTEN activity, PTEN immunoprecipitates were assayed as previously described (Georgescu et al., 1999) with minor modifications. Briefly, cells were lysed in lysis buffer containing 100 mm Tris-HCl, pH 7.2, 100 mm NaCl, 1 mm EDTA, 1% Triton X-100, and protease inhibitors (Thermo Scientific). Immunoprecipitation was then performed using anti-PTEN antibody and Dynabeads Protein A (Life Technologies). PTEN activity was measured with the Malachite Green Phosphatase Assay (Echelon Biosciences).
Crystallography.
For crystallographic studies, full-length authentic DJ-1 protein (i.e., lacking any purification tags) was expressed and purified essentially as described using the same construct (Wilson et al., 2003), a modified PET-15 vector, in E. coli strain BL21(DE3). The only differences in protocol were the use of 2 mm (rather than 1 mm) DTT throughout the purification, and a final (NH4)2SO4 cut at 90% saturation (rather than 70%). Crystals of DJ-1 were obtained at acidic pH by equilibrating against a reservoir comprising 28∼32% PEG 400 and 100 mm citrate, pH 4.6. They grew in ∼7 d, and for nitrosylation studies were transferred to the same buffer supplemented with 10 mm SNOC for 30–60 min before flash freezing in liquid nitrogen for data collection. Diffraction data for control and SNOC-treated crystals were collected at a temperature of 110 K to 1.5 Å resolution, using our in-house Rigaku FRE x-ray generator and an R-AXIS IV image plate detector. Data were processed and scaled using the HKL package (Otwinowski and Minor, 1997). In each case, the isomorphous DJ-1 structure [Protein Data Bank (PDB) ID: 1P5F] was used as the starting model for refinement using the CNS (Brünger et al., 1998) and model building with O (Jones and Kjeldgaard, 1997). Data collection and final refinement statistics are given in Table 1. Atomic coordinates and structure of _S_-nitrosylated DJ-1 have been deposited in the PDB (4RKW for the untreated protein; and 4RKY for the _S_-nitrosylated protein).
Table 1.
Crystallographic data collection and refinement
| Data collection | DJ-1 (control) | SNOC-treated DJ-1 |
|---|---|---|
| Space group | P3121 | P3121 |
| Cell dimensions | ||
| a | 74.87 | 74.95 |
| b | 74.87 | 74.95 |
| c | 75.01 | 75.06 |
| Total observations | 154,516 | 178,063 |
| Unique reflections | 38,720 | 39,454 |
| _R_merge | 0.054 (0.17)* | 0.043 (0.24) |
| I/σ_I_ | 47.3 (15.1) | 32.8 (8.2) |
| Completeness (%) | 98.2 (96.4) | 99.9 (99.6) |
| Refinement | ||
| Reflections | 37,881 | 38,523 |
| Non-hydrogen atoms | 1,372 | 1,373 |
| Water molecules | 194 | 176 |
| RWORK (%) | 20.2 | 20.7 |
| RFREE (%)† | 21.2 | 20.9 |
| RMS deviation on | ||
| Bond length (Å) | 0.004 | 0.005 |
| Bond angle (°) | 1.346 | 1.338 |
GST pull-down assay.
Purified GST or GST-DJ-1 bound on glutathione-Sepharose was incubated with purified PTEN for 3 h. After incubation, beads were washed three times with PBS. The samples were then resuspended in SDS-PAGE loading buffer and boiled for 5 min before Western blotting with anti-PTEN antibody.
Protein complex immunoprecipitation.
HEK293A cells in 10 cm dishes were harvested and solubilized by immunoprecipitation buffer [1% Triton X-100, 1% glycerol, protease inhibitors (Roche) in PBS]. Five hundred micrograms of cell lysate were incubated with a complex of protein A/G-agarose beads (Santa Cruz Biotechnology) and anti-PTEN or control antibody (or with antibody immobilized on the beads) for 3–5 h at 4°C. Beads were then washed with the immunoprecipitation buffer three times and subjected to immunoblotting.
Human brain samples.
Postmortem human brain samples from the frontal cortex were obtained from patients in whom Parkinson's disease had been diagnosed or age-matched, non-CNS disease control subjects (four males and three females). Patient data are summarized in Table 2. Human brain samples were obtained and analyzed with institutional permission following State of California and National Institutes of Health guidelines. Informed consent was obtained according to procedures approved by the Institutional Review Board at the University of California, San Diego.
Table 2.
List of human brain subjects in this study
| Subjects | Diagnosis | Brain region | PMI (h) | Age at time of death (years) | Gender |
|---|---|---|---|---|---|
| Control 1 | Myocardial infarction | Frontal cortex | N/A | 83 | M |
| Control 2 | Myocardial infarction | Frontal cortex | 8 | 63 | F |
| Control 3 | N/A | Frontal cortex | 8 | 74 | F |
| Control 4 | Myocardial infarction | Frontal cortex | 2 | 71 | M |
| Control 5 | N/A | Frontal cortex | 3 | 90 | F |
| Control 6 | N/A | Frontal cortex | 2.8 | 75 | F |
| Control 7 | N/A | Frontal cortex | 2.2 | 80 | M |
| Patient 1 | PD | Frontal cortex | 12 | 73 | M |
| Patient 2 | PD | Frontal cortex | 8 | 74 | F |
| Patient 3 | PD | Frontal cortex | 6 | 82 | M |
| Patient 4 | PD | Frontal cortex | 2.3 | 69 | M |
| Patient 5 | PD | Frontal cortex | 3 | 82 | F |
| Patient 6 | PD | Frontal cortex | 3.5 | 78 | F |
Assessment of SH-SY5Y cell death.
SH-SY5Y cells were cotransfected with constructs encoding wild-type (WT) PTEN, mutant PTEN(C83A), WT DJ-1, or mutant DJ-1(C106A). Twenty-four hours later, apoptotic cell death was assessed in a masked fashion via nuclear morphology using Hoechst 33258 staining, as we previously described (Uehara et al., 2006).
Results
_S_-Nitrosylation of PTEN inhibits its phosphatase activity
We previously reported that _S_-nitrosylation of PTEN at Cys83 inhibits its enzymatic activity (Kwak et al., 2010; Numajiri et al., 2011). To verify these findings in a cell-based context, we determined whether endogenous NO generated by nNOS could _S_-nitrosylate PTEN (forming SNO-PTEN). To this end, we exposed HEK cells stably expressing nNOS (HEK-nNOS cells) to the calcium ionophore A23187 to activate nNOS. We found by the biotin-switch assay that incubation in A23187 induced the formation of SNO-PTEN. SNO-PTEN was not detected when the cells were pretreated with the NOS inhibitor _N_G-nitro-l-arginine (NNA; Fig. 1A). Next, we examined the lipid phosphatase activity of recombinant PTEN using a standard protocol (Lacalle et al., 2004). Consistent with previous reports (Kwak et al., 2010; Numajiri et al., 2011), the physiological NO donor SNOC (25 μm) decreased PTEN activity by 40% (Fig. 1B). Moreover, nNOS-derived NO also decreased PTEN activity; this inhibitory effect was partially reversed by the NOS inhibitor _N_-ω-nitro-l-arginine methyl ester (l-NAME; Fig. 1C). These findings are consistent with the notion that _S_-nitrosylation represents a post-translational regulatory mechanism of PTEN activity.
Figure 1.

_S_-Nitrosylation of PTEN inhibits its phosphatase activity. A, HEK-nNOS cells were assayed for endogenous SNO-PTEN by the biotin-switch technique (top). Total PTEN levels (bottom). Activation of nNOS by Ca2+ ionophore A23187 (10 μm) resulted in the formation of SNO-PTEN, while the NOS inhibitor NNA (1 mm) blocked this effect. MMTS (10 mm) was used to block free thiols during the assay for _S_-nitrosylated protein. Vertical lines indicate images obtained from nonadjacent lanes of the same gel. B, In vitro purified PTEN was exposed to SNOC (25 μm) and subjected to the PTEN phosphatase activity assay. SNO-PTEN manifested decreased phosphatase activity. C, PTEN activity in HEK-nNOS cells after exposure to A23187 (10 μm) with or without pretreatment with the NOS inhibitor l-NAME (1 mm). *p < 0.05 by t test.
_S_-Nitrosylation of DJ-1 at Cys106
Previous studies suggested that high concentrations of NO donors (>0.25 mm) can _S_-nitrosylate DJ-1 in vitro at Cys46 and Cys53 (Ito et al., 2006). Such high concentrations of NO, however, can potentially _S_-nitrosylate cysteine thiols that would not normally react under physiological conditions (Stamler et al., 1997; Qu et al., 2011). Therefore, to characterize SNO-DJ-1 formation under more physiological conditions, we took an approach similar to that used above to analyze SNO-PTEN. Like PTEN, we found that DJ-1 could be _S_-nitrosylated by endogenous NO generated from nNOS in an NNA-sensitive manner (Fig. 2A).
Figure 2.

_S_-Nitrosylation of DJ-1 in vitro and in intact cells. A, HEK-nNOS cells were exposed to A23187 in the presence or absence of NNA, and cell lysates were subjected to the biotin-switch assay. Control experiments were performed without MMTS. The presence of DJ-1 proteins in the input samples was detected by standard immunoblot analysis (bottom). Vertical lines indicate images obtained from nonadjacent lanes of the same gel. B, _S_-Nitrosylation of Cys106 in DJ-1. HEK cells transfected with WT or cysteine mutant DJ-1(C46S or C106A) were exposed to 25 μm SNOC, and SNO-DJ-1 was detected by the biotin-switch assay (top). Mutation of Cys106 abolished the effect of SNOC on SNO-DJ-1 formation. C, HEK-nNOS cells were transfected with WT or DJ-1(C106A), exposed to A23187 (10 μm) for 2 h, and lysed for SNO-DJ-1 assay (top). D, Identification of the _S_-nitrosylation site on DJ-1 by x-ray crystallography. In crystals of DJ-1, SNOC exclusively modifies Cys106. Electron density (2Fo-Fc) maps in the vicinity of the three cysteine residues are superimposed on refined atomic models derived from control crystals (left) and SNOC-treated crystals (right) grown at pH 4.6. Both crystal structures were refined to 1.5 Å resolution. There is no evidence for modification of the sulfur at positions Cys46 or Cys56; additional density (labeled with *) emanating from the Cβ carbon of Cys53 is consistent with an alternate conformation of the side chain. Additional density from the Cys106 sulfur has been modeled as nitrosocysteine. Although some degree of hydrolysis to sulfenic acid cannot be ruled out, there is no evidence for oxidation to sulfinic acid, which has a distinctive bifurcated structure (Canet-Avilés et al., 2004). Atomic models are colored yellow (sulfur), blue (nitrogen), red (oxygen), and green (carbon).
Next, to determine the site of _S_-nitrosylation on DJ-1, we mutated Cys46 or Cys106, and assayed for SNO-DJ-1 formation by the biotin-switch assay in HEK cells exposed to 25 μm SNOC. We found that under these conditions the wild-type protein showed a detectable level of _S_-nitrosylation, while nitrosylation of the C106A mutant was not detected (Fig. 2B). Interestingly, however, the C46S mutant of DJ-1 actually showed a somewhat higher level of protein _S_-nitrosylation. Note that Cys46 is buried in a hydrophobic pocket, and thus it is likely that mutation of cysteine to serine, although representing a conservative change, creates a local perturbation of the structure. Such an occurrence may, for example, increase the accessibility of SNOC for Cys106. Furthermore, consistent with this notion, C106A mutation completely abrogated the formation of SNO-DJ-1 in response to endogenous NO in HEK-nNOS cells exposed to A23187 (Fig. 2C). Together, these results suggest that Cys106 is the predominant physiological target of _S_-nitrosylation on DJ-1.
Next, we investigated more directly the target sites of _S_-nitrosylation on DJ-1. To visualize SNO formation on wild-type human DJ-1, we analyzed the crystal structure of SNO-DJ-1 at a resolution of 1.5 Å. To minimize cysteine oxidation under ambient conditions, we used 2 mm DTT throughout the purification, then immediately set up crystallization drops, and harvested the crystals within 3–7 d before flash freezing in liquid nitrogen. We grew crystals at pH 8.5 and 4.6, and solved both structures at 1.5 Å resolution using our in-house x-ray source, since previous studies using a synchrotron source had reported cysteine oxidation mediated by free-radicals generated by the highly intense x-ray radiation. By contrast, we observed no significant modification of the cysteine residues in either case (Fig. 2D, left). At high pH, SNOC is known to be very unstable, quickly dissipating NO to generate other oxidizing species. Under such conditions, incubation of the crystals in SNOC at pH 8.5 for as little as 30 s led to selective oxidation of Cys106 on DJ-1 to sulfinic acid (data not shown), as previously described (Canet-Avilés et al., 2004). In contrast, at low pH, SNOC is more stable, allowing facile transfer of the NO group to protein thiols (Lipton and Stamler, 1994; Lipton et al., 1993). Accordingly, under such conditions at pH 4.6, we observed the selective modification of Cys106 on DJ-1, which could be most readily modeled as an _S_-nitrosocysteine (Fig. 2D, right). Collectively, the biochemical studies with the biotin-switch assay and the atomic resolution studies by x-ray crystallography provide strong evidence that Cys106 is the predominant physiological site of _S_-nitrosylation in DJ-1.
Transnitrosylation of PTEN by SNO-DJ-1
Evidence suggests that DJ-1 acts as a suppressor of PTEN activity (Kim et al., 2005; Yang et al., 2005; Aleyasin et al., 2010). Although the exact mechanism for this effect remains to be elucidated, there is speculation that oxidative/nitrosative stress may be involved because both PTEN and DJ-1 are redox-sensitive proteins. Along these lines, we and other groups have recently discovered that protein–protein transnitrosylation reactions (i.e., transfer of an NO group from one protein thiol to another) constitute a common mechanism in mammalian systems whereby one nitrosylated protein can _S_-nitrosylate another according to their respective Nernstian redox potentials (Kornberg et al., 2010; Nakamura et al., 2010; Qu et al., 2011).
Having shown that PTEN and DJ-1 can both be _S_-nitrosylated under physiologically relevant conditions, we hypothesized that SNO-DJ-1 might transnitrosylate PTEN to suppress PTEN phosphatase activity. To begin to test this possibility, we first asked whether PTEN interacts with DJ-1, since direct interaction of two proteins is generally required for the transnitrosylation reaction. For this, we initially used a GST pull-down assay in which the ability of PTEN to associate with GST-fused DJ-1 was examined. We found that PTEN interacted with GST-DJ-1 but not with GST alone (Fig. 3A). To further assess this PTEN–DJ-1 interaction, we next performed a coimmunoprecipitation assay in HEK293A cells. Anti-PTEN antibody coimmunoprecipitated an endogenous PTEN/DJ-1 protein complex, whereas control antibody failed to pull down DJ-1 (Fig. 3B), supporting the notion that endogenous PTEN and DJ-1 form a complex in cells, as previously predicted (Kim et al., 2009).
Figure 3.
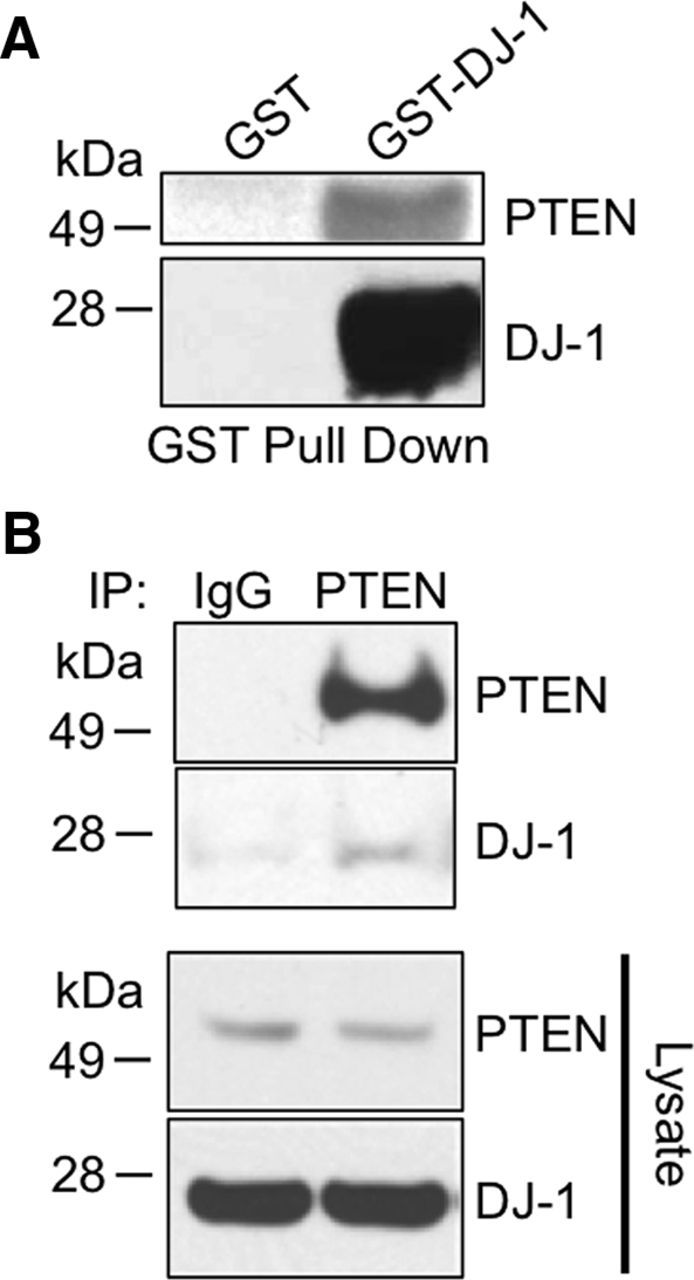
PTEN interacts with DJ-1 in vitro and in intact cells. A, GST pull-down assay. GST-fusion DJ-1 or GST alone was incubated with purified PTEN for 3 h and was pulled down with GSH beads. The precipitates were subjected to immunoblot and probed with anti-PTEN or anti-DJ-1 antibody. GST-DJ-1, but not GST alone, pulled down PTEN. B, Coimmunoprecipitation of DJ-1 and PTEN. HEK293A cells were lysed and coimmunoprecipitated with anti-PTEN antibody or control. Precipitates and total lysates were subjected to immunoblotting.
To test for the potential transfer of an NO group between PTEN and DJ-1, we next incubated purified recombinant DJ-1 protein with SNOC to produce SNO-DJ-1 protein in vitro. We then mixed PTEN with SNO-DJ-1 or unmodified DJ-1 to measure potential transnitrosylation. A biotin-switch assay demonstrated that SNO-DJ-1 acted as an NO donor to PTEN to form SNO-PTEN via transnitrosylation (Fig. 4A, left). In contrast, SNO-PTEN failed to transfer NO to DJ-1 since SNO-DJ-1 was not detected (Fig. 4A, right). Thus, these data suggest that NO is preferentially transferred from DJ-1 to PTEN but not vice versa. In control experiments, we found that non-nitrosylatable mutant DJ-1(C106A) could not transfer NO to PTEN since we did not observe the formation of SNO-PTEN (Fig. 4B). Consistent with our prior observations (Kwak et al., 2010; Numajiri et al., 2011), our mass spectrometry analysis identified Cys83 as the site of transnitrosylation on PTEN (Fig. 4C).
Figure 4.

SNO-DJ-1 transfers an NO group to PTEN via transnitrosylation. A, B, In vitro transnitrosylation assay. Transnitrosylation reactions were performed as described in the Materials and Methods. _S_-Nitrosylated PTEN and DJ-1 were detected by the biotin-switch assay. A, SNO-DJ-1 transnitrosylates PTEN (left), but SNO-PTEN did not mediate _S_-nitrosylation of DJ-1 (right). All images depicted were from the same blot. B, SNO-DJ-1(C46S), but not DJ-1(C106A), can transnitrosylate PTEN. C, Mass spectrum showing transnitrosylation of PTEN at Cys83. The MS/MS spectrum was acquired after performing the biotin-switch protocol on recombinant PTEN exposed to SNO-DJ-1. The presence of a biotin adduct (428.192 Da) on Cys83 indicates that this Cys was _S_-nitrosylated. D, SH-SY5Y cells transfected with control or DJ-1 siRNA were incubated for 3 d and exposed to 25 μm SNOC. Cell lysates were then subjected to the biotin-switch assay. Downregulation of endogenous DJ-1 abrogated the formation of SNO-PTEN. E, DJ-1 knockdown increases PTEN activity in SH-SY5Y cells. *p < 0.05 by t test.
Additionally, to evaluate the effect of endogenous DJ-1 on the generation of SNO-PTEN in intact neural cells, we used an RNAi technique to knock down endogenous DJ-1 in SH-SY5Y cells; transfection of DJ-1 siRNA led to a decrease in SNO-PTEN levels (Fig. 4D) and thus to an increase in PTEN activity (Fig. 4E). Importantly, using a modification of the Nernst equation that we recently developed (Nakamura et al., 2010), we calculated that a difference in the standard redox potential of 28.3 mV and a change in Gibbs free energy associated with transnitrosylation of NO from SNO-DJ-1 to PTEN of −2.7kJ/mol. These physical chemistry values are consistent with the notion that the transfer of NO from DJ-1 to PTEN can occur in intact cells as well as in vitro. Together, these findings support our hypothesis that SNO-DJ-1 can act as a nascent nitrosylase (i.e., an enzyme that donates an NO group to a protein thiol, in this case on a cysteine residue of PTEN via transnitrosylation).
Transnitrosylation inhibits neural cell death associated with PTEN activity
We and our colleagues have shown previously that (1) increased PTEN activity is associated with increased neuronal cell death, and (2) _S_-nitrosylation of PTEN inhibits its phosphatase activity to promote cell survival (Kwak et al., 2010; Numajiri et al., 2011). Hence, we reasoned that the formation of SNO-PTEN via transnitrosylation from DJ-1 may result in neuroprotection. To test this idea, we used a model system of neural SH-SY5Y cells because they express endogenous NOS and are amenable to facile transfection (Numajiri et al., 2011). Initially, we established experimental conditions whereby increased expression of DJ-1 resulted in elevated levels of SNO-PTEN, presumably via transnitrosylation (Fig. 5A,B). Previously, it had been reported (Li et al., 1998) that overexpression of PTEN promotes apoptotic cell death. Therefore, we next asked whether this PTEN-dependent cell death could be inhibited by transnitrosylation from DJ-1 in SH-SY5Y cells. As shown in Figure 5C, the overexpression of PTEN significantly increased cell death, but cotransfection with DJ-1 dramatically attenuated this effect. Strikingly, mutation of the redox-active site of _S_-nitrosylation/oxidation on DJ-1(Cys106) completely abrogated the neuroprotective effect of DJ-1. Additionally, WT DJ-1 failed to protect SH-SY5Y cells from the toxicity induced by non-nitrosylatable mutant PTEN(C83A). Importantly, when NO production was blocked with a NOS inhibitor, the protective activity of DJ-1 was partially reversed, consistent with the notion that NO contributed to the neuroprotective action of DJ-1 from PTEN-mediated injury. Together, these results are consistent with our hypothesis that DJ-1 exerts its neuroprotective activity at least in part via transnitrosylation and thus inhibition of PTEN.
Figure 5.

DJ-1 decreases PTEN-induced cell death in SH-SY5Y cells via transnitrosylation. A, B, DJ-1 promotes _S_-nitrosylation of PTEN in unstressed SH-SY5Y cells. SNO-PTEN levels were examined by the biotin-switch assay after transfection with WT DJ-1 or non-nitrosylatable mutant DJ-1(C106A). To control for nonspecific detection, ascorbate (Asc) was omitted from the assay. C, SH-SY5Y cells were transfected with the indicated constructs and incubated for 24 h in the presence or absence of 1 mm NNA. Cell death was measured by counting condensed nuclei after Hoechst staining. WT DJ-1 protected cells from PTEN-induced cell death, while non-nitrosylatable mutant DJ1(C106A) failed to protect cells. Cell death induced by non-nitrosylatable mutant PTEN(C83A) was not prevented by DJ-1. NNA partially abrogated the protective effects of WT DJ-1. *p < 0.02 by two-tailed Student's t test compared with “PTEN + DJ-1.”
_S_-Nitrosylation of PTEN in postmortem human brain
Previously, we and our colleagues reported that formation of endogenous SNO-PTEN could afford neuroprotection in rodent models of cerebral ischemia and Alzheimer's disease (Kwak et al., 2010; Numajiri et al., 2011). Here, we ascertained in human PD brains whether the level of SNO-PTEN found in vivo would be sufficient to offer a degree of neuroprotection. If transnitrosylation from DJ-1 to PTEN occurs in the human PD brain, then there should be an increased ratio of SNO-PTEN relative to SNO-DJ-1 compared with a control brain. Indeed, we observed by biotin-switch analysis of human postmortem PD versus control brains that SNO-PTEN levels were significantly increased in PD brains (∼1.75-fold, p = 0.022), while PD brains expressed slightly less SNO-DJ-1 compared with controls (Fig. 6A,B). Moreover, as predicted, the ratio of relative SNO-PTEN to SNO-DJ-1 levels was dramatically increased (Fig. 6C). Importantly, the level of SNO-PTEN to total PTEN was similar in human PD brain and SH-SY5Y cells under conditions of neuroprotection by DJ-1 (Fig. 6D). These results are consistent with the notion that in human PD brains, DJ-1 transnitrosylates PTEN, which would tend to detoxify neurotoxic levels of NO, but additional neurodegenerative signaling pathways (e.g., aberrantly increased production of NO generating SNO-Akt; Fig. 6E) would eventually overcome the neuroprotective effect of SNO-PTEN, leading to neuronal loss (Numajiri et al., 2011).
Figure 6.

Expression levels of SNO-PTEN are increased in human postmortem PD brains. A, B, SNO-DJ-1 (A) and SNO-PTEN (B) levels were assessed by biotin-switch assay in samples from control (non-CNS diseases) and PD brains. Formation of SNO-PTEN was increased in PD brains (B), while SNO-DJ-1 levels remain essentially unchanged (A). Omission of ascorbate (Asc) represents a negative control for the assay. C, Ratio of relative SNO-PTEN to SNO-DJ-1 levels. D, Similar levels of SNO-PTEN/total PTEN were formed in human PD brain and SH-SY5Y cells under our conditions. Patient information is listed in Table 2. *p < 0.05 by two-tailed Student's t test. E, SNO-Akt levels were assessed by biotin-switch assay in human control and PD brains.
Discussion
In a variety of neurodegenerative disorders, excessive nitrosative stress generates high concentrations of NO, which have been shown to contribute to neuronal damage and death (Dawson et al., 1991; Nakamura et al., 2013). In part, this neurodestructive effect is mediated by _S_-nitrosylation of Akt, thus inhibiting the neuroprotective cascade triggered by Akt (Numajiri et al., 2011). In contrast, at low concentrations, NO is generally considered to be a prosurvival factor, in part via formation of SNO-PTEN, which activates the neuroprotective Akt pathway (Kwak et al., 2010; Numajiri et al., 2011). Interestingly, prior work had also shown that DJ-1 can protect neurons from redox stress associated with ROS (Canet-Avilés et al., 2004), although the effect of DJ-1 on reactive nitrogen species (RNS)-mediated redox stress was heretofore unknown. In the present study, we mount evidence that, during mild nitrosative stress, transnitrosylation to form SNO-PTEN from SNO-DJ-1 represents an important molecular mechanism contributing to the neuroprotective effect of DJ-1. Specifically, we present evidence, including biochemical experiments and atomic structures at 1.5 Å resolution, that endogenous pathophysiologically relevant concentrations of NO result in the formation of SNO-DJ-1, which can then donate its NO group to PTEN to produce SNO-PTEN (Fig. 7).
Figure 7.

Proposed mechanism of transnitrosylation of PTEN by SNO-DJ-1. Schema illustrates SNO-PTEN formation by transnitrosylation. Pathophysiologically relevant levels of NO can _S_-nitrosylate DJ-1 at Cys106. DJ-1 interacts with the proapoptotic protein PTEN, and, when DJ-1 is _S_-nitrosylated, SNO-DJ-1 acts as an endogenous nitrosylase toward PTEN. This transnitrosylation reaction generates SNO-PTEN, resulting in the inhibition of PTEN phosphatase activity. Decreased phosphatase activity contributes to the survival of neurons (e.g., by relatively increasing neuroprotective phospho-Akt). Thus, transnitrosylation of PTEN by SNO-DJ-1 may mediate a neuroprotective NO signaling pathway.
Prior work had shown that DJ-1 exerts its antiapoptotic function, at least in part, via suppression of PTEN activity (Kim et al., 2005; Yang et al., 2005; Aleyasin et al., 2010). Here, we report a new mechanism for the inhibition of PTEN activity by DJ-1; namely, DJ-1 is _S_-nitrosylated predominantly at Cys106, and in turn transfers NO to Cys83 on PTEN. In support of this conclusion, we show that neural cell death induced by non-nitrosylatable mutant PTEN(C83A), unlike WT PTEN, cannot be rescued by DJ-1. Also consistent with this premise, a prior study (Zhang et al., 2012) demonstrated that mutant DJ-1(C106A) is incapable of increasing Akt phosphorylation/activation.
Overall, the modulation of the PTEN–PI3K–Akt pathway by DJ-1 appears to involve multiple cellular mechanisms. For example, DJ-1 can bind to cellular mRNAs, including those encoding components of the PTEN–PI3K–Akt cascade, to regulate their expression (van der Brug et al., 2008). Additionally, recent studies have shown that DJ-1 negatively regulates PTEN expression in several cancerous cell types, thus decreasing PTEN activity (Kim et al., 2005, 2009). Intriguingly, _S_-nitrosylation of PTEN also enhances ubiquitin-proteasome system-dependent degradation of PTEN (Kwak et al., 2010). Thus, our findings of PTEN transnitrosylation from SNO-DJ-1 pose a mechanistic model in which the transfer of NO from DJ-1 to PTEN not only inhibits PTEN phosphatase activity but also facilitates its degradation, both of which would contribute to neuroprotection.
Concerning the prosurvival function of DJ-1, dimerization is generally believed to be associated with this neuroprotective action. Ito et al. (2006) previously reported that a high concentration (>0.25 mm) of NO, derived from _S_-nitrosoglutathione, could _S_-nitrosylate DJ-1 in vitro at Cys46 and Cys53, potentially preventing dimerization of DJ-1 and abrogating the neuroprotective activity of DJ-1. In contrast, in the present study using intact cells we found that physiologically relevant levels of NO, derived from <25 μm SNOC or from endogenous nNOS, induce _S_-nitrosylation of DJ-1 at Cys106. The NO group is then transnitrosylated to PTEN to inhibit its activity and enhance neuroprotection. Our findings are thus consistent with previous observations that Cys106 plays an essential role in the neuroprotective activity of DJ-1 (Canet-Avilés et al., 2004; Aleyasin et al., 2007; Andres-Mateos et al., 2007; Blackinton et al., 2009; Hao et al., 2010). Importantly, Cys106 on DJ-1 is extremely susceptible to spontaneous oxidation in vitro, forming a cysteine sulfinate or sulfonate derivative, which would prevent _S_-nitrosylation of this cysteine thiol group. It is thus likely that Ito et al. (2006) failed to observe _S_-nitrosylation of Cys106 due to in vitro sulfination/sulfonation of Cys106 under their experimental conditions in ambient air.
The neuroprotective effect of DJ-1 is thought to involve its antioxidant activity. For example, RNAi-mediated knockdown of DJ-1 causes cellular accumulation of ROS and renders cells more susceptible to oxidative stress (Taira et al., 2004; Yang et al., 2005), suggesting that DJ-1 may possess antioxidant activity to detoxify ROS (Blackinton et al., 2009). In fact, the detoxification of ROS may involve the aforementioned formation of sulfinic or sulfonic acid at Cys106 on DJ-1. Here, we suggest that DJ-1 can also detoxify RNS by reaction with NO at the same cysteine residue. Moreover, subsequent transnitrosylation from DJ-1 to PTEN may represent a feedforward loop to further enhance survival via inhibition of PTEN (Fig. 7).
In summary, we report a molecular mechanism whereby DJ-1 acts as an NO-sensitive neuroprotective protein in addition to its known reactivity toward ROS. This neuroprotective _S_-nitrosylation occurs on the redox-sensitive residue Cys106 of DJ-1, with subsequent transnitrosylation to form SNO-PTEN. The work therefore elucidates a new mechanism for transferring NO from one protein to another, resulting in enhanced neuroprotection.
Footnotes
This work was supported in part by Michael J. Fox Foundation grants (to T.N. and S.A.L.) and the National Institutes of Health (Grants P01 ES016738, P01 HD29587, and P30 NS076411 to S.A.L.; and P41 GM103533, R01 MH067880, and National Heart, Lung, and Blood Institute Proteomics Center Award HHSN268201000035C to J.R.Y.). We thank Dr. Eliezer Masliah (University of California, San Diego) and the Brain and Body Donation Program of Banner Sun Health Research Institute for providing postmortem human brain tissues; and Dr. Mark Cookson for the gift of the plasmids and helpful discussions.
The authors declare no competing financial interests.
References
- Aleyasin H, Rousseaux MW, Phillips M, Kim RH, Bland RJ, Callaghan S, Slack RS, During MJ, Mak TW, Park DS. The Parkinson's disease gene DJ-1 is also a key regulator of stroke-induced damage. Proc Natl Acad Sci U S A. 2007;104:18748–18753. doi: 10.1073/pnas.0709379104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleyasin H, Rousseaux MW, Marcogliese PC, Hewitt SJ, Irrcher I, Joselin AP, Parsanejad M, Kim RH, Rizzu P, Callaghan SM, Slack RS, Mak TW, Park DS. DJ-1 protects the nigrostriatal axis from the neurotoxin MPTP by modulation of the AKT pathway. Proc Natl Acad Sci U S A. 2010;107:3186–3191. doi: 10.1073/pnas.0914876107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres-Mateos E, Perier C, Zhang L, Blanchard-Fillion B, Greco TM, Thomas B, Ko HS, Sasaki M, Ischiropoulos H, Przedborski S, Dawson TM, Dawson VL. DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc Natl Acad Sci U S A. 2007;104:14807–14812. doi: 10.1073/pnas.0703219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackinton J, Lakshminarasimhan M, Thomas KJ, Ahmad R, Greggio E, Raza AS, Cookson MR, Wilson MA. Formation of a stabilized cysteine sulfinic acid is critical for the mitochondrial function of the parkinsonism protein DJ-1. J Biol Chem. 2009;284:6476–6485. doi: 10.1074/jbc.M806599200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Canet-Avilés RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S, Baptista MJ, Ringe D, Petsko GA, Cookson MR. The Parkinson's disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci U S A. 2004;101:9103–9108. doi: 10.1073/pnas.0402959101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci U S A. 1991;88:6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Ruiz O, Zapata A, Shan L, Zhang Y, Tomac AC, Malik N, de la Cruz F, Bäckman CM. Selective deletion of PTEN in dopamine neurons leads to trophic effects and adaptation of striatal medium spiny projecting neurons. PLoS One. 2009;4:e7027. doi: 10.1371/journal.pone.0007027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu MM, Kirsch KH, Akagi T, Shishido T, Hanafusa H. The tumor-suppressor activity of PTEN is regulated by its carboxyl-terminal region. Proc Natl Acad Sci U S A. 1999;96:10182–10187. doi: 10.1073/pnas.96.18.10182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Görner K, Holtorf E, Waak J, Pham TT, Vogt-Weisenhorn DM, Wurst W, Haass C, Kahle PJ. Structural determinants of the C-terminal helix-kink-helix motif essential for protein stability and survival promoting activity of DJ-1. J Biol Chem. 2007;282:13680–13691. doi: 10.1074/jbc.M609821200. [DOI] [PubMed] [Google Scholar]
- Hao LY, Giasson BI, Bonini NM. DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc Natl Acad Sci U S A. 2010;107:9747–9752. doi: 10.1073/pnas.0911175107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito G, Ariga H, Nakagawa Y, Iwatsubo T. Roles of distinct cysteine residues in S-nitrosylation and dimerization of DJ-1. Biochem Biophys Res Commun. 2006;339:667–672. doi: 10.1016/j.bbrc.2005.11.058. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- Jones TA, Kjeldgaard M. Electron-density map interpretation. Methods Enzymol. 1997;277:173–208. doi: 10.1016/S0076-6879(97)77012-5. [DOI] [PubMed] [Google Scholar]
- Kim RH, Peters M, Jang Y, Shi W, Pintilie M, Fletcher GC, DeLuca C, Liepa J, Zhou L, Snow B, Binari RC, Manoukian AS, Bray MR, Liu FF, Tsao MS, Mak TW. DJ-1, a novel regulator of the tumor suppressor PTEN. Cancer Cell. 2005;7:263–273. doi: 10.1016/j.ccr.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Kim YC, Kitaura H, Taira T, Iguchi-Ariga SM, Ariga H. Oxidation of DJ-1-dependent cell transformation through direct binding of DJ-1 to PTEN. Int J Oncol. 2009;35:1331–1341. doi: 10.3892/ijo_00000451. [DOI] [PubMed] [Google Scholar]
- Kornberg MD, Sen N, Hara MR, Juluri KR, Nguyen JV, Snowman AM, Law L, Hester LD, Snyder SH. GAPDH mediates nitrosylation of nuclear proteins. Nat Cell Biol. 2010;12:1094–1100. doi: 10.1038/ncb2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak YD, Ma T, Diao S, Zhang X, Chen Y, Hsu J, Lipton SA, Masliah E, Xu H, Liao FF. NO signaling and S-nitrosylation regulate PTEN inhibition in neurodegeneration. Mol Neurodegener. 2010;5:49. doi: 10.1186/1750-1326-5-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacalle RA, Gómez-Moutón C, Barber DF, Jiménez-Baranda S, Mira E, Martínez AC, Carrera AC, Mañes S. PTEN regulates motility but not directionality during leukocyte chemotaxis. J Cell Sci. 2004;117:6207–6215. doi: 10.1242/jcs.01545. [DOI] [PubMed] [Google Scholar]
- Lei SZ, Pan ZH, Aggarwal SK, Chen HS, Hartman J, Sucher NJ, Lipton SA. Effect of nitric oxide production on the redox modulatory site of the NMDA receptor-channel complex. Neuron. 1992;8:1087–1099. doi: 10.1016/0896-6273(92)90130-6. [DOI] [PubMed] [Google Scholar]
- Li J, Simpson L, Takahashi M, Miliaresis C, Myers MP, Tonks N, Parsons R. The PTEN/MMAC1 tumor suppressor induces cell death that is rescued by the AKT/protein kinase B oncogene. Cancer Res. 1998;58:5667–5672. [PubMed] [Google Scholar]
- Lipton SA, Stamler JS. Actions of redox-related congeners of nitric oxide at the NMDA receptor. Neuropharmacology. 1994;33:1229–1233. doi: 10.1016/0028-3908(94)90021-3. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Choi YB, Pan ZH, Lei SZ, Chen HS, Sucher NJ, Loscalzo J, Singel DJ, Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- McDonald WH, Tabb DL, Sadygov RG, MacCoss MJ, Venable J, Graumann J, Johnson JR, Cociorva D, Yates JR., 3rd MS1, MS2, and SQT-three unified, compact, and easily parsed file formats for the storage of shotgun proteomic spectra and identifications. Rapid Commun Mass Spectrom. 2004;18:2162–2168. doi: 10.1002/rcm.1603. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Lipton SA. Emerging role of protein-protein transnitrosylation in cell signaling pathways. Antioxid Redox Signal. 2013;18:239–249. doi: 10.1089/ars.2012.4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Wang L, Wong CC, Scott FL, Eckelman BP, Han X, Tzitzilonis C, Meng F, Gu Z, Holland EA, Clemente AT, Okamoto S, Salvesen GS, Riek R, Yates JR, 3rd, Lipton SA. Transnitrosylation of XIAP regulates caspase-dependent neuronal cell death. Mol Cell. 2010;39:184–195. doi: 10.1016/j.molcel.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Tu S, Akhtar MW, Sunico CR, Okamoto S, Lipton SA. Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron. 2013;78:596–614. doi: 10.1016/j.neuron.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numajiri N, Takasawa K, Nishiya T, Tanaka H, Ohno K, Hayakawa W, Asada M, Matsuda H, Azumi K, Kamata H, Nakamura T, Hara H, Minami M, Lipton SA, Uehara T. On-off system for PI3-kinase-Akt signaling through S-nitrosylation of phosphatase with sequence homology to tensin (PTEN) Proc Natl Acad Sci U S A. 2011;108:10349–10354. doi: 10.1073/pnas.1103503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Premkumar L, Dobaczewska MK, Riedl SJ. Identification of an artificial peptide motif that binds and stabilizes reduced human DJ-1. J Struct Biol. 2011;176:414–418. doi: 10.1016/j.jsb.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu J, Nakamura T, Cao G, Holland EA, McKercher SR, Lipton SA. S-Nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by beta-amyloid peptide. Proc Natl Acad Sci U S A. 2011;108:14330–14335. doi: 10.1073/pnas.1105172108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler JS, Toone EJ, Lipton SA, Sucher NJ. (S)NO signals: translocation, regulation, and a consensus motif. Neuron. 1997;18:691–696. doi: 10.1016/S0896-6273(00)80310-4. [DOI] [PubMed] [Google Scholar]
- Taira T, Saito Y, Niki T, Iguchi-Ariga SM, Takahashi K, Ariga H. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 2004;5:213–218. doi: 10.1038/sj.embor.7400074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-Nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- van der Brug MP, Blackinton J, Chandran J, Hao LY, Lal A, Mazan-Mamczarz K, Martindale J, Xie C, Ahmad R, Thomas KJ, Beilina A, Gibbs JR, Ding J, Myers AJ, Zhan M, Cai H, Bonini NM, Gorospe M, Cookson MR. RNA binding activity of the recessive parkinsonism protein DJ-1 supports involvement in multiple cellular pathways. Proc Natl Acad Sci U S A. 2008;105:10244–10249. doi: 10.1073/pnas.0708518105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MA. The role of cysteine oxidation in DJ-1 function and dysfunction. Antioxid Redox Signal. 2011;15:111–122. doi: 10.1089/ars.2010.3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MA, Collins JL, Hod Y, Ringe D, Petsko GA. The 1.1-A resolution crystal structure of DJ-1, the protein mutated in autosomal recessive early onset Parkinson's disease. Proc Natl Acad Sci U S A. 2003;100:9256–9261. doi: 10.1073/pnas.1133288100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Venable J, Park SK, Cociorva D, Lu B, Liao L, Hewel J, Wong CC, Han X, Wohlschlegel J, Yates JR., III ProLuCID: using probability and statistical scores to improve sensitivity and specificity of CID and ETD database search results. Mol Cell Proteomics. 2006;5:S174. [Google Scholar]
- Yang Y, Gehrke S, Haque ME, Imai Y, Kosek J, Yang L, Beal MF, Nishimura I, Wakamatsu K, Ito S, Takahashi R, Lu B. Inactivation of Drosophila DJ-1 leads to impairments of oxidative stress response and phosphatidylinositol 3-kinase/Akt signaling. Proc Natl Acad Sci U S A. 2005;102:13670–13675. doi: 10.1073/pnas.0504610102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Wu J, Wu R, Ma J, Du G, Jiao R, Tian Y, Zheng Z, Yuan Z. DJ-1 promotes the proteasomal degradation of Fis1: implications of DJ-1 in neuronal protection. Biochem J. 2012;447:261–269. doi: 10.1042/BJ20120598. [DOI] [PubMed] [Google Scholar]