Antiangiogenic Therapy for Glioblastoma: Current Status and Future Prospects (original) (raw)
. Author manuscript; available in PMC: 2015 Nov 15.
Abstract
Glioblastoma is characterized by high expression levels of pro-angiogenic cytokines and microvascular proliferation, highlighting the potential value of treatments targeting angiogenesis. Antiangiogenic treatment likely achieves a beneficial impact through multiple mechanisms of action. Ultimately, however, alternative pro-angiogenic signal transduction pathways are activated leading to the development of resistance, even in tumors that initially respond. The identification of biomarkers or imaging parameters to predict response and to herald resistance is of high priority. Despite promising phase 2 clinical trial results and patient benefit in terms of clinical improvement and longer progression-free survival, an overall survival benefit has not been demonstrated in 4 randomized phase 3 trials of bevacizumab or cilengitide in newly diagnosed glioblastoma or cediranib or enzastaurin recurrent glioblastoma. However, future studies are warranted: predictive markers may allow appropriate patient enrichment, combination with chemotherapy may ultimately prove successful in improving overall survival, and novel agents targeting multiple pro-angiogenic pathways may prove effective.
Introduction
Glioblastoma, the most common primary malignant brain tumor, affects more than 3/100,000 individuals each year. Median survival is below one year in population-based studies. Older age and lower performance status are associated with less aggressive care and shorter survival. The current standard of care includes maximal safe resection followed by radiotherapy plus concomitant and adjuvant chemotherapy with temozolomide. Elderly patients, i.e. patients >65-70 years, not considered candidates for combined chemotherapy and radiation, mainly on the basis of comorbidities or impaired performance status, may be treated with radiation or temozolomide alone based on O6-methylguanine DNA methyltransferase (MGMT) promoter methylation status: in patients with tumors lacking MGMT promoter methylation radiotherapy alone is acceptable whereas in patients with MGMT promoter methylation temozolomide with or without radiation is acceptable(1).
Angiogenesis has emerged as a primary target of drug development for glioblastoma over the last decade. This development was triggered by the disappointing outcomes with cytotoxic drugs and the recognition that the extensive pathological vascularization should make this disease potentially susceptible to antiangiogenic therapy. Bevacizumab, a humanized antibody to vascular endothelial growth factor (VEGF), received accelerated approval for recurrent glioblastoma in the United States (US) and many other countries based on radiographic response rates(2, 3). In contrast, because of the lack of a controlled trial bevacizumab did not receive approval in the European Union (EU), resulting in different standards of care between the US and the EU.Although randomized trials in newly diagnosed glioblastoma patient have not demonstrated an overall survival benefit, the final status of bevacizumab in this setting has yet to be fully determined, as well be discussed subsequently. Other VEGFtargeting agents either have been or will continue to be explored in glioblastoma(4).
Mechanisms of Action and Resistance
The mechanisms of action of antiangiogenic therapies for solid tumors are multiple and may act in concert to delay tumor progression and ultimately prolong survival in several cancers. Folkman originally hypothesized that antiangiogenic agents confer an antitumor effect through induction of endothelial cell apoptosis, inhibition of new blood vessel growth, obliteration of small vessels, and decreased tumor perfusion, culminating in decreased delivery of oxygen and nutrients (“tumor starvation”)(5). However, during the initial stages of treatment, antiangiogenic agents may transiently “normalize” abnormal tumor vasculature by reducing blood vessel diameter and permeability which paradoxically improves tumor perfusion, reduces interstitial pressure, and improves tumor oxygenation(6-8), potentially sensitizing for radiotherapy and increasing tumor exposure to cytotoxic chemotherapy (Fig. 1)(7). Also, antiangiogenic therapy may prevent VEGF-mediated vascular regrowth following endothelial cell injury after genotoxic therapies(9-11). Second, antiangiogenic agents may exhibit intrinsic antitumor activity, e.g., against glioblastoma stem-like cells (GSC) residing in the perivascular niche(12, 13). Third, antiangiogenic agents may interfere with VEGF-mediated recruitment of tumor-infiltrating VEGFR1+ monocytes(14). Fourth, there is a potential role for antiangiogenic therapy in augmenting host immunity by reducing VEGFmediated immune suppression(15) thereby improving the efficacy of immunotherapy(16). The relative importance of these multiple mechanisms of action regarding the therapeutic benefit of antiangiogenic therapy is unknown and different mechanisms may be operative in distinct subsets of patients as well as at different stages of the disease.
Figure 1.
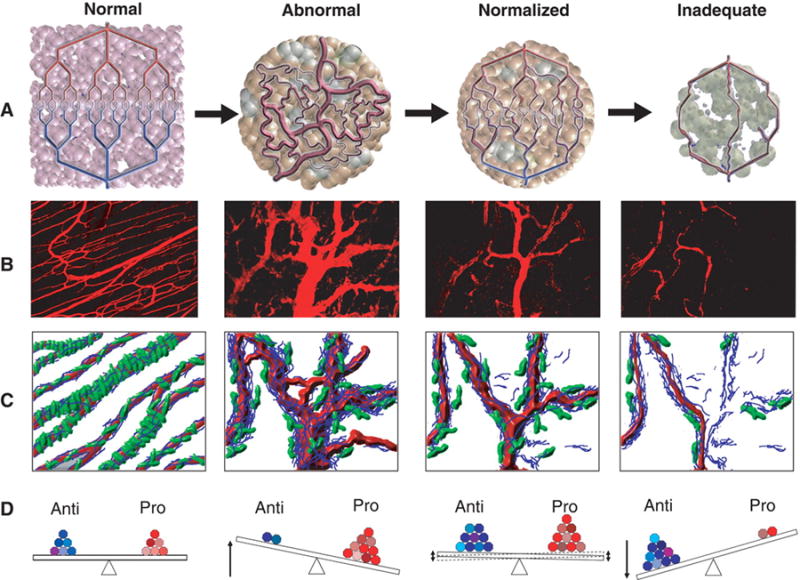
Normalization of tumor vasculature. (A) Tumor vasculature is structurally and functionally abnormal. One potential mechanism of action for antiangiogenic therapies is transient improvement in both the structure and the function of tumor vessels. However, sustained antiangiogenic treatment may eventually result in a vasculature that is both resistant to further treatment and inadequate for the delivery of drugs or oxygen. (B) Vessel structural patterns before, during and with sustained VEGFR2 blockade. (C) Diagram depicting the concomitant changes in pericyte coverage (green) and basement membrane thickness (blue) before, during and with sustained VEGFR2 blockage. (D) Changes in the balance of pro- and antiangiogenic factors leading to the phenotypic changes noted above. Reprinted from Jain (99).
The realization that antiangiogenic therapies provide transient clinical benefit and delay tumor progression prior to inevitable tumor progression has prompted an effort to better understand mechanisms of resistance to this class of therapeutic agent, as discussed subsequently. High rates of radiographic response rates and decrease in cerebral edema indicate a reduction in vascular permeability due to interruption of VEGF-A (originally termed vascular permeability factor) signaling(3, 6, 17). However, a lack of antitumor effect observed in some orthotopic rodent xenograft models of glioblastoma(18) suggests that angiogenesis inhibitors, e.g., cediranib, have limited intrinsic antitumor activity and that their main benefit may be limited to reductions in permeability and vasogenic cerebral edema(3, 6, 17). Notwithstanding a better understanding of the potential benefits of utilizing an optimal dose, schedule and drug combination, data from phase 3 clinical trials(19, 20)of bevacizumab suggest that some glioblastomas may be intrinsically resistant to antiangiogenic therapy. Inherent vessel insensitivity to the effect of VEGF inhibition could partially mediate this intrinsic resistance(21). Several adaptive resistance mechanisms may counteract any potential, initial benefit afforded by antiangiogenic therapy. In the setting of VEGF signaling inhibition the tumor and its microenvironment release alternative pro-angiogenic growth factors to promote VEGF-independent angiogenesis(22-24) which may be further augmented by the recruitment of pro-angiogenic myeloid cells such as monocytes, M2-skewed macrophages, granulocytes and myeloid-derived suppressor cells(14, 25, 26). Additionally, functional vessels are characteristically covered with pericytes which may protect endothelial cells from apoptosis in the face of VEGF blockade. Finally, adaptive resistance has been characterized by a transition to a mesenchymal and more invasive tumor phenotype(27-29). In the setting of antiangiogenic therapy, glioblastoma cells coopt normal blood vessels(30) as a route of invasion into the surrounding brain. Although initial reports implied that anti-VEGF therapies were associated with non-enhancing radiographic tumor progression(31) originally interpreted as an increase in tumor invasion, subsequent reports did not support this observation(19, 20, 32, 33).
Clinical Trials
Clinical trials evaluating antiangiogenic agents for glioblastoma initially lagged behind other cancer indications due to concern of potentially serious adverse events in brain tumor patients, notably intracranial hemorrhage or stroke. However, early clinical experience confirmed the rarity of such events and that the toxicity profile of antiangiogenic agents for glioblastoma was not significantly different from that in other cancer indications; thereafter, clinical study of antiangiogenic agents for glioblastoma accelerated. A multitude of antiangiogenic agents have been evaluated for glioblastoma including tyrosine kinase inhibitors(34-49), monoclonal antibodies against VEGFR, and a soluble decoy receptor(50) (Table 1). Since clinical development is most advanced for bevacizumab, we focus herein on the design, results and conclusions of the major bevacizumab trials for glioblastoma.
Table 1. Representative clinical trials of VEGF/VEGFR targeting therapeutics among recurrent glioblastoma patients.
| Agent | Mechanism | Dose | # Patients | ORR (%) | PFS-6 (%) | OS (median, months) | Reference |
|---|---|---|---|---|---|---|---|
| Aflibercept | Soluble decoy VEGFR | 4 mg/kg biweekly | 42 | 18 | 7.7 | 9.8 | 50 |
| Cediranib | VEGFR TKI | 30 mg daily | 118 | 15.3 | 16 | 8.0 | 98 |
| Nintedanib | VEGFR TKI | 200 mg twice a day | 13 | 0 | 4 | 8.1 | 48 |
| Pazopanib | VEGFR TKI | 800 mg daily | 35 | 5.7 | 3 | 8.8 | 34 |
| Pazopanib (+ lapatinib) | VEGFR TKI | 400 mg daily | 41 | 5 | 7.5 | NR | 47 |
| Sorafenib (+ daily TMZ) | VEGFR TKI | 400 mg daily | 32 | 3 | 9.4 | 10.4 | 35 |
| Sunitinib | VEGFR TKI | 37.5 mg daily | 32 | 10 | 10.4 | 9.4 | 42 |
| Vandetanib | VEGFR TKI | 300 mg daily | 32 | 12.5 | 6.5 | 6.3 | 41 |
| Bevacizumab | Humanized anti-VEGF mAb | 10mg/kg biweekly | 85 | 28 | 43 | 9.3 | 3 |
Bevacizumab for recurrent glioblastoma
Dramatic overall radiographic response (ORR) rates and reassuring safety data led to two phase II studies which subsequently became the basis of the US Food and Drug Administration accelerated approval of bevacizumab as monotherapy for recurrent glioblastoma in 2009 (Table 2)(51). Of note, both studies compared outcome to historical benchmarks and included independent radiologic review. The BRAIN study randomized patients to bevacizumab (n=85) or bevacizumab plus irinotecan (n=82) but was not designed to detect differences between the two treatment arms(3). Outcomes for the bevacizumab and bevacizumab plus irinotecan arms included ORR rates of 28.2% and 37.8%, PFS-6 rates of 42.6% and 50.3% and median overall survival (OS) of 9.2 and 8.7 months, respectively. A single-arm study of bevacizumab among 48 patients treated at the US National Cancer Institute (NCI) noted ORR and PFS-6 rates of 35% and 29%, respectively, and a median OS of 7.75 months(2). Although the BRAIN and NCI trials generated unprecedented ORR and PFS-6 rates the European Medicines Agency declined to approve bevacizumab for recurrent glioblastoma due to the absence of a non-bevacizumab control arm, a modest OS increment, inadequate elucidation of true antitumor effect, and challenges with radiographic response assessment(52).
Table 2. Landmark clinical trials of bevacizumab for glioblastoma (GBM).
| Trial | Regimen | # Patients | Median PFS (months) | PFS-6 (%) | Median OS (months) | Reference |
|---|---|---|---|---|---|---|
| RECURRENT GBM | ||||||
| BRAIN | BEV | 85 | 4.2 | 42.6 | 9.2 | 3 |
| BRAIN | BEV + irinotecan | 82 | 5.6 | 50.3 | 8.7 | 3 |
| NCI | BEV | 48 | 4.0 | 29 | 7.8 | 2 |
| BELOB | BEV | 50 | 3 | 18 | 8 | 69 |
| BELOB | Lomustine | 46 | 2 | 11 | 8 | 69 |
| BELOB | BEV + lomustine | 44 | 11 | 41 | 11 | 69 |
| NEWLY DIAGNOSED GBM | ||||||
| RTOG 0825 | BEV + TMZ/XRT | 312 | 10.7 (HR: 0.79; p=0.007) | NR | 15.7 | 79 |
| RTOG 0825 | TMZ/XRT | 309 | 7.3 | NR | 16.1 | 79 |
| AVAGlio | BEV + TMZ/XRT | 458 | 10.6 (HR:0.64, p<0.0001) | NR | 16.9 | 78 |
| AVAGlio | TMZ/XRT | 463 | 6.2 | NR | 16.8 | 78 |
Thereafter, attempts to augment the benefit of single-agent bevacizumab included studies evaluating bevacizumab combined with chemotherapeutics(31, 53-62), targeted therapies(63-65) and re-irradiation(66-68). Unfortunately all of these combinatorial regimens failed to improve outcome beyond that of bevacizumab monotherapy could be due to a decrease of drug delivery to the tumor(8). A single exception is a phase II study in which 148 recurrent GBM patients randomized to lomustine, bevacizumab or lomustine plus bevacizumab (Table 2)(69). Outcome was notably improved for the combination arm including PFS-6 of 41%, compared to 11% and 18% for lomustine and bevacizumab alone, respectively. The combination arm was also associated with improved overall survival at 9 months (OS9), the primary endpoint of this trial. The OS9 was 59% for the combination arm and 43% and 38% for lomustine and bevacizumab alone, respectively. Two aspects of this study warrant special comment. First this is the only study to date that incorporates a comparative, randomized statistical design with a non-bevacizumab control arm. Second, it is the first study to report a bevacizumab combination with improved outcome compared to bevacizumab monotherapy. Yet, the bevacizumab alone arm underperformed in this trial, and the differences between PFS and OS in all arms suggest that further interventions had a great impact on outcome in this trial. An ongoing phase III study to further evaluate these findings (EORTC 26101, NCT01290939) randomizes recurrent glioblastoma patients to lomustine or lomustine plus bevacizumab with a primary endpoint of OS.
Resistance to bevacizumab inevitably develops and such patients typically die rapidly due to ineffective therapies(2, 31, 54, 70-73). Retrospective data suggests that bevacizumab continuation beyond initial progression may modestly improve outcome(74). Prospective evaluation of this approach is forthcoming via an ongoing trial (TAMIGA). Nonetheless, effective therapies for bevacizumab refractory patients are desperately needed.
Bevacizumab for newly diagnosed glioblastoma
Initial single arm, phase II studies of bevacizumab in combination with temozolomide and radiation for newly diagnosed glioblastoma patients noted a near doubling of median PFS to 13-14 months compared to historical benchmarks and a nominal median OS increment to 20 months(75-77). Two randomized, placebo-controlled phase III studies, RTOG 0825 and AVAglio, reported extension of PFS but no difference in OS (Table 2)(78, 79). Specifically, median PFS was 47-71% longer for bevacizumab recipients compared to controls, but OS was not significantly different in the two treatment arms. Since30-40% of controls on each study received bevacizumab at progression, crossover is a potential confounder in terms of the impact on OS, although this remains a matter of speculation. Importantly, both studies assessed pre-defined clinical and molecular prognostic factors for association with outcome but failed to identify any of these patient subgroups more or less likely to benefit from bevacizumab; however, there is ongoing investigation in both trials to determine whether more complex genetic signatures may define subgroups more likely to benefit from bevacizumab in combination with chemoradiation, as discussed below and elsewhere.
Both RTOG 0825 and AVAglio assessed other measures of clinical benefit. AVAglio noted preserved Karnofsky performance status and lower corticosteroid requirement among bevacizumab recipients. Unexpectedly, results from validated measures of quality of life (QOL) including the EORTC QLQ-C30 and BN20 questionnaires that were incorporated by both studies yielded conflicting results. Among bevacizumab recipients, consistently improved QOL scores were reported across five prospectively defined domains on AVAglio, while lower scores were noted for several domains on RTOG 0825. The explanation for these discordant results remains unclear. Investigators in RTOG 0825 assessed radiographic response solely by enhancing tumor (Macdonald criteria(80)) and may have failed to identify early progression among bevacizumab recipients. In contrast, AVAglio assessed both enhancing and nonenhancing tumor (RANO criteria)(81). RTOG 0825, but not AVAglio, incorporated formal neurocognitive testing and noted diminished processing speed and executive function among bevacizumab recipients compared to controls. These notable findings warrant follow-up investigation. In summary, clinical trial data to date support improved PFS but lack of significant OS benefit with bevacizumab among recurrent and newly diagnosed glioblastoma patients.
Biological and Imaging Markers
The increased understanding of the molecular profile of glioblastoma suggests that subgroups of these patients may respond differentially to distinct classes of antiangiogenic agents. There are a number of tumor tissue and circulating candidate biomarkers for predicting the efficacy of antiangiogenic agents. Tumor tissue biomarkers that have been assessed, but not confirmed(79, 82) include a 9-gene signature representative of the mesenchymal subtype of glioblastoma(83, 84), VEGF expression(85, 86),O6-methylguanine methyltransferase promoter methylation status(79), epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor a (PDGFR-a) and c-KIT for VEGFR2 inhibition(6, 87). Negative predictive markers for the use of bevacizumab in newly diagnosed glioblastoma include an expanded set of mesenchymal genes(82), whereas the proneural molecular (tumors with IDH mutations excluded) subtype of glioblastoma specifically benefitted from bevacizumab(86). Independent, e.g. cross-trial confirmation of these putative predictive markers is needed, and their use in current clinical practice should be discouraged.
Circulating cytokines are attractive candidate biomarkers. However, in the AVAglio trial pretreatment plasma VEGF and sVEGFR2 levels were not associated with PFS or OS(20, 86, 88). Matrix metalloproteinase (MMP)-2 is a plasma candidate biomarker for efficacy of bevacizumab(89). Elevated soluble VEGFR1, a negative regulator of the VEGF signaling cascade, has been proposed as a resistance biomarker in other solid tumor types(90).
Radiographic response as defined by a reduction in tumor contrast-enhancement on brain CT or MRI scans may not reflect intrinsic antitumor activity since antiangiogenic treatment notably targeting VEGF signaling may rapidly reduce vessel permeability and contrast extravasation. This rapid and usually transient radiographic change is sometimes termed “ pseudoresponse.” Consequently, antiangiogenic treatments have compelled a focus on brain tumor imaging leading to the introduction of novel, candidate techniques to accurately define tumor response and tumor progression. Some of these MRI methods include apparent diffusion coefficient (ADC)(91), dynamic contrast-enhanced (DCE) and dynamic susceptibility-contrast (DSC) techniques to assess baseline and dynamic features of glioblastoma vasculature(92, 93), as well as vessel architectural imaging (VAI) that exploits a temporal shift in the magnetic resonance signal, forming the basis for vessel caliber estimation(94). VAI techniques demonstrate vessel-normalizing microcirculation during VEGF inhibition with cediranib, a pan-VEGF receptor tyrosine kinase inhibitor(94). The T1-derived parameter KTrans may reflect not only vessel normalization but also efficacy with VEGF inhibition. Cerebral blood flow may increase early after initiation of anti-VEGF therapy, identify responders, and is associated with improved tumor oxygenation status(87). Dopamine and amino acid positron emission tomography (PET) has been evaluated as an early imaging parameter of response to anti-VEGF therapy(95, 96). Further assessment of these imaging techniques and implementation of uniform imaging protocols in prospective, randomized trials is essential to determine their ultimate predictive value.
Future Directions
Significant effort and investment have been dedicated to the development of antiangiogenic therapies for glioblastoma. Consequently, new criteria for the assessment of disease by neuroimaging have been defined(81), new concepts of clinical trial design have been developed(97) and the quality of clinical trial design, conduct and analysis have been improved(20, 79, 98). Nevertheless, an overall survival benefit has yet to be identified afterfive randomized phase 3 trials in the newly diagnosed and recurrent glioblastoma setting. Where do we go from here?
First, future, pivotal, phase 3 trials of antiangiogenic agents should be conducted on the basis of data from well-designed, placebo-controlled, randomized, phase 2 trials when feasible(97). Second, it is highly likely that a future survival advantage is likely to come from the combination of antiangiogenic and cytotoxic treatments, similar to other solid tumor types. As noted, the only positive OS data from a randomized (phase 2) trial was observed for recurrent glioblastoma with chemotherapy plus bevacizumab. Third, although striking differences in patient response to and duration of benefit from VEGF inhibitors among patients with glioblastoma have been observed, none of the promising neuroimaging, histological and circulating markers associated with radiographic or clinical benefit have yet been validated. This until now missed opportunity of drug development is equally unfortunate for the field of neuro-oncology andfor the pharmaceutical industry and even more for patients who may derive benefit from this treatment approach. Thus, intensive effort should focus on the identification and validation of such predictive markers. Fourth, with improved cellular and rodent glioma models including patient-derived and stem-like cell models and feasible animal imaging techniques available, more preclinical studies focusing on predictive biomarkers and mechanisms of escape are feasible and should supplement the ongoing efforts of moving antiangiogenic agents forward.
Acknowledgments
Grant Support: T.T. Batchelor was supported by the NCI of the NIH under award numbers P50CA165962, R01CA129371, K24CA125440, K12CA090354, R13 CA124293.
T.T. Batchelor reports receiving commercial research grants from AstraZeneca, Millenium and Pfizer; speakers bureau honoraria from Research To Practice; and is a consultant/advisory board member for Agenus, Kirin, Merck, Oakstone, Proximagen, and UpToDate. D.A. Reardon reports receiving speakers bureau honoraria from Merck/Schering and Roche/Genentech. J.F. de Groot reports receiving a commercial research grant from AstraZeneca and is a consultant/advisory board member for Roche/Genentech. W. Wick is a consultant/advisory board member for Apogenix and Roche. M. Weller reports receiving commercial research support from Merck Serono and Roche.
Footnotes
Disclosure of Potential Conflicts of Interest: No other potential conflicts of interest were disclosed.
References
- 1.Weller M, Van den Bent M, Hopkins K, Tonn JC, Stupp R, Falini A, et al. The European Association for Neuro-Oncology (EANO) Task Force on Maliganant Glioma. EANO guideline on the diagnosis and treatment of malignant glioma. Lancet Oncol. 2014 Forthcoming. [Google Scholar]
- 2.Kreisl TN, Kim L, Moore K, Duic P, Royce C, Stroud I, et al. Phase II trial of singleagent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009 Feb 10;27(5):740–5. doi: 10.1200/JCO.2008.16.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009 Oct 1;27(28):4733–40. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 4.Wick W, Puduvalli VK, Chamberlain MC, van den Bent MJ, Carpentier AF, Cher LM, et al. Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J Clin Oncol. 2010 Mar 1;28(7):1168–74. doi: 10.1200/JCO.2009.23.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Folkman J. Tumor angiogenesis: Therapeutic implications. N Engl J Med. 1971 Nov 18;285(21):1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 6.Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DG, Cohen KS, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007 Jan;11(1):83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winkler F, Kozin SV, Tong RT, Chae SS, Booth MF, Garkavtsev I, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: Role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell. 2004 Dec;6(6):553–63. doi: 10.1016/j.ccr.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 8.Van der Veldt AA, Lubberink M, Bahce I, Walraven M, de Boer MP, Greuter HN, et al. Rapid decrease in delivery of chemotherapy to tumors after anti-VEGF therapy: Implications for scheduling of anti-angiogenic drugs. Cancer Cell. 2012 Jan 17;21(1):82–91. doi: 10.1016/j.ccr.2011.11.023. [DOI] [PubMed] [Google Scholar]
- 9.Duda DG, Jain RK, Willett CG. Antiangiogenics: The potential role of integrating this novel treatment modality with chemoradiation for solid cancers. J Clin Oncol. 2007 Sep 10;25(26):4033–42. doi: 10.1200/JCO.2007.11.3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shaked Y, Kerbel RS. Antiangiogenic strategies on defense: On the possibility of blocking rebounds by the tumor vasculature after chemotherapy. Cancer Res. 2007 Aug 1;67(15):7055–8. doi: 10.1158/0008-5472.CAN-07-0905. [DOI] [PubMed] [Google Scholar]
- 11.Gorski DH, Beckett MA, Jaskowiak NT, Calvin DP, Mauceri HJ, Salloum RM, et al. Blockage of the vascular endothelial growth factor stress response increases the antitumor effects of ionizing radiation. Cancer Res. 1999 Jul 15;59(14):3374–8. [PubMed] [Google Scholar]
- 12.Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007 Jan;11(1):69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 13.Folkins C, Man S, Xu P, Shaked Y, Hicklin DJ, Kerbel RS. Anticancer therapies combining antiangiogenic and tumor cell cytotoxic effects reduce the tumor stem-like cell fraction in glioma xenograft tumors. Cancer Res. 2007 Apr 15;67(8):3560–4. doi: 10.1158/0008-5472.CAN-06-4238. [DOI] [PubMed] [Google Scholar]
- 14.de Groot JF, Piao Y, Tran H, Gilbert M, Wu HK, Liu J, et al. Myeloid biomarkers associated with glioblastoma response to anti-VEGF therapy with aflibercept. Clin Cancer Res. 2011 Jul 15;17(14):4872–81. doi: 10.1158/1078-0432.CCR-11-0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Terme M, Pernot S, Marcheteau E, Sandoval F, Benhamouda N, Colussi O, et al. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res. 2013 Jan 15;73(2):539–49. doi: 10.1158/0008-5472.CAN-12-2325. [DOI] [PubMed] [Google Scholar]
- 16.Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010 Aug 1;70(15):6171–80. doi: 10.1158/0008-5472.CAN-10-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Batchelor TT, Duda DG, di Tomaso E, Ancukiewicz M, Plotkin SR, Gerstner E, et al. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J Clin Oncol. 2010 Jun 10;28(17):2817–23. doi: 10.1200/JCO.2009.26.3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamoun WS, Ley CD, Farrar CT, Duyverman AM, Lahdenranta J, Lacorre DA, et al. Edema control by cediranib, a vascular endothelial growth factor receptor-targeted kinase inhibitor, prolongs survival despite persistent brain tumor growth in mice. J Clin Oncol. 2009 May 20;27(15):2542–52. doi: 10.1200/JCO.2008.19.9356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilbert MR, Sulman EP, Mehta MP. Bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014 May 22;370(21):2048–9. doi: 10.1056/NEJMc1403303. [DOI] [PubMed] [Google Scholar]
- 20.Chinot OL, Wick W, Cloughesy T. Bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014 May 22;370(21):2049. doi: 10.1056/NEJMc1403303. [DOI] [PubMed] [Google Scholar]
- 21.Sitohy B, Nagy JA, Jaminet SC, Dvorak HF. Tumor-surrogate blood vessel subtypes exhibit differential susceptibility to anti-VEGF therapy. Cancer Res. 2011 Nov 15;71(22):7021–8. doi: 10.1158/0008-5472.CAN-11-1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeLay M, Jahangiri A, Carbonell WS, Hu YL, Tsao S, Tom MW, et al. Microarray analysis verifies two distinct phenotypes of glioblastomas resistant to antiangiogenic therapy. Clin Cancer Res. 2012 May 15;18(10):2930–42. doi: 10.1158/1078-0432.CCR-11-2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005 Oct;8(4):299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 24.Lu KV, Bergers G. Mechanisms of evasive resistance to anti-VEGF therapy in glioblastoma. CNS Oncol. 2013 Jan;2(1):49–65. doi: 10.2217/cns.12.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shojaei F, Wu X, Qu X, Kowanetz M, Yu L, Tan M, et al. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci U S A. 2009 Apr 21;106(16):6742–7. doi: 10.1073/pnas.0902280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Groot J, Liang J, Kong LY, Wei J, Piao Y, Fuller G, et al. Modulating antiangiogenic resistance by inhibiting the signal transducer and activator of transcription 3 pathway in glioblastoma. Oncotarget. 2012 Sep;3(9):1036–48. doi: 10.18632/oncotarget.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Piao Y, Liang J, Holmes L, Henry V, Sulman E, de Groot JF. Acquired resistance to anti-VEGF therapy in glioblastoma is associated with a mesenchymal transition. Clin Cancer Res. 2013 Aug 15;19(16):4392–403. doi: 10.1158/1078-0432.CCR-12-1557. [DOI] [PubMed] [Google Scholar]
- 28.Lu KV, Chang JP, Parachoniak CA, Pandika MM, Aghi MK, Meyronet D, et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell. 2012 Jul 10;22(1):21–35. doi: 10.1016/j.ccr.2012.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008 Mar;13(3):206–20. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Groot JF, Fuller G, Kumar AJ, Piao Y, Eterovic K, Ji Y, et al. Tumor invasion after treatment of glioblastoma with bevacizumab: Radiographic and pathologic correlation in humans and mice. Neuro Oncol. 2010 Mar;12(3):233–42. doi: 10.1093/neuonc/nop027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Norden AD, Young GS, Setayesh K, Muzikansky A, Klufas R, Ross GL, et al. Bevacizumab for recurrent malignant gliomas: Efficacy, toxicity, and patterns of recurrence. Neurology. 2008 Mar 4;70(10):779–87. doi: 10.1212/01.wnl.0000304121.57857.38. [DOI] [PubMed] [Google Scholar]
- 32.Pope WB, Xia Q, Paton VE, Das A, Hambleton J, Kim HJ, et al. Patterns of progression in patients with recurrent glioblastoma treated with bevacizumab. Neurology. 2011 Feb 1;76(5):432–7. doi: 10.1212/WNL.0b013e31820a0a8a. [DOI] [PubMed] [Google Scholar]
- 33.Wick A, Dorner N, Schafer N, Hofer S, Heiland S, Schemmer D, et al. Bevacizumab does not increase the risk of remote relapse in malignant glioma. Ann Neurol. 2011 Mar;69(3):586–92. doi: 10.1002/ana.22336. [DOI] [PubMed] [Google Scholar]
- 34.Iwamoto FM, Lamborn KR, Robins HI, Mehta MP, Chang SM, Butowski NA, et al. Phase II trial of pazopanib (GW786034), an oral multi-targeted angiogenesis inhibitor, for adults with recurrent glioblastoma (North American Brain Tumor Consortium Study 06-02) Neuro Oncol. 2010 Aug;12(8):855–61. doi: 10.1093/neuonc/noq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reardon DA, Vredenburgh JJ, Desjardins A, Peters K, Gururangan S, Sampson JH, et al. Effect of CYP3A-inducing anti-epileptics on sorafenib exposure: Results of a phase II study of sorafenib plus daily temozolomide in adults with recurrent glioblastoma. J Neurooncol. 2011 Jan;101(1):57–66. doi: 10.1007/s11060-010-0217-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pan E, Yu D, Yue B, Potthast L, Chowdhary S, Smith P, et al. A prospective phase II single-institution trial of sunitinib for recurrent malignant glioma. J Neurooncol. 2012 Oct;110(1):111–8. doi: 10.1007/s11060-012-0943-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerstner ER, Eichler AF, Plotkin SR, Drappatz J, Doyle CL, Xu L, et al. Phase I trial with biomarker studies of vatalanib (PTK787) in patients with newly diagnosed glioblastoma treated with enzyme inducing anti-epileptic drugs and standard radiation and temozolomide. J Neurooncol. 2011 Jun;103(2):325–32. doi: 10.1007/s11060-010-0390-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brandes AA, Stupp R, Hau P, Lacombe D, Gorlia T, Tosoni A, et al. EORTC study 26041-22041: Phase I/II study on concomitant and adjuvant temozolomide (TMZ) and radiotherapy (RT) with PTK787/ZK222584 (PTK/ZK) in newly diagnosed glioblastoma. Eur J Cancer. 2010 Jan;46(2):348–54. doi: 10.1016/j.ejca.2009.10.029. [DOI] [PubMed] [Google Scholar]
- 39.Hainsworth JD, Ervin T, Friedman E, Priego V, Murphy PB, Clark BL, et al. Concurrent radiotherapy and temozolomide followed by temozolomide and sorafenib in the first-line treatment of patients with glioblastoma multiforme. Cancer. 2010 Aug 1;116(15):3663–9. doi: 10.1002/cncr.25275. [DOI] [PubMed] [Google Scholar]
- 40.Lee EQ, Kuhn J, Lamborn KR, Abrey L, DeAngelis LM, Lieberman F, et al. Phase I/II study of sorafenib in combination with temsirolimus for recurrent glioblastoma or gliosarcoma: North american brain tumor consortium study 05-02. Neuro Oncol. 2012 Dec;14(12):1511–8. doi: 10.1093/neuonc/nos264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kreisl TN, McNeill KA, Sul J, Iwamoto FM, Shih J, Fine HA. A phase I/II trial of vandetanib for patients with recurrent malignant glioma. Neuro Oncol. 2012 Dec;14(12):1519–26. doi: 10.1093/neuonc/nos265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kreisl TN, Smith P, Sul J, Salgado C, Iwamoto FM, Shih JH, et al. Continuous daily sunitinib for recurrent glioblastoma. J Neurooncol. 2013 Jan;111(1):41–8. doi: 10.1007/s11060-012-0988-z. [DOI] [PubMed] [Google Scholar]
- 43.Drappatz J, Norden AD, Wong ET, Doherty LM, Lafrankie DC, Ciampa A, et al. Phase I study of vandetanib with radiotherapy and temozolomide for newly diagnosed glioblastoma. Int J Radiat Oncol Biol Phys. 2010 Sep 1;78(1):85–90. doi: 10.1016/j.ijrobp.2009.07.1741. [DOI] [PubMed] [Google Scholar]
- 44.Reardon DA, Egorin MJ, Desjardins A, Vredenburgh JJ, Beumer JH, Lagattuta TF, et al. Phase I pharmacokinetic study of the vascular endothelial growth factor receptor tyrosine kinase inhibitor vatalanib (PTK787) plus imatinib and hydroxyurea for malignant glioma. Cancer. 2009 May 15;115(10):2188–98. doi: 10.1002/cncr.24213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reardon DA, Conrad CA, Cloughesy T, Prados MD, Friedman HS, Aldape KD, et al. Phase I study of AEE788, a novel multitarget inhibitor of ErbB- and VEGF-receptorfamily tyrosine kinases, in recurrent glioblastoma patients. Cancer Chemother Pharmacol. 2012 Jun;69(6):1507–18. doi: 10.1007/s00280-012-1854-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reardon DA, Vredenburgh JJ, Coan A, Desjardins A, Peters KB, Gururangan S, et al. Phase I study of sunitinib and irinotecan for patients with recurrent malignant glioma. J Neurooncol. 2011 Dec;105(3):621–7. doi: 10.1007/s11060-011-0631-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reardon DA, Groves MD, Wen PY, Nabors L, Mikkelsen T, Rosenfeld S, et al. A phase I/II trial of pazopanib in combination with lapatinib in adult patients with relapsed malignant glioma. Clin Cancer Res. 2013 Feb 15;19(4):900–8. doi: 10.1158/1078-0432.CCR-12-1707. [DOI] [PubMed] [Google Scholar]
- 48.Muhic A, Poulsen HS, Sorensen M, Grunnet K, Lassen U. Phase II open-label study of nintedanib in patients with recurrent glioblastoma multiforme. J Neurooncol. 2013 Jan;111(2):205–12. doi: 10.1007/s11060-012-1009-y. [DOI] [PubMed] [Google Scholar]
- 49.Batchelor TT, Duda DG, di Tomaso E, Ancukiewicz M, Plotkin SR, Gerstner E, et al. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J Clin Oncol. 2010 Jun 10;28(17):2817–23. doi: 10.1200/JCO.2009.26.3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Groot JF, Lamborn KR, Chang SM, Gilbert MR, Cloughesy TF, Aldape K, et al. Phase II study of aflibercept in recurrent malignant glioma: A North American Brain Tumor Consortium Study. J Clin Oncol. 2011 Jul 1;29(19):2689–95. doi: 10.1200/JCO.2010.34.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cohen MH, Shen YL, Keegan P, Pazdur R. FDA drug approval summary: Bevacizumab (avastin) as treatment of recurrent glioblastoma multiforme. Oncologist. 2009 Nov;14(11):1131–8. doi: 10.1634/theoncologist.2009-0121. [DOI] [PubMed] [Google Scholar]
- 52.Wick W, Weller M, van den Bent M, Stupp R. Bevacizumab and recurrent malignant gliomas: A European perspective. J Clin Oncol. 2010 Apr 20;28(12):e188, 9. doi: 10.1200/JCO.2009.26.9027. author reply e190-2. [DOI] [PubMed] [Google Scholar]
- 53.Francesconi AB, Dupre S, Matos M, Martin D, Hughes BG, Wyld DK, et al. Carboplatin and etoposide combined with bevacizumab for the treatment of recurrent glioblastoma multiforme. J Clin Neurosci. 2010 Aug;17(8):970–4. doi: 10.1016/j.jocn.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 54.Reardon DA, Desjardins A, Peters KB, Gururangan S, Sampson JH, McLendon RE, et al. Phase II study of carboplatin, irinotecan, and bevacizumab for bevacizumab naive, recurrent glioblastoma. J Neurooncol. 2012 Mar;107(1):155–64. doi: 10.1007/s11060-011-0722-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reardon DA, Desjardins A, Vredenburgh JJ, Gururangan S, Sampson JH, Sathornsumetee S, et al. Metronomic chemotherapy with daily, oral etoposide plus bevacizumab for recurrent malignant glioma: A phase II study. Br J Cancer. 2009 Dec 15;101(12):1986–94. doi: 10.1038/sj.bjc.6605412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ali SA, McHayleh WM, Ahmad A, Sehgal R, Braffet M, Rahman M, et al. Bevacizumab and irinotecan therapy in glioblastoma multiforme: A series of 13 cases. J Neurosurg. 2008 Aug;109(2):268–72. doi: 10.3171/JNS/2008/109/8/0268. [DOI] [PubMed] [Google Scholar]
- 57.Bokstein F, Shpigel S, Blumenthal DT. Treatment with bevacizumab and irinotecan for recurrent high-grade glial tumors. Cancer. 2008 May 15;112(10):2267–73. doi: 10.1002/cncr.23401. [DOI] [PubMed] [Google Scholar]
- 58.Kang TY, Jin T, Elinzano H, Peereboom D. Irinotecan and bevacizumab in progressive primary brain tumors, an evaluation of efficacy and safety. J Neurooncol. 2008 Aug;89(1):113–8. doi: 10.1007/s11060-008-9599-0. [DOI] [PubMed] [Google Scholar]
- 59.Zuniga RM, Torcuator R, Jain R, Anderson J, Doyle T, Ellika S, et al. Efficacy, safety and patterns of response and recurrence in patients with recurrent high-grade gliomas treated with bevacizumab plus irinotecan. J Neurooncol. 2009 Feb;91(3):329–36. doi: 10.1007/s11060-008-9718-y. [DOI] [PubMed] [Google Scholar]
- 60.Hasselbalch B, Eriksen JG, Broholm H, Christensen IJ, Grunnet K, Horsman MR, et al. Prospective evaluation of angiogenic, hypoxic and EGFR-related biomarkers in recurrent glioblastoma multiforme treated with cetuximab, bevacizumab and irinotecan. APMIS. 2010 Aug;118(8):585–94. doi: 10.1111/j.1600-0463.2010.02631.x. [DOI] [PubMed] [Google Scholar]
- 61.Nghiemphu PL, Liu W, Lee Y, Than T, Graham C, Lai A, et al. Bevacizumab and chemotherapy for recurrent glioblastoma: A single-institution experience. Neurology. 2009 Apr 7;72(14):1217–22. doi: 10.1212/01.wnl.0000345668.03039.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Desjardins A, Reardon DA, Coan A, Marcello J, Herndon JE, 2nd, Bailey L, et al. Bevacizumab and daily temozolomide for recurrent glioblastoma. Cancer. 2012 Mar 1;118(5):1302–12. doi: 10.1002/cncr.26381. [DOI] [PubMed] [Google Scholar]
- 63.Sathornsumetee S, Desjardins A, Vredenburgh JJ, McLendon RE, Marcello J, Herndon JE, et al. Phase II trial of bevacizumab and erlotinib in patients with recurrent malignant glioma. Neuro Oncol. 2010 Dec;12(12):1300–10. doi: 10.1093/neuonc/noq099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Galanis E, Anderson SK, Lafky JM, Uhm JH, Giannini C, Kumar SK, et al. Phase II study of bevacizumab in combination with sorafenib in recurrent glioblastoma (N0776): A north central cancer treatment group trial. Clin Cancer Res. 2013 Sep 1;19(17):4816–23. doi: 10.1158/1078-0432.CCR-13-0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Drappatz J, Lee EQ, Hammond S, Grimm SA, Norden AD, Beroukhim R, et al. Phase I study of panobinostat in combination with bevacizumab for recurrent high-grade glioma. J Neurooncol. 2012 Mar;107(1):133–8. doi: 10.1007/s11060-011-0717-z. [DOI] [PubMed] [Google Scholar]
- 66.Cuneo KC, Vredenburgh JJ, Sampson JH, Reardon DA, Desjardins A, Peters KB, et al. Safety and efficacy of stereotactic radiosurgery and adjuvant bevacizumab in patients with recurrent malignant gliomas. Int J Radiat Oncol Biol Phys. 2012 Apr 1;82(5):2018–24. doi: 10.1016/j.ijrobp.2010.12.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gutin PH, Iwamoto FM, Beal K, Mohile NA, Karimi S, Hou BL, et al. Safety and efficacy of bevacizumab with hypofractionated stereotactic irradiation for recurrent malignant gliomas. Int J Radiat Oncol Biol Phys. 2009 Sep 1;75(1):156–63. doi: 10.1016/j.ijrobp.2008.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cabrera AR, Cuneo KC, Vredenburgh JJ, Sampson JH, Kirkpatrick JP. Stereotactic radiosurgery and bevacizumab for recurrent glioblastoma multiforme. J Natl Compr Canc Netw. 2012 Jun 1;10(6):695–9. doi: 10.6004/jnccn.2012.0072. [DOI] [PubMed] [Google Scholar]
- 69.Taal W, Oosterkamp HM, Walenkamp AM, Dubbink HJ, Beerepoot LV, Hanse MC, et al. Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): A randomised controlled phase 2 trial. Lancet Oncol. 2014 Jul 14; doi: 10.1016/S1470-2045(14)70314-6. [DOI] [PubMed] [Google Scholar]
- 70.Reardon DA, Desjardins A, Peters K, Gururangan S, Sampson J, Rich JN, et al. Phase II study of metronomic chemotherapy with bevacizumab for recurrent glioblastoma after progression on bevacizumab therapy. J Neurooncol. 2011 Jun;103(2):371–9. doi: 10.1007/s11060-010-0403-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lu-Emerson C, Norden AD, Drappatz J, Quant EC, Beroukhim R, Ciampa AS, et al. Retrospective study of dasatinib for recurrent glioblastoma after bevacizumab failure. J Neurooncol. 2011 Aug;104(1):287–91. doi: 10.1007/s11060-010-0489-x. [DOI] [PubMed] [Google Scholar]
- 72.Iwamoto FM, Abrey LE, Beal K, Gutin PH, Rosenblum MK, Reuter VE, et al. Patterns of relapse and prognosis after bevacizumab failure in recurrent glioblastoma. Neurology. 2009 Oct 13;73(15):1200–6. doi: 10.1212/WNL.0b013e3181bc0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Quant EC, Norden AD, Drappatz J, Muzikansky A, Doherty L, Lafrankie D, et al. Role of a second chemotherapy in recurrent malignant glioma patients who progress on bevacizumab. Neuro Oncol. 2009 Oct;11(5):550–5. doi: 10.1215/15228517-2009-006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Reardon DA, Herndon JE, 2nd, Peters KB, Desjardins A, Coan A, Lou E, et al. Bevacizumab continuation beyond initial bevacizumab progression among recurrent glioblastoma patients. Br J Cancer. 2012 Oct 23;107(9):1481–7. doi: 10.1038/bjc.2012.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lai A, Tran A, Nghiemphu PL, Pope WB, Solis OE, Selch M, et al. Phase II study of bevacizumab plus temozolomide during and after radiation therapy for patients with newly diagnosed glioblastoma multiforme. J Clin Oncol. 2011 Jan 10;29(2):142–8. doi: 10.1200/JCO.2010.30.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vredenburgh JJ, Desjardins A, Kirkpatrick JP, Reardon DA, Peters KB, Herndon JE, 2nd, et al. Addition of bevacizumab to standard radiation therapy and daily temozolomide is associated with minimal toxicity in newly diagnosed glioblastoma multiforme. Int J Radiat Oncol Biol Phys. 2012 Jan 1;82(1):58–66. doi: 10.1016/j.ijrobp.2010.08.058. [DOI] [PubMed] [Google Scholar]
- 77.Vredenburgh JJ, Desjardins A, Reardon DA, Peters KB, Herndon JE, 2nd, Marcello J, et al. The addition of bevacizumab to standard radiation therapy and temozolomide followed by bevacizumab, temozolomide, and irinotecan for newly diagnosed glioblastoma. Clin Cancer Res. 2011 Jun 15;17(12):4119–24. doi: 10.1158/1078-0432.CCR-11-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014 Feb 20;370(8):709–22. doi: 10.1056/NEJMoa1308345. [DOI] [PubMed] [Google Scholar]
- 79.Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014 Feb 20;370(8):699–708. doi: 10.1056/NEJMoa1308573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Macdonald DR, Cascino TL, Schold SC, Jr, Cairncross JG. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol. 1990 Jul;8(7):1277–80. doi: 10.1200/JCO.1990.8.7.1277. [DOI] [PubMed] [Google Scholar]
- 81.Wen PY, Macdonald DR, Reardon DA, Cloughesy TF, Sorensen AG, Galanis E, et al. Updated response assessment criteria for high-grade gliomas: Response assessment in neuro-oncology working group. J Clin Oncol. 2010 Apr 10;28(11):1963–72. doi: 10.1200/JCO.2009.26.3541. [DOI] [PubMed] [Google Scholar]
- 82.Sulman EP, Won M, Blumenthal DT, Vogelbaum MA, Colman H, Jenkins RB, et al. Molecular predictors of outcome and response to bevacizumab (BEV) based on analysis of RTOG 0825, a phase III trial comparing chemoradiation (CRT) with and without BEV in patients with newly diagnosed glioblastoma (GBM). J Clin Oncol; Proceedings of the American Society of Clinical Oncology; 2013 May 31-June 4; Chicago, Illinois. 2013. suppl; abstr LBA2010. [Google Scholar]
- 83.Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006 Mar;9(3):157–73. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 84.Gilbert MR, Wang M, Aldape KD, Stupp R, Hegi ME, Jaeckle KA, et al. Dose-dense temozolomide for newly diagnosed glioblastoma: A randomized phase III clinical trial. J Clin Oncol. 2013 Nov 10;31(32):4085–91. doi: 10.1200/JCO.2013.49.6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sathornsumetee S, Cao Y, Marcello JE, Herndon JE, 2nd, McLendon RE, Desjardins A, et al. Tumor angiogenic and hypoxic profiles predict radiographic response and survival in malignant astrocytoma patients treated with bevacizumab and irinotecan. J Clin Oncol. 2008 Jan 10;26(2):271–8. doi: 10.1200/JCO.2007.13.3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Phillips H, Sandmann T, Li C, Cloughesy T, Chinot O, Wick W, et al. Correlations of molecular subtypes with survival in AVAglio (bevacizumab [BV] plus radiotherapy [RT] and temozolomide [T] for newly diagnosed glioblastoma [GBM]). J Clin Oncol; Proceedings of the American Society of Clinical Oncology; 2014 May 30-June 3; Chicago, Illinois. 2014. p. 5s. suppl; abstr 2001. [Google Scholar]
- 87.Batchelor TT, Gerstner ER, Emblem KE, Duda DG, Kalpathy-Cramer J, Snuderl M, et al. Improved tumor oxygenation and survival in glioblastoma patients who show increased blood perfusion after cediranib and chemoradiation. Proc Natl Acad Sci U S A. 2013 Nov 19;110(47):19059–64. doi: 10.1073/pnas.1318022110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nishikawa R, Saran F, Mason W, Wick W, Cloughesy TF, Henriksson R, et al. Biomarker (BM) evaluations in the phase III AVAglio study of bevacizumab (Bv) plus standard radiotherapy (RT) and temozolomide (T) for newly diagnosed glioblastoma (GBM). J Clin Oncol; Proceedings of the American Society of Clinical Oncology; 2013 May 31-June 4; Chicago, Illinois. 2013. suppl; abstr 2023. [Google Scholar]
- 89.Tabouret E, Boudouresque F, Barrie M, Matta M, Boucard C, Loundou A, et al. Association of matrix metalloproteinase 2 plasma level with response and survival in patients treated with bevacizumab for recurrent high-grade glioma. Neuro Oncol. 2014 Mar;16(3):392–9. doi: 10.1093/neuonc/not226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Duda DG, Willett CG, Ancukiewicz M, di Tomaso E, Shah M, Czito BG, et al. Plasma soluble VEGFR-1 is a potential dual biomarker of response and toxicity for bevacizumab with chemoradiation in locally advanced rectal cancer. Oncologist. 2010;15(6):577–83. doi: 10.1634/theoncologist.2010-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pope WB, Kim HJ, Huo J, Alger J, Brown MS, Gjertson D, et al. Recurrent glioblastoma multiforme: ADC histogram analysis predicts response to bevacizumab treatment. Radiology. 2009 Jul;252(1):182–9. doi: 10.1148/radiol.2521081534. [DOI] [PubMed] [Google Scholar]
- 92.Waldman AD, Jackson A, Price SJ, Clark CA, Booth TC, Auer DP, et al. Quantitative imaging biomarkers in neuro-oncology. Nat Rev Clin Oncol. 2009 Aug;6(8):445–54. doi: 10.1038/nrclinonc.2009.92. [DOI] [PubMed] [Google Scholar]
- 93.O'Connor JP, Jackson A, Parker GJ, Roberts C, Jayson GC. Dynamic contrastenhanced MRI in clinical trials of antivascular therapies. Nat Rev Clin Oncol. 2012 Feb 14;9(3):167–77. doi: 10.1038/nrclinonc.2012.2. [DOI] [PubMed] [Google Scholar]
- 94.Emblem KE, Mouridsen K, Bjornerud A, Farrar CT, Jennings D, Borra RJ, et al. Vessel architectural imaging identifies cancer patient responders to anti-angiogenic therapy. Nat Med. 2013 Sep;19(9):1178–83. doi: 10.1038/nm.3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen W, Delaloye S, Silverman DH, Geist C, Czernin J, Sayre J, et al. Predicting treatment response of malignant gliomas to bevacizumab and irinotecan by imaging proliferation with [18F] fluorothymidine positron emission tomography: A pilot study. J Clin Oncol. 2007 Oct 20;25(30):4714–21. doi: 10.1200/JCO.2006.10.5825. [DOI] [PubMed] [Google Scholar]
- 96.Harris RJ, Cloughesy TF, Pope WB, Nghiemphu PL, Lai A, Zaw T, et al. 18F-FDOPA and 18F-FLT positron emission tomography parametric response maps predict response in recurrent malignant gliomas treated with bevacizumab. Neuro Oncol. 2012 Aug;14(8):1079–89. doi: 10.1093/neuonc/nos141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Galanis E, Wu W, Cloughesy T, Lamborn K, Mann B, Wen PY, et al. Phase 2 trial design in neuro-oncology revisited: A report from the RANO group. Lancet Oncol. 2012 May;13(5):e196–204. doi: 10.1016/S1470-2045(11)70406-5. [DOI] [PubMed] [Google Scholar]
- 98.Batchelor TT, Mulholland P, Neyns B, Nabors LB, Campone M, Wick A, et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol. 2013 Sep 10;31(26):3212–8. doi: 10.1200/JCO.2012.47.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jain RK. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science. 2005 Jan 7;307(5706):58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]