Bcl-xL sequesters its C-terminal membrane anchor in soluble, cytosolic homodimers (original) (raw)
Abstract
Bcl-xL is a potent inhibitor of apoptosis. While Bcl-xL can be bound to mitochondria, a substantial fraction, depending on the cell type or tissue, is found in the cytosol of healthy cells. Gel filtration and crosslinking experiments reveal that, unlike monomeric Bax, Bcl-xL migrates in a complex of approximately 50 kDa in the cytosol. Co-immunoprecipitation experiments indicate that Bcl-xL in the cytosol forms homodimers. The C-terminal hydrophobic tails of two Bcl-xL molecules are involved in homodimer formation, and analysis of mutants demonstrates that the C-terminal lysine residue and the G138 residue lining the BH3-binding pocket are required for homodimerization. The flexible loop preceding the C-terminal tail in Bcl-xL is longer than that of several monomeric Bcl-2 family members and is a requisite for the homodimer formation. Bad binding to Bcl-xL dissociates the homodimers and triggers Bcl-xL binding to mitochondrial membranes. The C-terminal tail of Bcl-xL is also required to mediate Bcl-xL/Bax heterodimer formation. Both mitochondrial import and antiapoptotic activity of different Bcl-xL mutants correlate with their ability to form homodimers.
Keywords: apoptosis, Bad, Bax, Bcl-2, Bcl-xL, dimerization
Introduction
The Bcl-2 family members are central regulators of apoptosis that promote or inhibit cell death (Adams and Cory, 1998; Rathmell and Thompson, 2002). An original model to explain the opposing bioactivity of close relatives was that homodimers were counteracted by heterodimers (Oltvai et al, 1993). However, the finding that Bax exists primarily as a monomer in the cytosol of healthy cells (Hsu and Youle, 1998) and oligomerizes during apoptosis (Tan et al, 1999; Antonsson et al, 2001) contributed to a current model that Bcl-2 and Bcl-xL heterodimerize with and sequester BH3-only proteins, preventing them from activating Bax and Bak oligomerization (Letai et al, 2002).
The BH3 domain in Bak, Bad and Bim has been proposed to mediate heterodimerization by fitting into a hydrophobic BH3-binding pocket of Bcl-xL (Sattler et al, 1997; Petros et al, 2000; Liu et al, 2003). Mutagenesis of amino acids lining the hydrophobic pocket in Bcl-xL confirms that this region is involved in heterodimer formation (Minn et al, 1999). The three-dimensional structure of Bax shows that the C-terminal hydrophobic tail (C-tail) normally occupies the homologous hydrophobic pocket in the soluble form of Bax found in healthy cells (Suzuki et al, 2000). Occupancy of the hydrophobic pocket by the C-terminal domain suggests that Bax binding to BH3 domains displaces the membrane anchor to initiate mitochondrial membrane insertion.
Bcl-xL is found both in the cytosol and attached to mitochondrial membranes in healthy cells and tissues (Hsu et al, 1997; Hausmann et al, 2000; Nijhawan et al, 2003). During apoptosis, the cytosolic fraction of Bcl-xL translocates and inserts into mitochondria (Hsu et al, 1997). The antiapoptotic Bcl-xL homologues, Bcl-w (O'Reilly et al, 2001; Wilson-Annan et al, 2003) and Mcl-1 (Nijhawan et al, 2003), also exist partially in the cytosol and translocate to mitochondria during apoptosis, whereas Bcl-2 is constitutively bound to mitochondrial membranes. We report here that cytosolic Bcl-xL exists as a homodimer. Bad displaces homodimer formation, indicating that the BH3-binding pocket of Bcl-xL is required for homodimerization and, like Bim binding does to Bcl-w (Wilson-Annan et al, 2003), triggers Bcl-xL mitochondrial translocation. Surprisingly, mutation in or deletion of the Bcl-xL C-tail eliminates dimer formation. These results suggest the new model that Bcl-xL sequesters its hydrophobic membrane anchor in the homodimer partner BH3-binding pocket, a model distinct from the self-sequestration mechanism of Bax, and that the dimeric form is important for Bcl-xL insertion into mitochondria and bioactivity.
Results
Bcl-xL in the cytosol forms homodimers
A substantial fraction of Bcl-xL, varying with different cell types, is found in the cytosol of healthy cells. As this subcellular distribution differs from that of Bcl-2, which is essentially all membrane bound, and that of Bax, which is fully cytosolic, we investigated the quaternary structure of Bcl-xL in the cytosol. Gel filtration of the cytosol of healthy HeLa cells followed by Western blotting of the eluted fractions for Bcl-xL shows that endogenous Bcl-xL migrates around 50 kDa on Superose 6 (Figure 1A) and around 40 kDa on Superdex 200 (data not shown), significantly higher than that expected for a 26 kDa monomer. In contrast, Bax migrates as a monomeric protein (Figure 1A). Crosslinking of Cos-7 and HeLa cell cytosolic extracts shows that most of the Bcl-xL migrates as a 52 kDa protein, while no changes in Bax are seen after crosslinking (Figure 1B). Thus, endogenous Bcl-xL does not appear as a monomer like Bax (Hsu and Youle, 1998) and Bcl-w (Wilson-Annan et al, 2003), but as a higher molecular weight complex in the cytosol.
Figure 1.
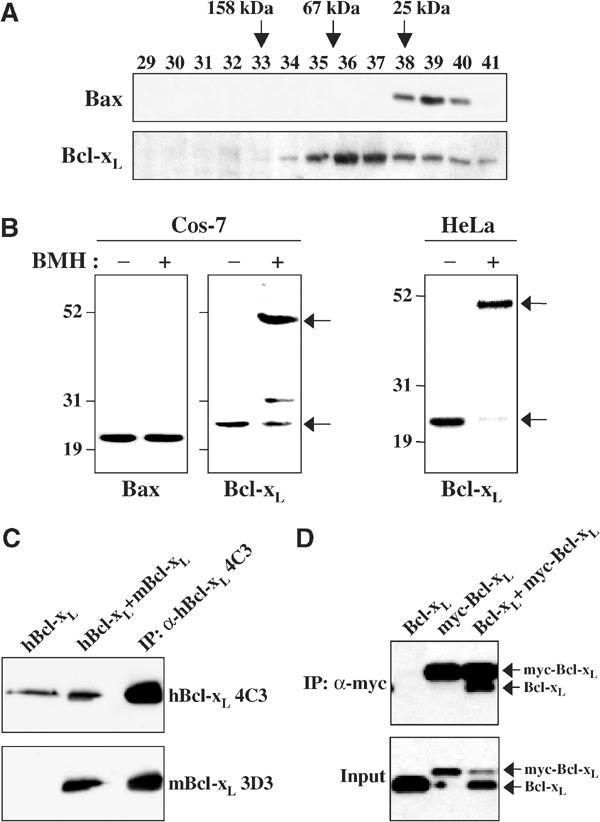
Cytosolic Bcl-xL is a homodimer. (A) Determination of endogenous cytosolic Bcl-xL and Bax quaternary structure by gel filtration. The cytosol fraction (S-100) isolated from HeLa cells was loaded onto a Superose 6 column previously calibrated with proteins of known molecular weight (aldolase: 158 kDa, albumin: 67 kDa and chymotrypsinogen A: 25 kDa). The fractions were collected and analyzed for Bcl-xL and Bax by Western blotting with anti-Bcl-xL L19 and anti-Bax 2D2 antibodies. While Bax was eluted below the 25 kDa marker as expected, Bcl-xL was found at a higher molecular weight. (B) Crosslinking of endogenous cytosolic Bcl-xL in Cos-7 and HeLa cells. The cytosol fractions from healthy Cos-7 and HeLa cells were treated with 1 mM BMH (Pierce) and incubated for 1 h at room temperature. Aliquots were applied to SDS–PAGE and analyzed for Bcl-xL and Bax by Western blotting with anti-Bcl-xL L19 and anti-Bax 2D2 antibodies. The arrows show monomeric and dimeric sizes of Bcl-xL. (C) Co-IP of human Bcl-xL and mouse Bcl-xL. HeLa cells stably expressing human Bcl-xL (hBcl-xL) were transiently transfected with mouse Bcl-xL (mBcl-xL). Human Bcl-xL was immunoprecipitated from the cytosol fraction (S-100) of cells by human specific Bcl-xL antibody, 4C3. The cytosolic fractions of HeLa cells expressing only human Bcl-xL (lane 1, hBcl-xL) and expressing both human and mouse Bcl-xL (lane 2, hBcl-xL+mBcl-xL) and the immunoprecipitated sample (lane 3) from cells expressing both human and mouse Bcl-xL were analyzed by Western blotting with the human specific α-hBcl-xL 4C3 antibody (top) and the mouse specific Bcl-xL antibody α-mBCL-xL 3D3 (bottom). (D) Co-IP of myc-Bcl-xL and Bcl-xL. Cos-7 cells were transfected with either Bcl-xL or myc-Bcl-xL alone, or with both (ratio, 1:1). Myc-Bcl-xL was immunoprecipitated from the S-100 by anti-myc 9B11 antibody. The immunoprecipitated samples were analyzed by Western blotting with anti-Bcl-xL 4C3 antibody.
To determine whether Bcl-xL formed homodimers in the cytosol, consistent with the molecular weight of the complex, species-specific anti-Bcl-xL antibodies (Hsu et al, 2003) were used for immunoprecipitation (IP) of human Bcl-xL from HeLa cells expressing human and mouse Bcl-xL simultaneously. Figure 1C shows that human Bcl-xL binds to and co-immunoprecipitates with mouse Bcl-xL. Lastly, myc-tagged Bcl-xL was found to co-immunoprecipitate with untagged Bcl-xL (Figure 1D). The co-IP results combined with the gel filtration and crosslinking results indicate that the soluble form of Bcl-xL exists as a homodimer. Bcl-xS, an alternative splice form of Bcl-xL, also has been reported to be a homodimer (Lindenboim et al, 2001).
Role of the C-terminal hydrophobic tail and the hydrophobic BH3-binding pocket of Bcl-xL in dimer formation
We examined the domains of Bcl-xL that could be involved in homodimer formation. Surprisingly, deletion of the C-terminal membrane anchor (C-terminal 21 amino acids) of Bcl-xL inhibited co-IP with wild-type (wt) myc-Bcl-xL (Figure 2A). A very small band of truncated Bcl-xL could be seen in the co-immunoprecipitate with wt myc-Bcl-xL, whereas no truncated Bcl-xL could be found in the co-immunoprecipitate with truncated myc-Bcl-xL. This indicates that deletion of one C-tail largely, but not completely, eliminates binding and elimination of both tails completely eliminates homodimer formation. Thus, the C-tail is required for the formation of stable homodimers. Detergents influence the oligomerization properties of proteins from the Bcl-2 family and, more specifically, Bcl-xL binding to Bax is stimulated by detergents (Hsu and Youle, 1997). We therefore examined Bcl-xL homodimer formation in two detergents with differential effects on Bcl-xL/Bax heterodimer formation and found that CHAPS did not affect homodimer formation, while Triton X-100 slightly inhibited the binding (Figure 2B).
Figure 2.
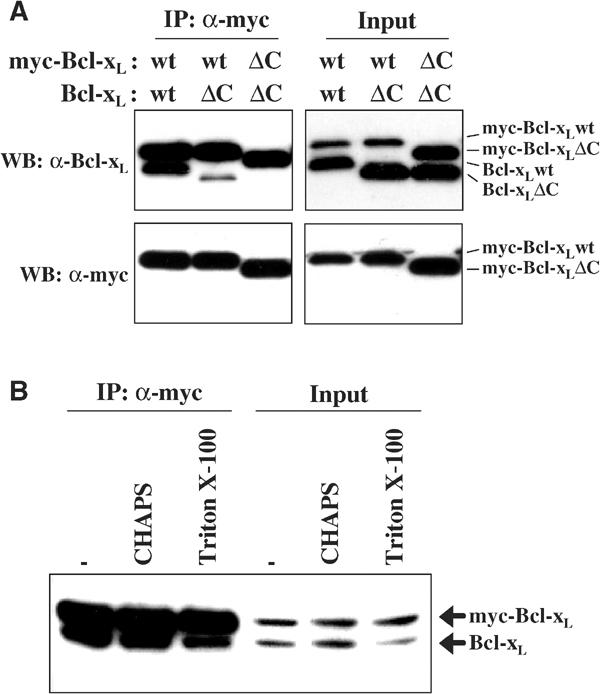
The BH3-binding pocket and the C-terminal hydrophobic anchor of Bcl-xL mediate homodimer formation. (A) Co-IP of myc-Bcl-xL and Bcl-xL. Cos-7 cells were co-transfected with full-length (wt) and the C-tail truncated form (ΔC) of Bcl-xL or myc-tagged Bcl-xL as indicated and incubated for 16 h. Myc-tagged Bcl-xL was immunoprecipitated from the cytosol fraction (S-100) of cells by the anti-myc antibody. The immunoprecipitated products and the input were visualized by Western blotting with antibodies against myc and Bcl-xL. (B) Detergent effects on Bcl-xL dimer formation. Cos-7 cells were co-transfected with Bcl-xL and myc-tagged Bcl-xL and cultured for 16 h. Myc-tagged Bcl-xL was immunoprecipitated from S-100 by the anti-myc antibody in the absence or presence of detergent (1% final concentration) as indicated. The immunoprecipitated products and the input were visualized by Western blotting with antibodies against myc and Bcl-xL.
The C-tails of the monomeric forms of Bax (Suzuki et al, 2000) and Bcl-w (Denisov et al, 2003; Hinds et al, 2003) fit into the hydrophobic BH3-binding pockets homologous to the pocket found in Bcl-xL that binds BH3-only proteins. Therefore, we hypothesized that the Bcl-xL tails may bind reciprocally into the BH3-binding pockets of the homodimer partner. To test this hypothesis, mutants in the C-terminal tail and BH3-binding pocket were analyzed.
Consequences of mutations in the C-terminal hydrophobic tail on dimer formation and on bioactivity
Mutagenesis of the C-tail was used to define the structural requirements for dimer formation and to explore the role of dimer formation in the biological activities of Bcl-xL (Figure 3A). Deletion of the C-terminal two amino acids, RK, significantly decreased homodimer formation and elimination of the four or five C-terminal amino acids completely eliminated homodimer formation (Figure 3B). This indicated some contribution of the two positively charged C-terminal amino acids and partial involvement of the next two, FS, residues. Consistent with this, an AAAAA C-terminal substitution mutant was less active in dimerization than either the Δ2C mutant or an AAARK mutant (Figure 3B). However, deleting the third, fourth and fifth residues (ΔLFS) or substituting them for alanine (AAARK) alone did not greatly decrease dimer formation. Interestingly, the AAAAK and AAARK mutants formed dimers significantly better than the AAARA mutant, showing again the critical role of the C-terminal lysine residue in dimer formation.
Figure 3.
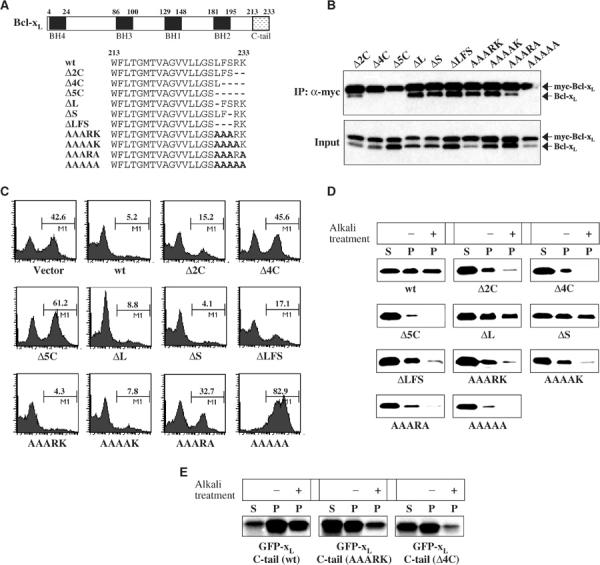
Antiapoptotic activity of Bcl-xL mutants correlates with their ability to form homodimers. (A) Schematic structure of human Bcl-xL and a C-terminal sequence representation of the different mutants of Bcl-xL studied. (B) Co-IP of myc-Bcl-xL mutants and the same untagged Bcl-xL mutants. Cos-7 cells were co-transfected with each myc-Bcl-xL and Bcl-xL mutant (ratio, 1:1). Myc-Bcl-xL was immunoprecipitated from the cytosol fraction (S-100) of cells by anti-myc antibody. The immunoprecipitated samples were analyzed by Western blotting with anti-Bcl-xL 4C3 antibodies. (C) Viability assays of wt Bcl-xL and Bcl-xL mutants. Jurkat cells were co-transfected with YFP-Bcl-xL constructs and Bax (ratio, 1:2) and incubated for 12 h and treated with 100 μM etoposide for 6 h. The amount of apoptosis among YFP-expressing cells was measured by Annexin V binding. The numbers in the figures are percentages of apoptotic cells among transfectants. (D) Only mutants capable of homodimer formation are imported into the mitochondria in vitro. Radiolabeled wt Bcl-xL and each of the C-tail mutants were incubated separately with mitochondria isolated from early-stage apoptotic HeLa cells for 15 min at 37°C. The analysis of one half of the samples shows the imported proteins (recovered in the mitochondrial pellet: P) compared to the nonimported proteins (remaining in the supernatant: S). The membrane insertion of the imported proteins was assessed by alkali (Na2CO3) treatment of the second half. Samples were analyzed by SDS–PAGE and fluorography. (E) The insertion into the mitochondrial membrane is impaired by mutations in the C-terminal tail of Bcl-xL. The C-loop and C-terminal tail of wt Bcl-xL (C-terminal 39 amino acids) and the same regions in mutants (AAARK and Δ4C) were fused to the C-terminus of GFP, used as a reporter protein. The radiolabeled fusion proteins were incubated separately with mitochondria isolated from early-stage apoptotic HeLa cells, and the import experiments were performed as described in Figure 3D.
We examined the biological activity of this series of mutants and found that apoptosis inhibition activity correlated with the ability to form dimers (Figure 3C, Table I). We also explored mitochondrial import of these mutants because some of the same amino acids that have been shown previously to be important for mitochondrial targeting (Kaufmann et al, 2003) are important for dimer formation. Native Bcl-xL fails to import efficiently into healthy HeLa cell mitochondria (see Figure 5B), but does insert into mitochondria isolated from early-stage apoptotic HeLa cells (Figure 3D). However, all the Bcl-xL mutants unable to dimerize (Δ4C, Δ5C, AAAAA) were also unable to be imported into isolated mitochondria in this in vitro system (Figure 3D). While the ΔL and ΔS mutants were able to insert into the membrane as assessed by alkali treatment of the mitochondrial pellet following the import experiment, the other mutants, Δ2C, ΔLFS, AAARK, AAAAK, and AAARA, weakly inserted into the membrane even though they form dimers (Figure 3D). The C-terminal tail contains all the information for the mitochondrial targeting as GFP fused at the C-terminus to the 39 C-terminal amino acids of Bcl-xL inserts efficiently into membranes (Figure 3E). Two of the C-tail mutations directly affect insertion as fusion of GFP with the AAARK mutant tail was less efficient than the wt tail and GFP fused to the tail lacking the last four amino acids was even less active (Figure 3E). This correlates with a previous report that the presence of basic amino acids in the C-tail is required for Bcl-xL to the mitochondrial targeting membrane (Kaufmann et al, 2003). Those results indicate a more stringent requirement of the C-tail integrity for mitochondrial import than for dimer formation. It is nonetheless noteworthy that all the mutants unable to dimerize were also unable to be imported into mitochondria and lost the ability to protect against induced cell death.
Table 1.
Summary of results for Bcl-xL mutagenesis
| Bcl-xL | Sequence | Antiapoptosis | Homodimer | Import |
|---|---|---|---|---|
| wt | GVVLLGSLFSRK233 | +++++ | ++++ | +++ |
| C-tail mutants | ||||
| Δ2C | GVVLLGSLFS- - | +++ | ++ | + |
| Δ4C | GVVLLGSL- - - - | − | − | − |
| Δ5C | GVVLLGS- - - - - | − | − | − |
| ΔL | GVVLLGS-FSRK | ++++ | ++++ | +++ |
| ΔS | GVVLLGSLF-RK | +++++ | ++++ | +++ |
| ΔLFS | GVVLLGS- - -RK | +++ | ++++ | + |
| AAARK | GVVLLGSAAARK | +++++ | ++++ | + |
| AAAAK | GVVLLGSAAAAK | ++++ | ++++ | + |
| AAARA | GVVLLGSAAARA | + | ++ | + |
| AAAAA | GVVLLGSAAAAA | − | − | − |
| BH3-pocket mutants | ||||
| Y101K | +++ | ++++ | +++ | |
| G138A | − | ± | − |
Figure 5.
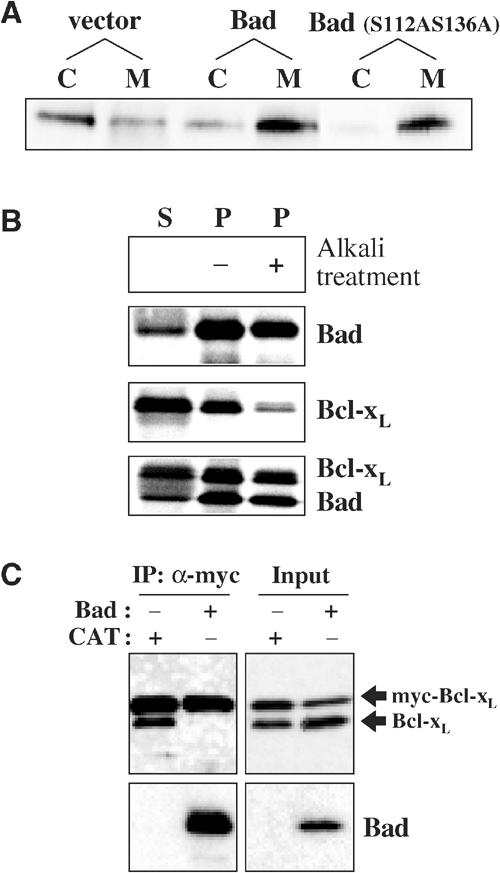
Bad promotes Bcl-xL redistribution to the mitochondria and disrupts Bcl-xL homodimers. (A) Bcl-xL redistributes from the cytosol to the mitochondria upon overexpression of Bad. HeLa cells were transfected with pcDNA3 vector, mouse Bad or mutant mouse Bad (S112AS136A). The cells were subfractionated as described in Materials and methods, and the cytosolic fraction (C) and the mitochondrial fraction (M) were analyzed by SDS–PAGE and Western blotting for endogenous Bcl-xL. (B) Bad triggers the translocation and insertion of Bcl-xL into the mitochondria in vitro. Radiolabeled human Bad and Bcl-xL were incubated separately with mitochondria isolated from healthy HeLa cells for 15 min at 37°C. The import experiment was performed as described in Figure 3D. In the lower panels, Bad was pre-incubated with Bcl-xL for 10 min at 37°C prior to the import experiment. Bcl-xL (upper band) redistributed mainly to the mitochondrial pellet and became resistant to carbonate extraction upon co-import with Bad. (C) Bad inhibits Bcl-xL dimer formation. Myc-Bcl-xL and Bcl-xL were co-transfected into Cos-7 cells with human Bad or chloramphenicol transferase (CAT). At 16 h after transfection, staurosporine was added into the media to a final concentration of 1.2 μM and further incubated for 4 h. The mitochondrial fraction was incubated with 1% Triton X-100 for 20 min on ice. After centrifugation at 12 000 g for 10 min at 4°C, the supernatant was used for the co-IP of myc-Bcl-xL and Bcl-xL. After IP with anti-myc antibody in the presence of 1% Triton X-100, the immunoprecipitated samples and the input were analyzed by Western blotting with anti-Bcl-xL 4C3 and anti-Bad antibodies.
Mutations in the BH3-binding pocket of Bcl-xL correlate dimer formation with mitochondrial import and bioactivity
To investigate the involvement of the BH3-binding pocket in the homodimerization of Bcl-xL, we constructed mutations in two amino acids, G138A and Y101K, lining the BH3-binding pocket (Sattler et al, 1997) that have been reported to inhibit Bcl-xL binding to Bax. We found that the Y101K mutant retained homodimerization activity, membrane insertion activity and antiapoptotic bioactivity, whereas the G138A mutant lacked all the three activities (see Supplementary Data (Sedlak et al, 1995)). Thus, in contrast to their ability to heterodimerize with Bax, the ability of the Bcl-xL mutants in the BH3-binding pocket to homodimerize correlates with the ability to insert into the mitochondrial membrane and with bioactivity. However, the G138A mutation occurs in the α-6 helix that may be directly involved in membrane insertion. Thus, the mutation may directly inhibit membrane insertion independently of the effect on dimer formation.
The length of C-terminal flexible loop in Bcl-xL is critical for dimer formation and bioactivity
While the related Bcl-2 family members Bcl-w and Bax have been found to exist as monomers in the cytosol, the Bcl-xL conformation seems to favor homodimerization. However, Bcl-xL may also be able to form monomers as the gel filtration profile shows that the protein migrates asymmetrically to the right of the peak (Figure 1A). Bax has a five-amino-acid loop and Bcl-w has a six-amino-acid flexible loop (C-loop) prior to the C-tail, whereas Bcl-xL has an 18-amino-acid linker (Figure 4A). This extra length may allow the C-tail of Bcl-xL to fit across into the dimer partner BH3-binding pocket. To examine this proposal, we constructed two chimeras by switching the C-loop and C-tail of Bcl-xL for those of Bcl-w, respectively (Figure 4B). Chimera 1, having a short C-loop, failed to inhibit apoptosis and failed to form dimers, indicating that the C-loop in Bcl-xL is critical for dimer formation and bioactivity (Figure 4C and D). Chimera 2 retained bioactivity, but failed to form homodimers, suggesting that the C-tail of Bcl-w (37 amino acids), which is longer than that of Bcl-xL (21 amino acids), is not able to fit into the BH3 pocket of Bcl-xL and may constitutively bind mitochondria. In vitro import experiments and the subcellular localization by confocal microscopy of YFP fusion proteins in living cells demonstrate that chimera 1 does not insert into the mitochondrial membrane (Figure 4E and F). The C-tail of chimera 1 could be sequestered into its own BH3-pocket like Bcl-w and thereby locate to the cytosol. Chimera 2 inserted into the mitochondrial membrane in vitro and the yellow fluorescence protein (YFP)-tagged version was predominantly bound to mitochondria in cells, consistent with the hypothesis that its constitutive mitochondrial localization mediates bioactivity.
Figure 4.
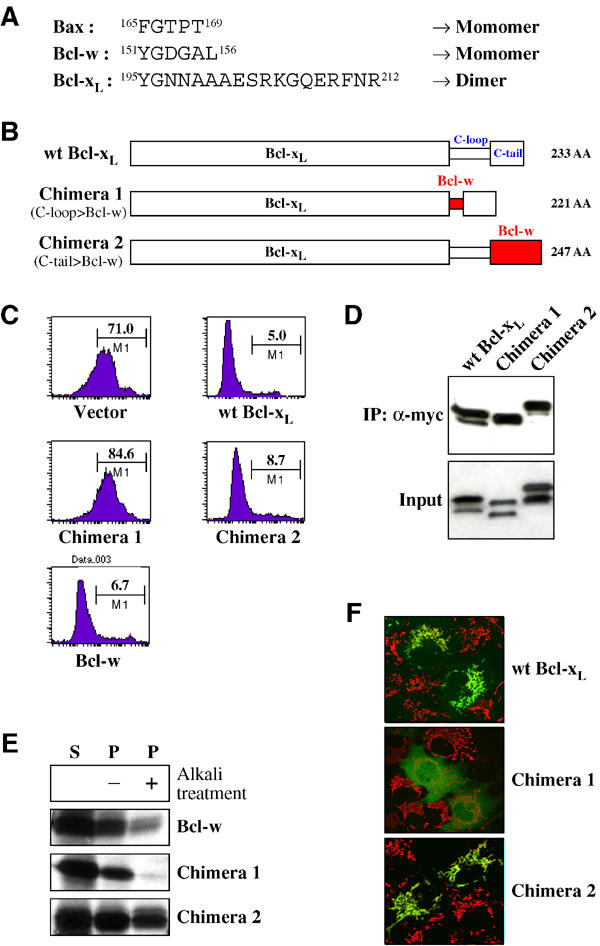
The involvement of the Bcl-xL C-terminal loop in dimer formation and bioactivity. (A) Comparison of the size and sequence of the loop preceding the C-tail (C-loop) among monomeric (Bax and Bcl-w) and the dimeric (Bcl-xL) members of the Bcl-2 family. (B) Schematic representation of Bcl-xL/Bcl-w chimeras. Chimera 1 is made by switching the short loop prior to C-tail of Bcl-w with the long loop of Bcl-xL. Chimera 2 is constructed by replacing the C-tail of Bcl-xL with that of Bcl-w. The total amino-acid number of each chimera is shown on the right side. (C) Viability assays of wt Bcl-xL, Bcl-w, and chimeras. Jurkat cells were co-transfected with the YFP-Bcl-xL constructs and Bax (ratio, 1:2) and incubated for 12 h and treated with 100 μM etoposide for 6 h. The viability assay experiment was performed as described in Figure 3C. (D) Homodimerization of chimeras. Cos-7 cells were co-transfected with each myc-tagged chimera and chimera as indicated in the figure (ratio, 1:1), and myc-tagged chimera was immunoprecipitated from the cytosol fraction (S-100) of cells by anti-myc antibody. The immunoprecipitated and the input samples were analyzed by Western blotting with anti-Bcl-xL 4C3 antibodies. (E) Mitochondrial import of chimeras in vitro. Radiolabeled Bcl-w and chimeras were incubated separately with isolated mitochondria isolated from early-stage apoptotic HeLa cells for 15 min at 37°C and the import experiment was performed as described in Figure 3D. (F) Subcellular localization of Bcl-xL chimeras. Cos-7 cells were transfected with plasmids encoding YFP fused to wt Bcl-xL, chimera 1 or chimera 2. After overnight incubation, cells were treated with MitoTracker Red CMXRos. Location of YFP-fusion proteins (green) and mitochondria (red) in cells was visualized by confocal microscopy. Images were overlaid to examine co-localization (yellow) of YFP-fusion protein and mitochondria.
Bad triggers Bcl-xL to the mitochondrial membrane in vivo and in vitro by disrupting Bcl-xL homodimers
Although the mitochondrial redistribution of various proapoptotic Bcl-2 proteins is induced by many cell death stimuli, the molecular basis for membrane translocation remains unknown (Cory and Adams, 2002), except in the case of Bcl-w where Bim binding initiates integral membrane insertion (Wilson-Annan et al, 2003). As the proapoptotic Bad protein was shown to bind Bcl-xL (Petros et al, 2000), we examined the effect of Bad activation on the subcellular localization of Bcl-xL. HeLa cells were separated into cytosolic and mitochondria-containing membrane fractions and the endogenous Bcl-xL localization was probed. Overexpression of Bad shifted endogenous Bcl-xL into the mitochondrial fraction (Figure 5A). Bad can be sequestered in the cytosol by phosphorylation-dependent binding to 14-3-3 (Zha et al, 1996). Upon overexpression of a constitutively active Bad mutant, Bad (S112AS136A), which cannot bind 14-3-3 as the major phosphorylation sites are mutated, endogenous Bcl-xL is completely redistributed to the membrane pellet (Figure 5A). Therefore, Bad expression triggers the relocalization of endogenous Bcl-xL to membranes.
As Bad activation and binding provokes the translocation of Bcl-xL to membranes, we examined the mitochondrial import of Bcl-xL and Bad in vitro to assess whether any other factor(s) may be required for their translocation. Radiolabeled proteins were synthesized by in vitro transcription and translation using rabbit reticulocyte lysates. As the synthesized proteins do not undergo post-translational modifications such as phosphorylation, and other cytosolic proteins including potential binding partners are absent, this in vitro system provides a convenient tool to study their import in isolation. Bad imported very efficiently into isolated mitochondria (Figure 5B) and appeared to insert into the membrane as the protein remained in the pellet fraction even after alkali extraction. In contrast, the in vitro synthesized Bcl-xL, like its in vivo counterpart, was found predominantly in the supernatant and was not integrally inserted into the membrane (Figure 5B). As no cytosolic binding partners are present in these in vitro experiments, Bcl-xL remains in the cytosol probably due to a conformation that hides its mitochondrial targeting sequence. However, Bcl-xL was efficiently imported and became integrally inserted into mitochondria when pre-incubated with Bad prior to the import experiment (Figure 5B).
We examined the dimer status of Bcl-xL in the membrane fraction early after the induction of apoptosis with and without overexpression of Bad (Figure 5C). Bcl-xL in the membrane fraction extracted with Triton X-100 was dimeric, although the exact status on membranes remains unclear as the membrane-bound Bcl-xL could have reformed dimers after extraction. However, upon overexpression of Bad, the membrane-bound Bcl-xL was not homodimeric (Figure 5C). Bad binds to Bcl-xL in the BH3-binding pocket (Petros et al, 2000) and prevents dimer formation or dissociates dimers, consistent with the reciprocal C-tail exchange model, as both the C-tail and the BH3-binding pocket appear to be involved in Bcl-xL homodimer formation. Bad binding to Bcl-xL triggers mitochondrial binding in cells (Figure 5A), induces deep membrane insertion of Bcl-xL in vitro (Figure 5B), and dissociates or inhibits Bcl-xL dimer formation (Figure 5C), linking the dimer status of Bcl-xL to mitochondrial association and bioactivity.
The C-terminal hydrophobic tail of Bcl-xL is required for binding of Bcl-xL to Bax
Although Bcl-xL and Bax do not normally bind one another in the cytosol of healthy cells, nonionic detergents such as Triton X-100 will initiate heterodimer formation (Hsu and Youle, 1997). We examined whether the C-tail of Bcl-xL was involved in this detergent-dependent process. Cos-7 cells were co-transfected with Bax and either full-length wt Bcl-xL or the C-tail truncated form of Bcl-xL lacking the last 21 amino acids (Bcl-xL ΔC). We performed IP of the cytosol from these cells in the presence of Triton X-100 with the anti-Bax antibody, and then performed Western blotting with the anti-Bax and anti-Bcl-xL antibodies. In contrast to wt Bcl-xL, Bcl-xL ΔC was not co-immunoprecipitated with Bax (Figure 6A left). To confirm this result, we performed the reciprocal IP experiment. Using the same cell lysates, we immunoprecipitated with the anti-Bcl-xL 4C3 antibody and then performed Western blotting. Again, Bax co-precipitated with wt Bcl-xL but not with Bcl-xL ΔC (Figure 6A right). We reproduced the same results using the 2H12 anti-Bcl-xL antibody for IP (data not shown). These results indicate that the C-tail of Bcl-xL is essential not only for Bcl-xL homodimer formation in the absence of detergents but also for the interaction between Bcl-xL and Bax in the presence of detergents. In order to investigate the molecular basis of Bcl-xL binding to Bax, we constructed several Bcl-xL C-terminal tail deletion mutants. Five distinct deletion mutants were constructed based on the occurrence of hydrophobic amino-acid residues (Figure 6B). We carried out the IPs with the anti-Bax antibody in the presence of Triton X-100. The removal of up to six amino acids from the C-terminus had no effect on binding, whereas the deletion of nine amino acids largely disrupted the interaction between Bax and Bcl-xL, indicating that the sequence LLG in Bcl-xL is critical for heterodimerization (Figure 6B). Fewer residues of the Bcl-xL tail are required for Bax binding than for Bcl-xL homodimer formation (Figures 6B and 3B).
Figure 6.
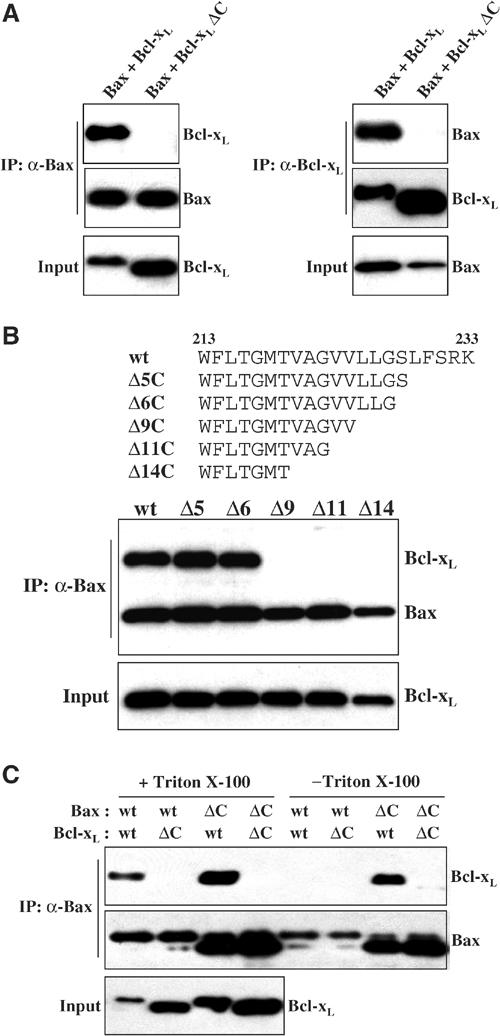
The C-terminal hydrophobic anchor of Bcl-xL is required for Bcl-xL/Bax heterodimerization. (A) Bax was co-transfected with wt Bcl-xL or the C-tail truncated Bcl-xL (Bcl-xL ΔC) into Cos-7 cells. Bax (left) and Bcl-xL (right) were immunoprecipitated from the cytosol fraction (S-100) of cells by anti-Bax 1F6 and anti-Bcl-xL 4C3 antibodies, respectively, in the presence of Triton X-100. (B) Co-IP of Bax and Bcl-xL C-tail deletion mutants shown above. Cos-7 cells were co-transfected with Bax and each Bcl-xL deletion mutant. Bax was immunoprecipitated from the S-100 by anti-Bax 1F6 antibody in the presence of Triton X-100. The immunoprecipitated samples were analyzed by Western blotting with either anti-Bax 2D2 or anti-Bcl-xL 2H12 antibodies. (C) Detergent-dependent Bax/Bcl-xL heterodimerization. Full-length Bax (wt Bax) or C-tail truncated Bax (Bax ΔC) was co-transfected with wt Bcl-xL or the C-tail truncated (Bcl-xL ΔC) in Cos-7 cells. Bax was immunoprecipitated from the S-100 by anti-Bax 1F6 antibody either in the presence or in the absence of Triton X-100. The immunoprecipitated samples were analyzed by Western blotting with anti-Bax 2D2 and anti-Bcl-xL 2H12 antibodies.
Exposure of the hydrophobic BH3-binding pocket in Bax activates Bax/Bcl-xL heterodimerization
The C-tail of Bax is homologous in sequence to that of Bcl-xL and fits into a hydrophobic pocket in Bax, which is comparable in structure to the hydrophobic, BH3-binding pocket in Bcl-xL. To test whether the Bcl-xL C-tail was binding to the corresponding pocket in Bax, we deleted the C-tail of Bax to empty the pocket of the Bax tail. Cos-7 cells were co-transfected with wt Bax or Bax ΔC lacking the C-terminal 23 amino acids and wt or truncated Bcl-xL, separately, and the cytosol fractions were immunoprecipitated using an anti-Bax antibody in both the presence and absence of the nonionic detergent Triton X-100. As revealed by Western blotting, wt Bax was co-immunoprecipitated with wt Bcl-xL only in the presence of detergent (Figure 6C) as reported previously (Hsu and Youle, 1997). We also examined the interaction between the C-tail truncated form of Bax and Bcl-xL. Surprisingly, Bax ΔC was co-immunoprecipitated with wt Bcl-xL even in the absence of detergents (Figure 6C). The degree of binding was only slightly increased in the presence of Triton X-100. However, no binding was observed between Bax ΔC and Bcl-xL ΔC. We also found that wt Bcl-xL but not Bcl-xL ΔC would coimmunoprecipitate with truncated Bak (data not shown). These results confirm that the Bcl-xL C-terminal hydrophobic tail, but not that of Bax, is required for the binding of Bcl-xL to Bax. These results also indicate that the stimulation of dimer formation by detergent occurs by facilitating or allowing the release of the Bax C-tail from the hydrophobic pocket.
Discussion
Bcl-xL exists partially in the cytosol and partially attached to the mitochondria, and translocates to the mitochondria during apoptosis in primary cells and in several cell lines (Hsu et al, 1997; Hausmann et al, 2000; Nijhawan et al, 2003), although one cell line has been reported to have constitutively mitochondrial-associated Bcl-xL (Kaufmann et al, 2003). Bcl-w (O'Reilly et al, 2001; Wilson-Annan et al, 2003) and Mcl-1 (Nijhawan et al, 2003) also exist partially in the cytosol and translocate to mitochondria during cell death. Interestingly, Bcl-w binding to Bim (Wilson-Annan et al, 2003) and Bcl-xL binding to Bad (Figure 5) trigger the mitochondrial translocation step. The Bcl-w structure, like that of Bax, reveals that the C-terminal hydrophobic tail of the protein is located within its own hydrophobic BH3-binding pocket. The binding of the BH3 domain of Bim into this pocket would thereby displace the C-tail from the pocket, allowing its insertion into the mitochondrial membrane. By analogy, the BH3 domain of Bad appears to displace the C-terminal domain of Bcl-xL to initiate Bcl-xL insertion into the membrane.
In contrast to Bax (Hsu and Youle, 1998) and Bcl-w (Wilson-Annan et al, 2003) that were found to be monomeric, soluble proteins, we find that Bcl-xL can exist in the cytosol as a homodimer. The mechanism of dimer formation may occur through the exchange of the C-terminal membrane anchor domains bound into the BH3-binding pockets of the reciprocal dimer partner. The C-terminal four amino-acid residues appear to play a crucial role in this docking process, especially the final two charged residues. Evidence that the tails fit into the reciprocal BH3-binding pocket include: (1) the Bax (Suzuki et al, 2000) and Bcl-w (Denisov et al, 2003; Hinds et al, 2003) C-tails occupy this pocket in the monomer form, (2) the Bcl-xL tails are required for dimer formation, (3) Bad binding to Bcl-xL inhibits dimer formation and at least one mutation in the BH3-binding pocket, G138A, prevents dimer formation, (4) the longer loop in Bcl-xL that could allow for reciprocal C-tail exchange is required for dimer formation and (5) Bcl-xL binds to Bax in the presence of detergents via the same C-tail, suggesting the capacity of the tail to cross over and engage other Bcl-2 family members. This engagement appears to require the same pocket in Bax normally occupied by the Bax C-tail, because elimination of the Bax tail allows Bcl-xL tail-mediated binding to Bax even in the absence of detergents. Together, these results all suggest the reciprocal C-tail exchange model of homodimer formation. In addition to the reciprocal tail exchange model, dimers or even higher molecular weight oligomers of Bcl-xL could be formed by a head-to-tail chain formation, with the C-tail linking to the BH3-binding pockets of the other protein. However, our results cannot rule out other models where the C-tail is important for the other domain or additional domain exchanges between Bcl-xL monomers.
Exchange of subunits or helices is found in other dimeric proteins. Ribonuclease A is a monomer, whereas the close homologue, bovine seminal ribonuclease, is a homodimer with the N-terminal helixes exchanged to form two catalytic active sites (D'Alessio, 1999). Diphtheria toxin has two forms where a monomeric form can exchange subunits to form homodimers (Bennett et al, 1994).
Mutagenesis analysis reveals a correlation between the capability of homodimer formation and the antiapoptotic activity of Bcl-xL. Even though the correlation between the capability of homodimer formation and import competence is more complex as the C-tail is directly involved in both processes (Figure 3), all the mutants that were able to insert into the mitochondrial membrane were found to homodimerize in the cytosol of healthy cells. The sequestration of the hydrophobic part of the protein might be crucial to prevent aggregation of the protein in living cells, keeping protein import competent at all times. Upon apoptosis, the tails presumably disengage, likely due to competition for the BH3-binding pocket with BH3-only proteins, and insert into mitochondrial membranes. Thus far, all the Bcl-xL structures available were determined with C-terminal truncated versions of Bcl-xL, leaving the BH3-binding pocket empty (Sattler et al, 1997; Petros et al, 2000; Liu et al, 2003). Interestingly, it seems that BH3 domains of various BH3-only proteins, namely Bak, Bad and Bim, interact differently with the BH3-binding pocket of Bcl-xL (Liu et al, 2003).
The recent structure of the Bim/Bcl-xL complex shows that a long helix of Bim fits into the BH3-binding pocket of C-terminally truncated Bcl-xL (Liu et al, 2003). The hydrophobic residues of the Bim helix fit across one side of the pocket adjacent to the Bcl-xL hydrophobic residues in the pocket. As the C-terminal domain of Bcl-xL is homologous to that of Bax, it is plausible that it forms a helix that, like those of Bax and Bcl-w, could fit into this pocket. What we propose is that the C-tail of Bcl-xL may also fit across into the pocket of another Bcl-xL protein or Bax. Interestingly, Liu et al (2003) also report that Bcl-xL binds full-length Bax in the absence of detergents upon deletion of the C-terminal helices 8 and 9 of Bax. This is similar to but distinct from our results. We found binding of Bcl-xL to Bax in the absence of detergents upon the deletion of helix 9 without deletion of helix 8. The use of His tags or baculovirus/insect cell expression of Bcl-xL and Bax by Liu et al (2003) may explain this discrepancy. However, the conclusion that Bcl-xL binding to Bax occurs through the Bax hydrophobic pocket remains the same. The additional finding of ours is that this interaction between Bcl-xL and Bax requires the Bcl-xL C-tail. Our results are also compatible with the finding that the BH3 domain of Bax is required for heterodimer formation (Wang et al, 1998) as this domain lines the hydrophobic pocket in Bax and the mutations in the BH3 domain of Bax that prevent binding to Bcl-xL could also disrupt the pocket in Bax, so it cannot receive the C-tail of Bcl-xL. More difficult to reconcile with our results are those of Minn et al (1999). We have confirmed the results in that paper that a mutation in the Bcl-xL pocket, Y101K, prevents Bcl-xL binding to Bax but not Bcl-xL bioactivity. This construct contains the Bcl-xL C-terminal tail and could engage Bax according to our model. One possible explanation for the failure of this mutant to bind Bax is that the mutation could allow tighter binding of the C-tail of Bcl-xL into the Bcl-xL pocket, similar to the localization of the Bax hydrophobic tail in the homologous pocket (Suzuki et al, 2000), and this sequestration of the C-tail could prevent the C-tail from engaging the Bax pocket. Alternately, as we find that more than the C-tail is required for Bcl-xL to bind Bax because the Bcl-xL tail fused to GFP does not bind Bax (data not shown), the Y101K mutation may disrupt this additional interaction.
The mechanism of sequestration of the hydrophobic C-tail into a BH3-binding pocket to allow the proteins to remain cytosolic appears to be conserved within the Bcl-2 family, as exemplified by Bax and Bcl-w previously and by Bcl-xL in this study. However, Bcl-xL is the only member of the family as of yet found to form homodimers in healthy cells. Analysis of chimeras reveals that the length of C-terminal flexible loop in Bcl-xL preceding the C-tail appears to play a crucial role in this homodimerization, as well as the C-tail itself. The dimeric nature may explain the especially potent antiapoptotic activity of Bcl-xL. Upon apoptosis, a BH3-only protein may disrupt a Bcl-xL homodimer by binding into one of the BH3-binding pockets, releasing two Bcl-xL proteins for mitochondrial import, thereby creating a two-to-one ratio of the antiapoptotic Bcl-xL to the BH3-only protein at the mitochondrial membrane.
Bax and Bak appear to be the final mediators of the apoptotic signal within the Bcl-2 family, based on the powerful loss of apoptosis function seen in the double knockout mice (Wei et al, 2001). Three different models have been proposed recently to account for the role of Bcl-2 family member heterodimer formation in the regulation of Bax and Bak. One model is that certain BH3-only proteins can directly activate Bax and Bak by heterodimer formation. Bcl-2/Bcl-xL/Bcl-w are proposed to sequester these proteins to prevent apoptosis (Cheng et al, 2001; Rathmell and Thompson, 2002). Alternatively or additionally, a subset of BH3-only proteins could activate apoptosis by displacing other BH3-only proteins from Bcl-2/Bcl-xL/Bcl-w that in turn activate Bax and Bak (Letai et al, 2002). A third recent model proposes that, following BH3-only protein activation of Bak, Bak can cascade into self-induced oligomerization. Bcl-2 is proposed to block this by binding specifically to the activated conformation of Bak and halt further oligomerization steps (Ruffolo and Shore, 2003). Our finding reported here, that the C-tail of Bcl-xL not only mediates the membrane docking step but also can mediate both homo- and heterodimer formation, adds greatly to the complexity of potential models that could be considered for how Bcl-2 family member dimer formation regulates cell death.
Materials and methods
Cell culture and transfection
Cos-7 green monkey renal epithelial cells and HeLa cells (American Type Culture Collection, Rockville, MD, USA) were grown in Dulbecco's modified Eagle's complete medium (DMEM) supplemented with 10% heat-treated fetal calf serum (FCS), 2 mM glutamine, non-essential amino acids, 2.5 mM sodium pyruvate, 100 U/ml penicillin and 100 μg/ml streptomycin. Jurkat cells (clone E6-1) were obtained from the American Type Culture Collection (Manassas, VA, USA) and were cultured in RPMI 1640 medium supplemented with 10% FCS, 2 mM L-glutamine and 50 μM 2-ME. Cells were cultured in T75 flasks at 37°C in 5% CO2. HeLa and Cos-7 cells were transfected using the FuGENE 6 (Roche) and Jurkat cells were transfected by electroporation (200 V, 1600 μF).
Construction of Bcl-xL mutants and chimeras
Various human Bcl-xL C-tail mutants and chimeras were constructed using PCR with P_fu turbo_ polymerase (Stratagene). Bcl-xL point mutants, Y101K and G138A, were constructed using QuikChange Site-Directed Mutagenesis Kit (Stratagene). PCR products were cloned into pCMV-Tag 3B (Stratagene), pcDNA 3.1 (Invitrogen) and pEYFP-C1 (Clontech). Chimeras were constructed by ligation of two PCR fragments. For construction of chimera 1, fragment A (encoding N-terminal 195 amino acids of Bcl-xL) was amplified with primer #1 (5′-TCGAGATCTACCATGTCTCAGAGCAACCGGGAGCT-3′) and primer #2 (5′-CATGAGCTCCACAAAAGTATCCCAGCCG-3′), and fragment B (encoding 27 amino acids of Bcl-w C-loop and Bcl-xL C-tail) was amplified with primer #3 (5′-ATGAGCTCTACGGGGACGGGGCCCTGTGGTTCCTGACGGG CATGACTGTG-3′) and primer #4 (5′-CTGGAATTCATTTCCGACTGAAGAGTGAGCCCAG), using human Bcl-xL cDNA as templates. Fragment A and fragment B were digested with _Bgl_II and _Sac_I and with _Sac_I and _Eco_RI, respectively, and then ligated into _Bgl_II and _Eco_RI sites of vectors. For construction of chimera 2, fragment C (encoding N-terminal 213 amino acids of Bcl-xL) was amplified with primer #1 and primer #5 (5′-ACCATCGATTGAAGCGTTCCTGGCCCT3′) using human Bcl-xL cDNA as a template, and fragment D (encoding 37 amino acid of Bcl-w C-tail) was amplified with primer #6 (5′-ATATCGAGAGGAGGCGCGGCGTCTGC) and primer #7 (5′-CATGAATTCACTTGCTAGCAAAAAAGGCCCCTACAG-3′) using human Bcl-w cDNA as a template. Fragments C and D were digested with _Bgl_II and _Taq_I and with _Taq_I and _Eco_RI, respectively, and then ligated into _Bgl_II and _Eco_RI sites of vectors.
Subcellular fractionation and gel filtration
Subcellular fractionation of HeLa cells was performed as described previously (Yang et al, 1997). Briefly, cells were harvested by centrifugation at 750 g for 10 min at 4°C. The cell pellets were washed with ice-cold PBS and resuspended with five volumes of cell lysis buffer (20 mM Hepes-KOH, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol and 0.1 mM phenylmethylsulfonyl fluoride (PMSF)) containing 250 mM sucrose. The cells were homogenized with 50 strokes using a Teflon homogenizer, and the homogenates were centrifuged at 750 g for 10 min at 4°C. The supernatants were centrifuged at 10 000 g for 15 min at 4°C, and the resulting mitochondria pellets were subjected to further analysis. The supernatant was re-centrifuged at 100 000 g for 1 h at 4°C and the resulting supernatant (S-100) containing the cytosol (1 mg/200 μl) was loaded on a 25 ml Superose 6 column (Amersham-Biosciences) and 0.5 ml fractions were collected. The proteins of each fractions were precipitated with 12% (w/v) TCA.
Immunoprecipitation and Western blotting
Transiently transfected Cos-7 cells or HeLa cells were lysed in cell lysis buffer (10 mM HEPES, pH 7.4, 150 mM NaCl) supplemented with protease inhibitors (25 μg/ml PMSF, 2 μg/ml leupeptin and 5 μg/ml aprotinin). The supernatant cytosolic fraction (S-100) was prepared as described above and used for IP unless otherwise indicated. Monoclonal anti-myc antibody 9B11 (Cell Signaling) was used for IP of myc-Bcl-xL according to the manufacturer's instructions. Briefly, the S-100 was mixed with 9B11 antibody (1:1000 dilution) in IP buffer (10 mM HEPES, pH 7.4, 150 mM NaCl and 0.1% BSA supplemented with protease inhibitors) and incubated overnight at 4°C with gentle rocking. Protein A Sepharose beads were added and allowed to incubate for 1 h. The unbound proteins were removed by washing the beads three times in IP buffer. The bound proteins were then eluted from the beads with SDS–PAGE sample buffer. Monoclonal anti-Bax 1F6 and anti-Bcl-xL 4C3 antibodies immobilized onto CNBr-activated Sepharose 4B beads were used for IP of Bax and Bcl-xL. The S-100 was mixed with antibody beads either in the absence or in the presence of 1.0% Triton X-100 and allowed to incubate for 1 h.
The immunoprecipitated samples were analyzed by Western blotting. SDS–PAGE was carried out on 10–20% polyacrylamide gradient gels. The gels were electroblotted onto Immobilon-P membranes (Milipore). Antibodies used for immunoblotting analysis were: anti-myc antibody (Clontech, 1:100 dilution), anti-Bcl-xL 2H12 antibody (1:10 diluted culture fluid), anti-human Bcl-xL 4C3 antibody (1:10 diluted culture fluid), anti-mouse Bcl-xL 3D3 antibody (1:10 diluted culture fluid), anti-Bax 2D2 antibody (1:10 diluted culture fluid), anti-Bcl-xL L19 antibody (Santa Cruz, 1:2000 dilution) and anti-Bad antibody (Cell Signaling, 1:1000 dilution).
Assessment of apoptosis by Annexin V binding
Jurkat cells were co-transfected with GFP-Bcl-xL constructs and Bax. At 12 h after transfection, Jurkat cells were treated with 100 μM etoposide (Sigma) for 6 h, stained with biotin-conjugated Annexin V, followed by streptavidin-Cy-chrome (PharMingen), and analyzed by flow cytometry (FACSCaliber) using CellQuest software (Becton Dickinson, San Jose, CA, USA). Transfectants (GFP positive cells) were first gated (FL1 positive cells) and then Annexin V binding among them was analyzed by FL3 (emission of Cy-chrome: 670 nm).
In vitro mitochondrial import
Mitochondria (1 mg/ml protein) were isolated from either healthy HeLa cells or from early apoptotic HeLa cells incubated for 4 h with 1.2 μM staurosporine and incubated in import buffer (220 mM mannitol, 70 mM sucrose, 10 mM Hepes/KOH, pH 7.2, 1 mM MgCl2, 1 mM EDTA, 1 mM dithiothreitol) containing 1 mM ATP, 0.1 mM GTP and 5 mM NADH. Import reactions were started by addition of 35S-labeled protein synthesized by in vitro transcription and translation in rabbit reticulocyte lysate (TNT Coupled system, Promega). Alternatively, for co-import experiments, the 35S-labeled proteins were pre-incubated in import buffer and the import reactions were started by addition of isolated mitochondria. After incubation, the samples were split as described. The mitochondria of one aliquot were recovered by centrifugation (10 min, 10 000 g, 4°C) and resuspended in sample buffer, while the proteins of the resulting supernatant were precipitated with 12% (w/v) TCA. For carbonate extraction, the recovered mitochondria of another aliquot were resuspended in a 0.1 M Na2CO3 buffer as described previously (Fujiki et al, 1982), incubated on ice for 30 min and the pellet was recovered by a 30 min centrifugation step at 100 000 g in a Beckman TLA 120.2 rotor.
Confocal microscopy
Images were captured with a confocal microscope (LSM 510, Carl Zeiss). The excitation wavelengths for YFP and Mitotracker Red CMXRos (Molecular Probes, Inc.) were 514 and 543 nm, respectively.
Supplementary Material
Supplemental Figure 1
Supplementary Material
Acknowledgments
We thank Dr Motoshi Suzuki (Biochemistry Section, NINDS) and Dr Juanita C Sharpe (Biochemistry Section, NINDS) for expert technical assistance, and Dr David Huang (The WEHI Institute, Melbourne, Australia) for kindly providing the Bcl-w clone. We also thank Dr Carolyn L Smith (Light Imaging Facility, NINDS) for confocal microscopy, and Drs James W Nagle and Deborah Kauffman (NINDS DNA sequencing facility) for DNA sequencing.
References
- Adams JM, Cory S (1998) The Bcl-2 protein family: arbiters of cell survival. Science 281: 1322–1326 [DOI] [PubMed] [Google Scholar]
- Antonsson B, Montessuit S, Sanchez B, Martinou JC (2001) Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J Biol Chem 276: 11615–11623 [DOI] [PubMed] [Google Scholar]
- Bennett MJ, Choe S, Eisenberg D (1994) Domain swapping: entangling alliances between proteins. Proc Natl Acad Sci USA 91: 3127–3131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ (2001) BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell 8: 705–711 [DOI] [PubMed] [Google Scholar]
- Cory S, Adams JM (2002) The Bcl-2 family: regulator of the cellular life-or-death switch. Nat Rev Cancer 2: 647–656 [DOI] [PubMed] [Google Scholar]
- D'Alessio G (1999) Evolution of oligomeric proteins. The unusual case of a dimeric ribonuclease. Eur J Biochem 266: 699–708 [DOI] [PubMed] [Google Scholar]
- Denisov AY, Madiraju MS, Chen G, Khadir A, Beauparlant P, Attardo G, Shore GC, Gehring K (2003) Solution structure of human BCL-w: modulation of ligand binding by the C-terminal helix. J Biol Chem 278: 21124–21128 [DOI] [PubMed] [Google Scholar]
- Fujiki Y, Hubbard AL, Fowler S, Lazarow PB (1982) Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J Cell Biol 93: 97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausmann G, O'Reilly LA, van Driel R, Beaumont JG, Strasser A, Adams JM, Huang DC (2000) Pro-apoptotic apoptosis protease-activating factor 1 (Apaf-1) has a cytoplasmic localization distinct from Bcl-2 or Bcl-x(L). J Cell Biol 149: 623–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinds MG, Lackmann M, Skea GL, Harrison PJ, Huang DC, Day CL (2003) The structure of Bcl-w reveals a role for the C-terminal residues in modulating biological activity. EMBO J 22: 1497–1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YT, Hou Q, Cymbalyuk E, Youle RJ (2003) Generation and characterization of species-specific anti-Bcl-X(L) monoclonal antibodies. Hybrid Hybridomics 22: 91–95 [DOI] [PubMed] [Google Scholar]
- Hsu YT, Wolter KG, Youle RJ (1997) Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc Natl Acad Sci USA 94: 3668–3672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YT, Youle RJ (1997) Nonionic detergents induce dimerization among members of the Bcl-2 family. J Biol Chem 272: 13829–13834 [DOI] [PubMed] [Google Scholar]
- Hsu YT, Youle RJ (1998) Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. J Biol Chem 273: 10777–10783 [DOI] [PubMed] [Google Scholar]
- Kaufmann T, Schlipf S, Sanz J, Neubert K, Stein R, Borner C (2003) Characterization of the signal that directs Bcl-x(L), but not Bcl-2, to the mitochondrial outer membrane. J Cell Biol 160: 53–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ (2002) Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2: 183–192 [DOI] [PubMed] [Google Scholar]
- Lindenboim L, Borner C, Stein R (2001) Bcl-x(S) can form homodimers and heterodimers and its apoptotic activity requires localization of Bcl-x(S) to the mitochondria and its BH3 and loop domains. Cell Death Differ 8: 933–942 [DOI] [PubMed] [Google Scholar]
- Liu X, Dai S, Zhu Y, Marrack P, Kappler JW (2003) The Structure of a Bcl-x(L)/Bim fragment complex. Implications for Bim function. Immunity 19: 341–352 [DOI] [PubMed] [Google Scholar]
- Minn AJ, Kettlun CS, Liang H, Kelekar A, Vander Heiden MG, Chang BS, Fesik SW, Fill M, Thompson CB (1999) Bcl-xL regulates apoptosis by heterodimerization-dependent and -independent mechanisms. EMBO J 18: 632–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, Wang X (2003) Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev 17: 1475–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly LA, Print C, Hausmann G, Moriishi K, Cory S, Huang DC, Strasser A (2001) Tissue expression and subcellular localization of the pro-survival molecule Bcl-w. Cell Death Differ 8: 486–494 [DOI] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, Korsmeyer SJ (1993) Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 74: 609–619 [DOI] [PubMed] [Google Scholar]
- Petros AM, Nettesheim DG, Wang Y, Olejniczak ET, Meadows RP, Mack J, Swift K, Matayoshi ED, Zhang H, Thompson CB, Fesik SW (2000) Rationale for Bcl-xL/Bad peptide complex formation from structure, mutagenesis, and biophysical studies. Protein Sci 9: 2528–2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathmell JC, Thompson CB (2002) Pathways of apoptosis in lymphocyte development, homeostasis, and disease. Cell 109 (Suppl): S97–S107 [DOI] [PubMed] [Google Scholar]
- Ruffolo SC, Shore GC (2003) BCL-2 selectively interacts with the BID-induced open conformer of BAK, inhibiting BAK auto-oligomerization. J Biol Chem 278: 25039–25045 [DOI] [PubMed] [Google Scholar]
- Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, Thompson CB, Fesik SW (1997) Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science 275: 983–986 [DOI] [PubMed] [Google Scholar]
- Sedlak TW, Oltvai ZN, Yang E, Wang K, Boise LH, Thompson CB, Korsmeyer SJ (1995) Multiple Bcl-2 family members demonstrate selective dimerizations with Bax. Proc Natl Acad Sci USA 92: 7834–7838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Youle RJ, Tjandra N (2000) Structure of Bax: coregulation of dimer formation and intracellular localization. Cell 103: 645–654 [DOI] [PubMed] [Google Scholar]
- Tan YJ, Beerheide W, Ting AE (1999) Biophysical characterization of the oligomeric state of Bax and its complex formation with Bcl-xL. Biochem Biophys Res Commun 255: 334–339 [DOI] [PubMed] [Google Scholar]
- Wang K, Gross A, Waksman G, Korsmeyer SJ (1998) Mutagenesis of the BH3 domain of BAX identifies residues critical for dimerization and killing. Mol Cell Biol 18: 6083–6089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson-Annan J, O'Reilly LA, Crawford SA, Hausmann G, Beaumont JG, Parma LP, Chen L, Lackmann M, Lithgow T, Hinds MG, Day CL, Adams JM, Huang DC (2003) Proapoptotic BH3-only proteins trigger membrane integration of prosurvival Bcl-w and neutralize its activity. J Cell Biol 162: 877–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X (1997) Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 275: 1129–1132 [DOI] [PubMed] [Google Scholar]
- Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ (1996) Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not Bcl-X(L). Cell 87: 589–592 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1
Supplementary Material