Effect of high-fat and high-carbohydrate diets on pulmonary O2 uptake kinetics during the transition to moderate-intensity exercise (original) (raw)
Abstract
Mitochondrial pyruvate dehydrogenase (PDH) regulates the delivery of carbohydrate-derived substrate to the mitochondrial tricarboxylic acid cycle and electron transport chain. PDH activity at rest and its activation during exercise is attenuated following high-fat (HFAT) compared with high-carbohydrate (HCHO) diets. Given the reliance on carbohydrate-derived substrate early in transitions to exercise, this study examined the effects of HFAT and HCHO on phase II pulmonary O2 uptake (V̇o2p) kinetics during transitions into the moderate-intensity (MOD) exercise domain. Eight active adult men underwent dietary manipulations consisting of 6 days of HFAT (73% fat, 22% protein, 5% carbohydrate) followed immediately by 6 days of HCHO (10% fat, 10% protein, 80% carbohydrate); each dietary phase was preceded by a glycogen depletion protocol. Participants performed three MOD transitions from a 20 W cycling baseline to work rate equivalent to 80% of estimated lactate threshold on days 5 and 6 of each diet. Steady-state V̇o2p was greater (P < 0.05), and respiratory exchange ratio and carbohydrate oxidation rates were lower (P < 0.05) during HFAT. The phase II V̇o2p time constant (τV̇o2p) [HFAT 40 ± 16, HCHO 32 ± 19 s (mean ± SD)] and V̇o2p gain (HFAT 10.3 ± 0.8, HCHO 9.4 ± 0.7 ml·min−1·W−1) were greater (P < 0.05) in HFAT. The overall adjustment (effective time constant) of muscle deoxygenation (Δ[HHb]) was not different between diets (HFAT 24 ± 4 s, HCHO 23 ± 4 s), which coupled with a slower τV̇o2p, indicates a slowed microvascular blood flow response. These results suggest that the slower V̇o2p kinetics associated with HFAT are consistent with inhibition and slower activation of PDH, a lower rate of pyruvate production, and/or attenuated microvascular blood flow and O2 delivery.
Keywords: O2 uptake kinetics, high-fat diet, high-carbohydrate diet, muscle deoxygenation, near-infrared spectroscopy, substrate oxidation
during step-transitions in work rate (WR) initiated either from rest or light-intensity exercise, to heavier exercise intensities requiring a greater rate of ATP turnover, pulmonary O2 uptake (V̇o2p), and muscle O2 utilization (V̇o2m) increase with a finite time course toward a new, higher steady-state level (14, 34). The delay in attaining a steady state for V̇o2p and V̇o2m (and mitochondrial oxidative phosphorylation) is considered to reflect a metabolic capacitance (represented by total creatine) and resistance, which are inherent to the energy-producing systems (mitochondria) (24, 12). More specifically, this delay has been attributed to a sluggish activation of rate-limiting enzymes and metabolic pathways responsible for providing oxidizable substrates (other than O2) to the mitochondrial tricarboxylic acid (TCA) cycle and electron transport chain (ETC) to a slow adjustment of microvascular blood flow and O2 delivery to the exercising muscle, or to a combination of both factors [for reviews see (31, 33, 45)]. The consequence of a delay in meeting the increased ATP demands at the onset of exercise through oxidative phosphorylation is that there is greater reliance on substrate-level phosphorylation with a consequent fall in metabolic stability (15) within the active muscle as evidenced by an intensity-dependent increase in O2 deficit and breakdown of phosphocreatine (PCr) and glycogen; accumulation of lactate−, hydrogen ions (H+), ADPfree, AMPfree, free inosine 5′-monophosphate (IMPfree), and inorganic phosphate (Pi2−); and fall in the Gibbs free energy release associated with ATP hydrolysis.
The mitochondrial pyruvate dehydrogenase (PDH) complex catalyzes the rate-limiting, irreversible oxidative decarboxylation of pyruvate to acetyl CoA and therefore, regulates the entry of carbohydrate-derived substrate into the mitochondria for provision of acetyl CoA and reducing equivalents (NADH; FADH2) to the TCA cycle and ETC, respectively (37). The activity of PDH is determined in part by covalent phosphorylation/dephosphorylation, and thus by the activities of the associated PDH kinase (PDK; phosphorylation/inhibition) and PDH phosphatase (dephosphorylation/activation) enzymes (37). For example, when exercise was initiated from a baseline of elevated PDH activity either by means of dichloroacetate (DCA; an inhibitor of PDK) infusion or subsequent to a priming bout of heavy-intensity exercise, the reliance on substrate-level phosphorylation was reduced (16, 44) and, in the case of priming exercise, the adjustment of V̇o2p was faster (16). Alternatively, when the activation of PDH was attenuated [via hyperventilation-induced hypocapnic alkalosis (22)], physiological and metabolic responses consistent with a slower activation of oxidative phosphorylation were observed (6, 22).
It is established that PDH activation and whole-body rates of carbohydrate and fat oxidation during exercise are dependent on the availability of carbohydrate and fat within the muscle [for review see (38)]. Previous studies examining increased lipid (or decreased carbohydrate) availability following high-fat (low-carbohydrate) diets or intralipid infusions alone or in combination with prior glycogen depletion (GD) have shown that PDH kinase was upregulated and steady-state PDH activity was downregulated (30, 32, 46), whereas increased carbohydrate availability (or reduced fatty acid availability) resulted in greater PDH activation (48).
Additionally, a high-fat meal was found to impair flow-mediated dilation (47), a consequence of impaired endothelial function (1). Given the importance of the vascular endothelium in blood flow regulation (7), dietary-induced endothelial dysfunction may limit the adjustment of microvascular blood flow and O2 delivery at the onset of exercise, and thus possibly impact the activation of mitochondrial oxidative phosphorylation. Muscle deoxygenation as measured using near-infrared spectroscopy (NIRS) reflects the dynamic relationship between local muscle O2 delivery and muscle O2 utilization (9), and when combined with measures of V̇o2p kinetics (reflecting muscle O2 utilization), can provide information on the dynamics of microvascular blood flow during transitions to exercise.
Therefore, given the importance of carbohydrates in energy production early in exercise and the possible role of PDH in limiting the provision of carbohydrate-derived substrate and thus the activation of mitochondrial oxidative phosphorylation, the primary purpose of this study was to examine the effects of a high fat/low carbohydrate (HFAT) and high carbohydrate/low fat (HCHO) dietary intervention on the adjustment of phase II V̇o2p (reflecting muscle O2 utilization) during the transition to exercise. In addition, given the possible effects of diet on blood flow regulation, a secondary purpose was to examine the effects of diet on muscle deoxygenation and thus the dynamic relationship between local microvascular blood flow (and O2 delivery) and muscle O2 utilization. It was hypothesized that 1) phase II V̇o2p kinetics would be slower during HFAT compared with HCHO, reflecting a slower activation of mitochondrial PDH and thus oxidative phosphorylation, and 2) muscle deoxygenation kinetics would be faster during HFAT, reflecting attenuated muscle microvascular blood flow and O2 delivery dynamics, and an attenuated muscle microvascular blood flow-to-muscle O2 utilization relationship, which also may contribute to slowed V̇o2p during transitions to exercise.
METHODS
Subjects.
Eight male subjects volunteered for this study. All subjects were healthy nonsmokers and had no known respiratory, cardiovascular, or metabolic disease. The protocol and procedures associated with this study were approved by The University of Western Ontario Research Ethics Board for Health Sciences Research Involving Human Subjects, in accordance with the Declaration of Helsinki.
Study design.
During the initial visit to the laboratory subjects performed a ramp incremental (RI) test (25 W/min) to volitional fatigue on a cycle ergometer (H-300-R; Lode, Groningen, Netherlands) for determination of V̇o2max and estimated lactate threshold (θ̂l). Subjects were instructed to keep a 2-day dietary record of exact amounts of food and liquids consumed. The dietary records were analyzed for nutrient and energy intake using a commercially available computer software program (Diet Analysis Plus 8.0.1; Thomson-Nelson, Scarborough, ON, Canada), and individualized isocaloric diets (relative to the normal mixed diet) were designed and given to each subject. The dietary intervention protocols were modified on the basis of previous studies [e.g., (29, 32)] for the HFAT (∼75% fat, ∼20% protein, ∼5% carbohydrate) and HCHO (∼10% fat, ∼10% protein, ∼80% carbohydrate) conditions.
The dietary intervention protocol began with the HFAT diet and was followed immediately by the HCHO diet with each dietary phase lasting 6 days. Before the start of each diet, subjects performed a GD protocol that consisted of 60 min of constant-load leg cycling exercise at a WR corresponding to ∼70% V̇o2max. This was followed by five repeated bouts of cycling at a WR corresponding to ∼110% of the peak WR achieved during the RI test with each bout lasting 1 min and separated by 4 min of cycling at a light intensity WR (=20 W). Variations of this protocol have been shown previously to be effective in lowering muscle glycogen content by approximately 55–90% (5, 32). This GD protocol was included to amplify the shift in fuel selection for oxidation by maximally decreasing carbohydrate availability for oxidation during HFAT (thereby increasing fat oxidation) and maximally increasing carbohydrate availability during HCHO through glycogen supercompensation (thereby increasing carbohydrate oxidation).
Moderate-intensity exercise testing.
On days 5 and 6 of each of the HFAT and HCHO diets, subjects reported to the laboratory having been advised to eat their final meal at least 2 h prior to arriving at the laboratory, and to not engage in strenuous physical activity or consume alcohol or caffeinated beverages during the previous 24-h period. Subjects then performed a series of constant-load, step exercise transitions on an electronically braked cycle ergometer. Each transition began with 6 min of baseline cycling at 20 W followed by an instantaneous increase in WR to an intensity equivalent to ∼80% θ̂l lasting 8 min. Three repetitions of this protocol were repeated on each of days 5 and 6 of the HFAT and HCHO diets, with each repetition separated by ∼30 min of resting recovery to allow physiological and metabolic variables to return back toward pretransition, baseline levels.
On day 5 of each of the dietary phases a percutaneous Teflon catheter (Angiocath, 21 gauge) was inserted into a dorsal hand vein for blood sampling. The venous blood was arterialized by wrapping the hand and forearm with a heating pad, with additional heating provided by a tungsten heating lamp. Blood samples (∼5 ml) were drawn at rest, during 20 W of baseline cycling, and at specific times (1, 2, 3, 4, 6, and 8 min) during the transition to the moderate intensity (MOD) WR. One portion of whole blood (100 μl) was deproteinized 1:4 with 500 μl of 0.5 M perchloric acid (PCA), placed on ice for 5 min, and then neutralized with 125 μl of 1 M K2CO3. The neutralized sample was centrifuged for 3 min and the PCA extract was removed, placed into a labeled Eppendorf tube, and stored at −80°C for later analysis of β-hydroxybutyrate, glucose, and lactate as previously described (4). Another portion of whole blood (∼3 ml) was centrifuged for 5 min, and the supernatant transferred to a labeled Eppendorf tube and stored at −80°C for later analysis of plasma concentration of free fatty acids (FFA) (NEFA C test kit; Wako Chemicals, Richmond VA). The remaining plasma portion of the blood sample was stored at −80°C for later analysis of plasma concentrations of insulin (Coat-a-Count insulin test kit; Diagnostics Products, Los Angeles, CA).
Measurements.
Gas-exchange measurements were similar to those previously described (36). Briefly, inspired and expired flow rates were measured using a low dead space (90 ml) bidirectional turbine (VMM 110; Alpha Technologies). Inspired and expired gases were sampled continuously (every 20 ms) at the mouth and analyzed for concentrations of O2, CO2, and N2 by mass spectrometry (Amis 2000; Innovision, Odense, Denmark). Data collected every 20 ms were transferred to a computer, which aligned concentrations with volume information to build a profile of each breath. Breath-by-breath alveolar gas exchange was calculated using the algorithms outlined by Beaver et al. (2).
Heart rate (HR) was monitored continuously by electrocardiogram (ECG) using PowerLab ML132/ML880 (ADInstruments, Colorado Springs, CO) and using a three-lead ECG arrangement. Data were recorded on a separate data collection computer using LabChart v4.2 (ADInstruments).
Local muscle oxygenation and deoxygenation profiles of the vastus lateralis were made using NIRS (NIRO 300; Hamamatsu Photonics, Hamamatsu, Japan). Optodes were placed on the belly of the muscle midway between the lateral epicondyle and greater trochanter of the femur using an interoptode spacing of 5 cm. The optodes were housed in an optically dense plastic holder thus ensuring that the position of the optodes, relative to the skin, was fixed and invariant. The thigh, with the attached optodes and covering, was wrapped with an elastic bandage to minimize movement of the optodes while still permitting freedom of movement for cycling.
The physical principles of tissue spectroscopy and the manner in which these are applied were described in detail by Elwell (10). Briefly, one fiber optic bundle carried the near infrared light produced by the laser diodes to the tissue of interest while a second fiber optic bundle returned the transmitted light from the tissue to a photon detector (photomultiplier tube) in the spectrometer. The intensity of the incident and transmitted light were recorded continuously at 2 Hz and, along with the specific extinction coefficients and optical path length, used for online estimation and display of the concentration changes of oxy-(Δ[O2Hb]), deoxy-(Δ[HHb]), and total (Δ[HbTOT]) hemoglobin-myoglobin relative to the steady-state, resting baseline; subjects rested quietly for at least 5 min to ensure steady-state conditions before the NIRS unit was initialized and the signals set to zero. Because of the uncertainty of the actual path length traveled by the photons in muscle tissue and the stability of the path length during the transition to exercise, values are reported in arbitrary units (au). In this study, changes in [HHb] from a zero baseline prior to the onset of exercise are reported as a change (Δ) in au.
Data analysis.
Gas exchange data for each trial were edited by removing aberrant data points that lay outside 4 SD of the local mean. The data for each transition were linearly interpolated to 1-s intervals and time-aligned such that time zero represented the onset of exercise. Data from each transition were ensemble-averaged to yield a single, averaged response for each subject. This transition was further time-averaged into 5-s bins to provide a single time-averaged response for each subject. The on-transient response for V̇o2p was modeled using a monoexponential of the form Y(t) = YBsl + Amp (1 − e−(t − TD)/τ) (equation 1), where Y(t) represents V̇o2p at any time (t); YBsl is the baseline V̇o2p during 20 W cycling; Amp is the steady-state increase in V̇o2p above the baseline value; τ is the time constant defined as the time required for V̇o2p to increase to 63% of the steady-state increase; and TD is the time delay. Data were modeled from the beginning of phase I-phase II transition to 8 min (480 s) of exercise as previously described (34). The model parameters were estimated by least-squares nonlinear regression (Origin; OriginLab, Northampton, MA) in which the best fit was defined by minimization of the residual sum of squares and minimal variation of residuals around the _y_-axis (y = 0). The 95% confidence interval (CI95%) for the estimated time constant was determined after preliminary fit of the data with Bsl, Amp, and TD constrained to the best-fit values and the τ allowed to vary.
The rate of carbohydrate and fat oxidation were calculated for steady states of baseline and end-exercise according to the equations presented in previous studies (27). Carbohydrate oxidation rate (g/min) = 4.585 (V̇co2p) − 3.226 (V̇o2p). Fat oxidation rate (g/min) = 1.695 (V̇o2p) − 1.701 (V̇co2p).
HR data were determined from the R-R interval on a second-by-second basis, edited by removing data lying outside 4 SD of the mean response, and modeled using equation 1, as described above. The on-transient HR response was modeled from the onset of exercise to end-exercise (480 s), with the time delay fixed to the onset of the exercise transition (i.e., TD = 0).
The NIRS-derived Δ[HHb] data were time-aligned and ensemble-averaged to 5-s bins to yield a single response for each subject. The Δ[HHb] profile has been described to consist of a time delay at the onset of exercise followed by an exponential-like increase in the signal toward a new steady-state exercise value (16). The time delay for the Δ[HHb] response (TD-Δ[HHb]) was estimated for each subject using the second-by-second data (i.e., data before they were ensemble-averaged into 5-s bins) and corresponded to the time after the onset of exercise where the Δ[HHb] signal showed a consistent increase above the baseline or nadir value, as described previously (16). Determination of the TD-Δ[HHb] was made on individual trials and averaged to yield a single value for each individual. The Δ[HHb] data were modeled from the end of the TDΔ[HHb] to the end of exercise using an exponential model as described in equation 1. The τΔ[HHb] described the time course for the increase in Δ[HHb], whereas the overall change of the effective time constant for Δ[HHb] (τ′Δ[HHb] = TDΔ[HHb] + τΔ[HHb]) described the overall time course of the Δ[HHb] from the onset of exercise.
The Δ[O2Hb] and Δ[HbTOT] signals did not approximate an exponential response and thus were not modeled during the exercise transition. The Δ[O2Hb] and Δ[HbTOT] values were reported as the average responses calculated during the steady state at baseline (−30 s to 0 s) and end-exercise (450 s to 480 s).
Statistics.
Differences in dependent variables and model parameter estimates between HFAT and HCHO diets were analyzed using paired _t_-tests and repeated measures ANOVA. Statistical analyses were performed using SPSS (v. 17; SPSS, Chicago, IL). Statistical significance was accepted at P < 0.05, with significant interactions and main effects analyzed using Tukey's LSD post hoc test. All data are presented as means ± SD.
RESULTS
Subject characteristics and glycogen depletion protocol.
Subject characteristics and results of the initial RI test are presented in Table 1. Body mass was lower (P < 0.05) during the HFAT (82 ± 8 kg) diet compared with either the normal mixed (84 ± 8 kg) or HCHO (84 ± 8 kg) diets despite there being no differences in caloric intake among the diets. Analysis of the dietary composition confirmed differences (P < 0.05) between the HCHO and HFAT diets (Table 2).
Table 1.
Physical characteristics and response to initial ramp incremental test
| Age, yr | Body Mass, kg | Height, cm | V̇o2peak, liter/min | V̇o2peak, ml·kg−1·min−1 | θ̇l, liter/min | 80% θ̇l, liter/min | 80% θ̇l, W | WRmax, W |
|---|---|---|---|---|---|---|---|---|
| 24 ± 1 | 84 ± 8 | 181 ± 6 | 3.77 ± 0.37 | 45 ± 4 | 2.11 ± 0.29 | 1.69 ± 0.23 | 93 ± 20 | 334 ± 37 |
Table 2.
Dietary characteristics for 2-day normal mixed diet and 6-day averaged high-fat and high-carbohydrate diets
| Mixed Diet | HFAT Diet | HCHO Diet | |
|---|---|---|---|
| Caloric intake, kcal | 2,920 ± 472 | 2,973 ± 431 | 2,980 ± 470 |
| Carbohydrate, % | 45.6 ± 8.1 | 5.3 ± 0.5*† | 79.6 ± 0.5* |
| Fat, % | 31.0 ± 5.5 | 73.0 ± 0.0*† | 10.0 ± 0.0* |
| Protein, % | 23.3 ± 4.2 | 21.8 ± 0.5† | 9.9 ± 0.4* |
During the GD protocol, WRs corresponding to the heavy intensity, constant-load exercise bout at 70% V̇o2peak and to the severe-intensity, intermittent exercise bouts at 110% WRmax were 189 ± 24 W and 369 ± 42 W, respectively. Subjects successfully completed the entire GD protocol before the start of the HFAT diet, but were able to complete only 33 ± 2 min of the heavy intensity constant-load exercise bout prior to the start of the HCHO diet, and although they were able to complete all five of the severe-intensity interval exercise bouts, they were unable to maintain the required WR (due either to a decrease in cadence or to a decrease in imposed load) despite strong verbal encouragement.
Substrate oxidation rates and blood metabolites.
Calculated respiratory exchange ratio (RER) and fat and carbohydrate oxidation rates for steady-state baseline and the steady state of the moderate-intensity exercise transition are presented in Fig. 1. The steady-state RER was lower (P < 0.001) during the HFAT than in the HCHO diet at baseline (HFAT 0.82 ± 0.09, HCHO 0.95 ± 0.14) and end-exercise (HFAT 0.83 ± 0.02, HCHO 0.96 ± 0.04). The steady-state fat oxidation rates were higher (P < 0.05) in HFAT than in HCHO at baseline (HFAT 0.29 ± 0.14 g/min, HCHO 0.18 ± 0.08 g/min) and at end-exercise (HFAT 0.48 ± 0.09 g/min, HCHO 0.12 ± 0.06 g/min). Also, the steady-state carbohydrate oxidation rates were lower (P < 0.001) in HFAT than in HCHO at baseline (HFAT 0.51 ± 0.36 g/min, HCHO 0.95 ± 0.54 g/min) and end-exercise (HFAT 0.99 ± 0.25 g/min, HCHO 1.81 ± 0.42 g/min).
Fig. 1.
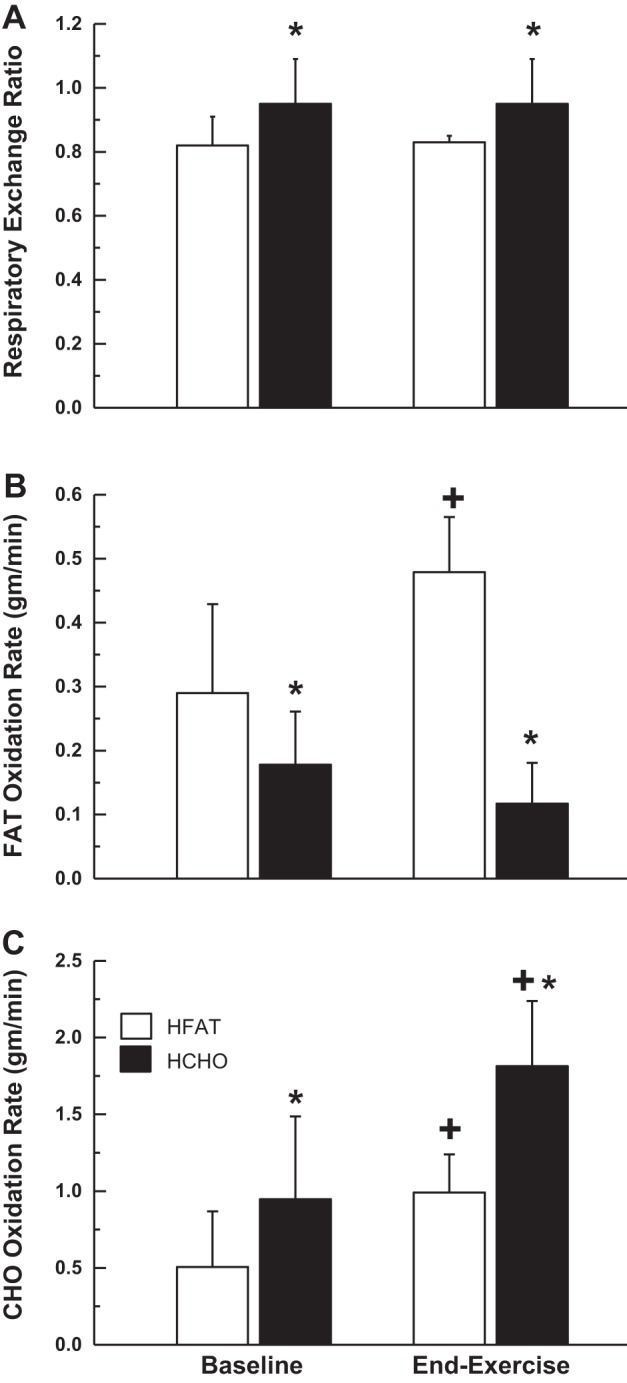
Calculated respiratory exchange ratio (RER) (A), and fat and carbohydrate oxidation rates (B and C, respectively) for steady-state baseline (20 W) cycling, and moderate-intensity exercise following the high fat (HFAT, open bars) and high carbohydrate (HCHO, solid bars) diets. Values are means ± SD. *Significant difference (P < 0.05) between the high-fat and high-carbohydrate diets; +significant difference (P < 0.05) from baseline within a given dietary condition.
Blood [glucose] was higher (P < 0.05) at rest in HCHO (5.02 ± 1.52 mmol/liter) than HFAT (2.74 ± 0.98 mmol/liter), but was similar between conditions during exercise (end-exercise HCHO 4.19 ± 0.93 mmol/liter, end-exercise HFAT 3.59 ± 0.92 mmol/liter). Blood [lactate] was higher (P < 0.05) in HCHO than in HFAT from the second minute to end-exercise. Plasma [FFA] and [β-hydroxybutyrate] were both higher (P < 0.05) in HFAT (both at rest and throughout exercise) than in HCHO. Blood [insulin] was higher (P < 0.05) at rest in HCHO (3.52 ± 1.76 μIU/ml) than in HFAT (2.39 ± 1.05 μIU/ml).
Gas exchange (V̇o2 V̇co2) kinetics.
The pulmonary V̇o2 responses during the transitions to MOD exercise are presented in Fig. 2 and the parameter estimates for V̇o2p kinetics are presented in Table 3. During MOD exercise, the end-exercise steady-state V̇o2p corresponded to 80 ± 4% (HFAT) and 72 ± 4% (HCHO) of the θ̇l estimated during the initial RI test when subjects consumed their normal mixed diet, or 45 ± 6 (HFAT) and 40 ± 6 (HCHO) % V̇o2peak.
Fig. 2.
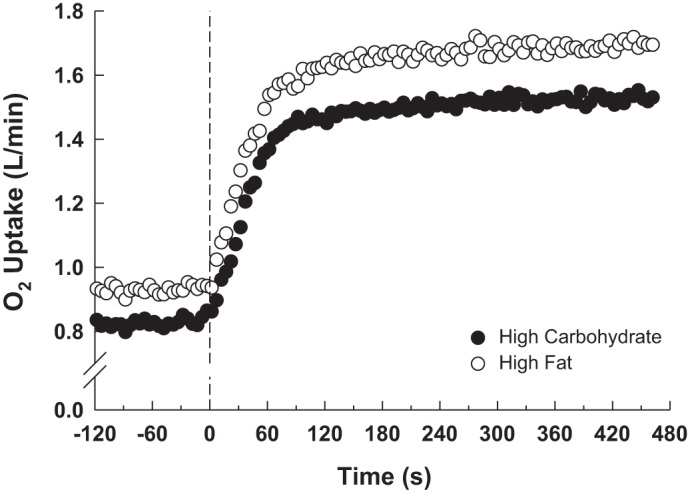
Group mean pulmonary O2 uptake (V̇o2p) response at baseline and throughout the transition to moderate-intensity exercise during the HFAT (open symbols) and HCHO (closed symbol) diets.
Table 3.
Summary of parameter estimates for V̇o2p on-transients to moderate-intensity exercise during high-fat and high-carbohydrate diets
| HFAT Diet | HCHO Diet | |
|---|---|---|
| Baseline V̇o2p, liter/min | 0.93 ± 0.07* | 0.83 ± 0.08 |
| End-exercise V̇o2p, liter/min | 1.68 ± 0.23*† | 1.52 ± 0.25† |
| τV̇o2p, s | 40 ± 16* | 32 ± 19 |
| Time delay, s | 5 ± 10* | 13 ± 8 |
| Mean response time, s‡ | 45 ± 8 | 46 ± 12 |
| ΔV̇o2p/ΔWR, ml·min−1·W−1 | 10.3 ± 0.8* | 9.4 ± 0.7 |
Baseline and end-exercise V̇o2p, V̇o2p amplitude (ΔV̇o2p), and V̇o2p gain (ΔV̇o2p/ΔWR) were greater (P < 0.001) in HFAT than in HCHO (Table 3). The phase II V̇o2p time constant (τV̇o2p) was ∼25% greater (P < 0.015) in HFAT (40 ± 16 s; CI95%, 3.6 s) than in HCHO (32 ± 19 s; CI95%, 2.5 s, Fig. 3).
Fig. 3.
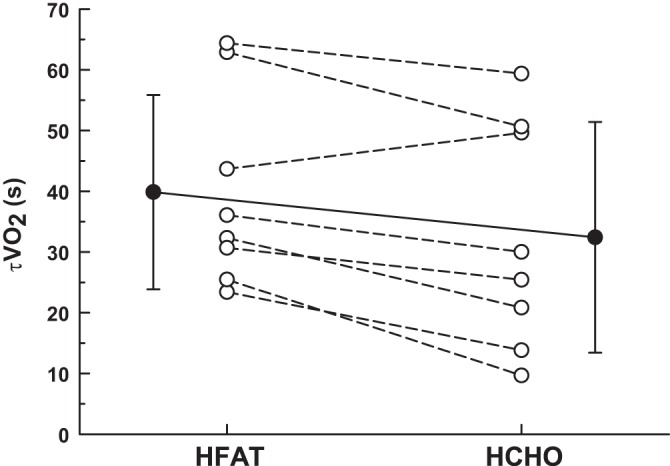
The time constant for the phase II V̇o2p response (τV̇o2p) during the HFAT and HCHO diets; individual responses (open symbols); group mean (± SD) response (closed symbol). Values are means ± SD; τV̇o2p was significantly greater (P < 0.015) in HFAT vs. HCHO.
Baseline V̇co2p (HFAT 0.80 ± 0.13 liter/min, HCHO 0.83 ± 0.15 liter/min, P < 0.105) and end-exercise V̇co2p (HFAT 1.41 ± 0.20 liter/min, HCHO 1.45 ± 0.24 liter/min, P < 0.048) were lower, and the rate of adjustment of V̇co2p during the exercise transition (τV̇co2p HFAT 105 ± 42 s, τV̇co2p HCHO 79 ± 42 s, P < 0.040) was greater in HFAT than in HCHO.
HR kinetics.
The HR responses to MOD exercise are presented in Table 4. Baseline (P < 0.024) and end-exercise HRs (P < 0.003) were ∼10 beats per minute higher in HFAT than in HCHO, with no difference in HR amplitude between diets. The τHR was greater (P < 0.014) in the HFAT (31 ± 10 s) than in the HCHO diet (24 ± 8 s).
Table 4.
Summary of parameter estimates for heart rate kinetics during the transition to moderate-intensity exercise in high-fat and high-carbohydrate diets
| HFAT Diet | HCHO Diet | |
|---|---|---|
| Heart rate | ||
| Baseline, bpm | 85 ± 13* | 76 ± 10 |
| End-exercise, bpm | 110 ± 9*† | 98 ± 8† |
| τHR, s | 31 ± 10* | 24 ± 8 |
NIRS-derived deoxy-(Δ[HHb]), oxy-(Δ[O2Hb]), and total (Δ[HbTOT]) hemoglobin concentration change.
Parameter estimates for Δ[HHb] exercise response are presented in Table 5. The delay prior to a sustained increase in the Δ[HHb] (TDΔ[HHb]) was ∼12 s, with no difference between conditions. Although the τΔ[HHb] was greater (P < 0.05) in the HFAT (13 ± 4 s) than HCHO (11 ± 4 s) diets, the overall response time (τ′Δ[HHb] = TDΔ[HHb] + τΔ[HHb]) was not different between diets (HFAT 24 ± 4 s, HCHO 23 ± 4 s) despite slower V̇o2p kinetics in the HFAT diet (Fig. 4); the end-exercise steady-state Δ[HHb]/ΔV̇o2p ratio was not different between the HFAT (10 ± 4 au·liter−1·min−1) and HCHO (12 ± 5 au·liter−1·min−1) diets.
Table 5.
Summary of parameter estimates for near-infrared spectroscopy-derived oxyhemoglobin, deoxyhemoglobin, and total hemoglobin concentration changes during the transition to moderate-intensity exercise in the high-fat and high-carbohydrate diets
| HFAT Diet | HCHO Diet | |
|---|---|---|
| Δ[O2Hb] | ||
| Baseline, au | −0.8 ± 2.6 | 0.7 ± 4.1 |
| End-exercise, au | −2.8 ± 4.6 | −1.9 ± 7.1 |
| [HbTOT] | ||
| Baseline, au | −0.9 ± 2.8 | 0.1 ± 3.5 |
| End-exercise, au | 5.4 ± 2.9† | 7.0 ± 5.0† |
| Δ[HHb] | ||
| Baseline, au | −0.2 ± 2.8 | −0.7 ± 3.3 |
| End-exercise, au | 8.0 ± 6.4† | 8.4 ± 7.9† |
| τΔ[HHb], s | 13 ± 4* | 11 ± 4 |
| TDΔ[HHb], s | 11 ± 4 | 12 ± 4 |
| MRTΔ[HHb], s | 24 ± 4 | 23 ± 4 |
| Δ[HHb]/ΔV̇o2p, au·liter−1·min−1 | 10 ± 4 | 12 ± 5 |
Fig. 4.
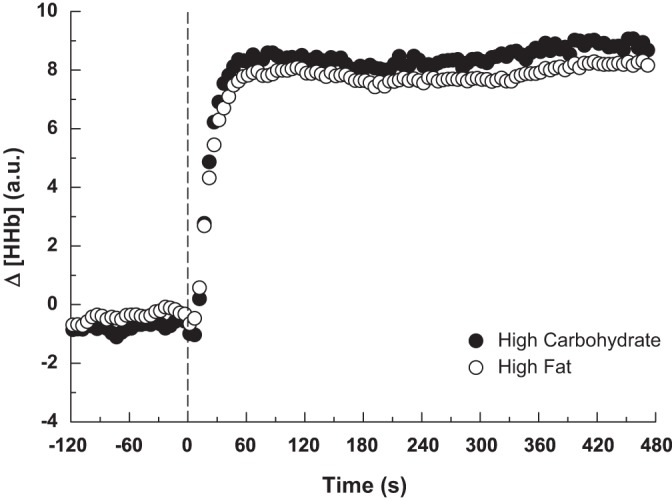
Group mean muscle deoxygenation (Δ[HHb]) response at baseline and throughout the transition to moderate-intensity exercise during the HFAT (open symbols) and HCHO (closed symbols) diets.
DISCUSSION
This is the first study to examine the effects of a sequential 6-day HFAT diet followed by a 6-day HCHO diet (with each preceded by a GD protocol) on phase II pulmonary O2 uptake (V̇o2p) kinetics during transitions to MOD exercise. In addition, this is the first study to report NIRS-derived quadriceps muscle deoxygenation kinetics during HFAT and HCHO dietary interventions, which provides a reflection of local muscle O2 extraction and thus the dynamic relationship between local microvascular blood flow and O2 delivery and muscle O2 utilization during transitions to MOD exercise. Together, these measurements provide information on the role of muscle substrate availability on constraining the adjustment of muscle O2 utilization (as reflected by V̇o2p) and microvascular blood flow during the transition to exercise.
The major new findings presented in this study are 1) during the transition to MOD exercise, the time constants for the phase II V̇o2p (τV̇o2p) and V̇co2p (τV̇co2p) responses were ∼25% and ∼30% greater, respectively, in the HFAT compared with the HCHO diet; 2) the V̇o2p gain (i.e., ΔV̇o2p/ΔWR), a reflection of the O2 cost of exercise and related inversely to exercise efficiency, was greater during the HFAT compared with the HCHO diet; 3) muscle deoxygenation kinetics (i.e., MRTΔ[HHb]) were similar between diets despite slower V̇o2p kinetics in the HFAT condition; and 4) steady-state baseline and end-exercise HRs and HR kinetics (τHR) during the transition to exercise were greater in the HFAT diet.
Effectiveness of the dietary-GD intervention.
The sequence of a 6-day HFAT followed by a 6-day HCHO dietary intervention used in the present study, with each preceded by a GD exercise protocol, has been shown previously to minimize (HFAT) and then to maximize (HCHO) muscle carbohydrate (glycogen supercompensation) availability [as modified from Bergström et al. (5)]. Variations of this protocol were previously shown to be effective in lowering muscle glycogen content by approximately 55–90% (5, 32), and simultaneously to downregulate and attenuate the exercise-induced activation of PDH in HFAT compared with HCHO (8, 29, 32). Although muscle glycogen content was not measured in the present study, the inclusion of heavy- and severe-intensity exercise bouts immediately before starting each of the dietary protocols, the extremes in fat/carbohydrate composition of the diets (HFAT ∼75% fat, ∼5% carbohydrate; HCHO ∼10% fat, ∼80% carbohydrate), and the duration of the dietary intervention (6 days) ensured that muscle glycogen content and carbohydrate availability were lower [and muscle triglyceride levels were higher (21, 41)] in the HFAT than in the HCHO portions of the protocol. The effectiveness of the dietary intervention is supported by the following: 1) steady-state RER and V̇co2p were higher (by ∼16% and ∼3%, respectively), and absolute V̇o2p and V̇o2p gain were lower (by ∼11% and ∼9%, respectively) in HCHO; 2) steady-state fat oxidation rates were higher (by approximately twofold to fourfold) in HFAT, whereas carbohydrate oxidation rates were higher (by approximately twofold) in HCHO, which is in accordance with previous studies showing similar changes (8, 42); 3) blood [glucose] was higher in HCHO, while blood [FFA] and [β-hydroxybutyrate] were elevated in HFAT; and 4) steady-state HR was higher (by ∼11%) in HFAT perhaps to enhance bulk O2 delivery to muscle in support of the increased O2 requirement of fatty acid oxidation. Thus the dietary-induced effects on fat and carbohydrate oxidation rates suggests that reduced availability of carbohydrate during HFAT, and greater carbohydrate availability during HCHO, resulted in adaptations within muscle, which shifted the substrate being preferentially oxidized during exercise.
Effect of diet on V̇o2p, HR, and muscle deoxygenation kinetics.
The main novel finding of the present study was that phase II V̇o2p kinetics were slowed (P < 0.015) in the HFAT compared with the HCHO diet during the transition to MOD exercise, reflecting a slower activation and adjustment of muscle O2 utilization and mitochondrial oxidative phosphorylation in the HFAT diet. In addition, HR kinetics (τHR), a measure of central blood flow (cardiac output) adjustment, also was slower (P < 0.014) in HFAT than in HCHO diets. In a previous study, Hughson and Kowalchuk (19) examined the relationship between V̇co2p and ventilation after a 72-h period of HFAT or HCHO dietary restrictions in females, and although it was not a focus of their study, they reported no differences in τV̇o2p between diets. However, in that study, only a single exercise transition was performed within each of the dietary conditions, and gas exchange variables were calculated less frequently (15-s intervals during the first 5 min of the test, 30-s intervals during the final 5 min of the 10-min exercise protocol), which would reduce the confidence of estimating the kinetic parameters for the gas exchange variables. Additionally, participants were all females and there was no control for their menstrual cycle phase; the prediet GD protocol was less severe, being performed at a lower exercise intensity and without inclusion of severe-intensity exercise bouts; the prescribed diets were not monitored or analyzed for caloric intake and dietary composition, and the diets were each separated by 5-day period of consuming a balanced, mixed diet.
Molé and Hoffmann (25) proposed that the initial changes in substrate flux and fat and carbohydrate oxidation rates would determine the phase II dynamics of muscle V̇o2p. Using assumptions related to control and time course of activation of metabolic flux rates, the authors used computer modeling to deconstruct the phase II and III V̇o2p profiles into individual components related to rate of activation of fat and carbohydrate oxidation-dependent components in relation to RER during exercise. The major finding of that paper, as related to our present study, was that the time constant for fat oxidation (τFAT) was independent of RER, whereas the time constant for carbohydrate oxidation (τCHO) was inversely related to RER. We further analyzed their data [presented in Table 4 on p. 2,101 in Molé and Hoffmann (25)] by calculating an overall mean response time (MRT) for V̇o2p (calculated as a weighted sum of the asymptotes and time constant; the reported time delay was ignored for simplicity) for each of the fat and carbohydrate components [similar to that described in Hughson et al. (18)]. According to this calculation, the overall MRT was inversely related to RER (i.e., MRT of ∼15 s and ∼60 s at an RER of 0.97 and 0.78, respectively), a trend similar to that reported in the present study. In their discussion, Molé and Hoffmann state that while their results were “encouraging, [they] must reserve judgment until a more complete assessment of dietary effects can be made and other experimental tests of the model are undertaken and reported” [p. 2,103 (25)]. Our current study, which specifically altered dietary intake and muscle substrate availability, extends the findings of Molé and Hoffmann (25) by including a more rigorous estimation of V̇o2p kinetics during defined dietary and muscle substrate interventions.
The slower V̇o2 adjustment in HFAT compared with HCHO may be attributed in part to an inhibition and attenuated activation of the mitochondrial PDH complex. Previous studies reported that after 3–6 days of a HFAT diet, or after 30 min of intralipid infusion to raise plasma [FFA], PDH activity was lower at rest (28, 30, 32, 39, 46) and during exercise (32), and the rate of PDH activation during the transition to exercise was slowed (32). Also, the resting PDH kinase (PDK) activity responsible for catalyzing the covalent phosphorylation and thus inhibition of the PDH complex, was elevated early (within 1–3 days) on a HFAT diet in humans (30, 29, 46), and within 24 h of fasting in rats (28). Additionally, both PDK mRNA and mitochondrial PDK protein content were elevated (within 1 day) on the HFAT diet (28, 29). Thus a fat-induced inhibition of PDH activity may delay the activation of PDH during transitions to exercise and attenuate the delivery of carbohydrate-derived substrate (acetyl CoA, reducing equivalents) to the mitochondrial TCA and ETC pathways.
In the present study, each of the diets was preceded by a heavy/severe-intensity GD protocol to lower existing muscle glycogen levels. By design, carbohydrate ingestion in HFAT compared with HCHO was minimized, thereby ensuring that muscle glycogen content would remain lower during the HFAT than during the HCHO phase of the diet. It was expected that establishing and maintaining low muscle glycogen levels in combination with an HFAT diet would reduce the rate of muscle glycogenolysis and attenuate muscle pyruvate production and oxidation during a subsequent bout of exercise. Putman and coworkers (32) reported that a combined GD and HFAT dietary protocol was associated with reduced PDH activity at rest, and at the onset of exercise, glycogen breakdown, pyruvate formation, and PDH activation were lower in HFAT than HCHO. In contrast, St. Amand and coworkers (39) reported that HFAT alone (i.e., without prior GD) was associated with a lower PDH activity at rest, but during MOD exercise, muscle PDH activation was not different between the HFAT and higher carbohydrate diet, presumably because of higher rates of glycogen breakdown and pyruvate production. Importantly, pyruvate is both a substrate for the PDH-catalyzed reaction and an allosteric inhibitor of the PDH kinase, therefore a low rate of pyruvate formation during exercise in HFAT (a consequence of a low resting muscle glycogen content) would prevent or slow activation of the PDH complex through its combined effects as a substrate for the PDH-catalyzed reaction and as an allosteric inhibitor of PDH kinase (32).
Pharmacological activation of PDH before exercise with DCA administration (35, 44) was shown to reduce glycogen and PCr breakdown, reduce muscle lactate accumulation, reduce substrate-level phosphorylation (44), and increase the rate of fall of muscle intracellular Po2 (17). Also performing a prior bout of heavy-intensity exercise to prime the muscle (16) was associated with a higher pretransition level of PDH activation and faster V̇o2p kinetics during the transition to exercise (16). Taken together, these responses are consistent with a faster activation of mitochondrial oxidative phosphorylation. Given the regulatory (rate-limiting) role played by PDH in providing carbohydrate-derived substrate to the mitochondrial oxidative pathways and the reported downregulation of PDH activity in high-fat, low-carbohydrate conditions, it is not unreasonable to suggest that inhibition of PDH activation at the onset of exercise may impose a metabolic limitation and constrain the activation of oxidative phosphorylation that may contribute to the slower V̇o2p kinetics during the HFAT diet in the present study. However, a measurable speeding of V̇o2p kinetics following DCA-induced PDH activation has not always been observed (13, 35). Therefore, although PDH can impose a limitation under some circumstances or in some individuals, in normal circumstances it may not always be limiting for the adjustment of oxidative phosphorylation and V̇o2p kinetics.
In the present study, NIRS was used to measure oxygenation and deoxygenation changes within the vastus lateralis muscle. Increases in muscle deoxygenation (i.e., Δ[HHb]) are related in part to muscle fractional O2 extraction and thus the ratio of local muscle microvascular blood flow-to-O2 utilization (Q̇m-to-V̇o2m), and when used in combination with phase II V̇o2p kinetics, the Δ[HHb] provides an estimate of local microvascular blood flow changes during the exercise transition (11). In the present study, the overall Δ[HHb] kinetics as defined by the τ′Δ[HHb] was similar between dietary conditions despite the slower V̇o2p kinetics in the HFAT condition. Combined, these data suggest possible differences in microvascular blood flow between dietary conditions, with the adjustment of microvascular blood flow being attenuated and/or slowed relative to the increase in muscle O2 utilization in the HFAT than in the HCHO diet.
It was previously demonstrated that high fat content in the diet or meals, or elevated intralipid-infusion-induced plasma FFA levels were associated with the development of insulin resistance, endothelial dysfunction, impaired endothelial nitric oxide production, and reduced microvascular perfusion (23, 26, 43). However, studies have shown that exercise/contraction-induced increases in blood flow are intact in models that display endothelial dysfunction (20, 49), indicating that exercise/contraction follows endothelium-independent mechanisms for increasing blood flow. In particular, St.-Pierre et al. (40) showed that contraction-mediated increases in microvascular blood flow were not altered by high fat feeding in rats. However, the similar kinetics of muscle deoxygenation in the two dietary conditions despite a slower rate of adjustment of muscle O2 utilization may reflect an impaired adjustment of muscle microvascular blood flow in the HFAT condition (i.e., the slow microvascular blood flow and O2 delivery limits the activation of muscle O2 utilization) (3). Alternatively, a slower muscle blood flow response in HFAT may simply be consequent to the slower adjustment of muscle O2 utilization (i.e., consistent with a metabolic limitation). Although these data cannot distinguish between the two mechanisms, in either case, during the exercise transient, a given change in WR (and presumably ATP utilization) is associated with a lower V̇o2m and higher HHb-to-V̇o2m in HFAT, implicating a lower microvascular blood flow response in the HFAT condition.
Conclusion.
This study demonstrated that a GD plus 6-day HFAT dietary intervention followed by an HCHO dietary intervention was effective in altering fuel selection during exercise, with carbohydrate oxidation rate being lower and fat oxidation rate being greater in the HFAT than in the HCHO condition. Additionally, the steady-state baseline and end-MOD exercise V̇o2p and HR, and the increase in V̇o2p gain (ΔV̇o2p/ΔWR) were greater in the HFAT than in the HCHO condition, consistent with the higher O2 cost of fat compared with carbohydrate oxidation, whereas the higher V̇o2p gain in HFAT suggests that exercise efficiency was lower compared with the HCHO condition. The novel findings of the present study were that the phase II pulmonary V̇o2p time constant (τV̇o2p) was ∼25% greater in the HFAT (40 s) compared with the HCHO condition (32 s), suggesting that the adjustment of muscle O2 utilization and mitochondrial oxidative phosphorylation were slowed during the transition to MOD exercise during the HFAT compared with the HCHO diet. In spite of slower V̇o2p kinetics in the HFAT condition, muscle deoxygenation kinetics were similar in the two dietary conditions, suggesting that the adjustment of microvascular blood flow during the exercise transition was lower and/or slower during the HFAT diet. Therefore, the slower V̇o2p kinetics during the transition to MOD exercise in the HFAT compared with the HCHO diet may reflect a downregulation of PDH activity at baseline and delayed activation during the exercise transition, and/or a slower rate of glycogenolysis and pyruvate production, which would attenuate the delivery of carbohydrate-derived substrate [i.e., acetyl-CoA; reducing equivalents (NADH; FADH2)] into the mitochondrial TCA cycle and ETC pathways. A slower adjustment of microvascular blood flow could slow O2 diffusion from the red blood cell to the mitochondria, thereby limiting availability of O2 at the mitochondrial terminal oxidase. Thus attenuated deliveries of oxidative substrate required in oxidative phosphorylation (O2, NADH, H+) in the HFAT compared with the HCHO condition could contribute to a slower V̇o2m and thus slow the adjustment of pulmonary V̇o2p during the transition to MOD exercise.
GRANTS
This study was supported by grants from the Natural Sciences and Engineering Research Council of Canada. Additional support was provided by grants from The University of Western Ontario Academic Development Fund, the Canadian Foundation for Innovation, and Ontario Innovation Trust.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
J.R., D.H.P., S.J.P., G.J.H., and J.M.K. conception and design of research; J.R. performed experiments; J.R., L.K.L., S.J.P., and J.M.K. analyzed data; J.R., L.K.L., S.J.P., G.J.H., and J.M.K. interpreted results of experiments; J.R. and L.K.L. prepared figures; J.R., L.K.L., and J.M.K. drafted manuscript; J.R., L.K.L., D.H.P., S.J.P., G.J.H., and J.M.K. edited and revised manuscript; J.R., L.K.L., D.H.P., S.J.P., G.J.H., and J.M.K. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the subjects who participated in the study and Brad Hansen for technical support. We also thank Nelson Education for kindly allowing us to use their Diet Analysis Plus software to analyze and design the diets used in this study.
REFERENCES
- 1.Bae JH, Schwemmer M, Lee IK, Lee HJ, Park KR, Kim KY, Bassenge E. Postprandial hypertriglyceridemia-induced endothelial dysfunction in healthy subjects is independent of lipid oxidation. Int J Cardiol 87: 259–267, 2003. [DOI] [PubMed] [Google Scholar]
- 2.Beaver WL, Wasserman K, Whipp BJ. Breath-by-breath measurement of true alveolar gas exchange. J Appl Physiol 51: 1662–1675, 1981. [DOI] [PubMed] [Google Scholar]
- 3.Behnke BJ, Kindig CA, McDonough P, Poole DC, Sexton WL. Dynamics of microvascular oxygen pressure during rest-contraction transition in skeletal muscle of diabetic rats. Am J Physiol Heart Circ Physiol 283: H926–H932, 2002. [DOI] [PubMed] [Google Scholar]
- 4.Bergmeyer HU. Methods in Enzymatic Analysis. New York: Academic Press, 1974. [Google Scholar]
- 5.Bergström J, Hermansen L, Hultman E, Saltin B. Diet, muscle glycogen and physical performance. Acta Physiol Scand 71: 140–150, 1967. [DOI] [PubMed] [Google Scholar]
- 6.Chin LM, Leigh RJ, Heigenhauser GJ, Rossiter HB, Paterson DH, Kowalchuk JM. Hyperventilation-induced hypocapnic alkalosis slows the adaptation of pulmonary O2 uptake during the transition to moderate-intensity exercise. J Physiol 583: 351–364, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clifford PS, Hellsten Y. Vasodilatory mechanisms in contracting skeletal muscle. J Appl Physiol 97: 393–403, 2004. [DOI] [PubMed] [Google Scholar]
- 8.Constantin-Teodosiu D, Constantin D, Stephens F, Laithwaite D, Greenhaff PL. The role of FOXO and PPAR transcription factors in diet-mediated inhibition of PDC activation and carbohydrate oxidation during exercise in humans and the role of pharmacological activation of PDC in overriding these changes. Diabetes 61: 1017–1024, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeLorey DS, Kowalchuk JM, Paterson DH. Relationship between pulmonary O2 uptake kinetics and muscle deoxygenation during moderate-intensity exercise. J Appl Physiol 95: 113–120, 2003. [DOI] [PubMed] [Google Scholar]
- 10.Elwell C. A Practical User's Guide to Near Infrared Spectroscopy. London: Hamatsu Photonics KK, 1995, p. 1–155. [Google Scholar]
- 11.Ferreira LF, Townsend DK, Lutjemeier BJ, Barstow TJ. Muscle capillary blood flow kinetics estimated from pulmonary O2 uptake and near-infrared spectroscopy. J Appl Physiol 98: 1820–1828, 2005. [DOI] [PubMed] [Google Scholar]
- 12.Glancy B, Barstow T, Willis WT. Linear relation between time constant of oxygen uptake kinetics, total creatine, and mitochondrial content in vitro. Am J Physiol Cell Physiol 294: C79–C87, 2008. [DOI] [PubMed] [Google Scholar]
- 13.Grassi B, Hogan MC, Greenhaff PL, Hamann JJ, Kelley KM, Aschenbach WG, Constantin-Teodosiu D, Gladden LB. Oxygen uptake on-kinetics in dog gastrocnemius in situ following activation of pyruvate dehydrogenase by dichloroacetate. J Physiol 538: 195–207, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grassi B, Poole DC, Richardson RS, Knight DR, Erickson BK, Wagner PD. Muscle O2 uptake kinetics in humans: implications for metabolic control. J Appl Physiol 80: 988–998, 1996. [DOI] [PubMed] [Google Scholar]
- 15.Grassi B, Porcelli S, Salvadego D, Zoladz JA. Slow VO2 kinetics during moderate-intensity exercise as markers of lower metabolic stability and lower exercise tolerance. Eur J Appl Physiol 111: 345–355, 2011. [DOI] [PubMed] [Google Scholar]
- 16.Gurd BJ, Peters SJ, Heigenhauser GJ, LeBlanc PJ, Doherty TJ, Paterson DH, Kowalchuk JM. Prior heavy exercise elevates pyruvate dehydrogenase activity and speeds O2 uptake kinetics during subsequent moderate-intensity exercise in healthy young adults. J Physiol 577: 985–996, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Howlett RA, Hogan MC. Dichloroacetate accelerates the fall in intracellular Po2 at onset of contractions in Xenopus single muscle fibers. Am J Physiol Regul Integr Comp Physiol 284: R481–R485, 2003. [DOI] [PubMed] [Google Scholar]
- 18.Hughson RL, Cochrane JE, Butler GC. Faster O2 uptake kinetics at onset of supine exercise with than without lower body negative pressure. J Appl Physiol 75: 1962–1967, 1993. [DOI] [PubMed] [Google Scholar]
- 19.Hughson RL, Kowalchuk JM. Influence of diet on CO2 production and ventilation in constant-load exercise. Respir Physiol 46: 149–160, 1981. [DOI] [PubMed] [Google Scholar]
- 20.Inyard AC, Clerk LH, Vincent MA, Barrett EJ. Contraction stimulates nitric oxide-independent microvascular recruitment and increases muscle insulin uptake. Diabetes 56: 2194–2200, 2007. [DOI] [PubMed] [Google Scholar]
- 21.Jansson E, Kaijser L. Effect of diet on the utilization of blood and intramuscular substrates during exercise in man. Acta Physiol Scand 115: 19–30, 1982. [DOI] [PubMed] [Google Scholar]
- 22.LeBlanc PJ, Parolin ML, Jones NL, Heigenhauser GJ. Effects of respiratory alkalosis on human skeletal muscle metabolism at the onset of submaximal exercise. J Physiol 544: 303–313, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lind L, Fugmann A, Branth S, Vessby B, Millgard J, Berne C, Lithell H. The impairment in endothelial function induced by non-esterified fatty acids can be reversed by insulin. Clin Sci (Lond) 99: 169–174, 2000. [DOI] [PubMed] [Google Scholar]
- 24.Meyer RA. A linear model of muscle respiration explains monoexponential phosphocreatine changes. Am J Physiol Cell Physiol 254: C548–C553, 1988. [DOI] [PubMed] [Google Scholar]
- 25.Molé PA, Hoffmann JJ. V̇o2 kinetics of mild exercise are altered by RER. J Appl Physiol 87: 2097–2106, 1999. [DOI] [PubMed] [Google Scholar]
- 26.Ng CK, Chan AP, Cheng A. Impairment of endothelial function–a possible mechanism for atherosclerosis of a high-fat meal intake. Ann Acad Med Singapore 30: 499–502, 2001. [PubMed] [Google Scholar]
- 27.Peronnet F, Massicotte D. Table of nonprotein respiratory quotient: an update. Can J Sport Sci 16: 23–29, 1991. [PubMed] [Google Scholar]
- 28.Peters SJ, Harris RA, Heigenhauser GJ, Spriet LL. Muscle fiber type comparison of PDH kinase activity and isoform expression in fed and fasted rats. Am J Physiol Regul Integr Comp Physiol 280: R661–R668, 2001. [DOI] [PubMed] [Google Scholar]
- 29.Peters SJ, Harris RA, Wu P, Pehleman TL, Heigenhauser GJ, Spriet LL. Human skeletal muscle PDH kinase activity and isoform expression during a 3-day high-fat/low-carbohydrate diet. Am J Physiol Endocrinol Metab 281: E1151–E1158, 2001. [DOI] [PubMed] [Google Scholar]
- 30.Peters SJ, St Amand TA, Howlett RA, Heigenhauser GJ, Spriet LL. Human skeletal muscle pyruvate dehydrogenase kinase activity increases after a low-carbohydrate diet. Am J Physiol Endocrinol Metab 275: E980–E986, 1998. [DOI] [PubMed] [Google Scholar]
- 31.Poole DC, Jones AM. Oxygen uptake kinetics. Compr Physiol 2: 933–996, 2012. [DOI] [PubMed] [Google Scholar]
- 32.Putman CT, Spriet LL, Hultman E, Lindinger MI, Lands LC, McKelvie RS, Cederblad G, Jones NL, Heigenhauser GJ. Pyruvate dehydrogenase activity and acetyl group accumulation during exercise after different diets. Am J Physiol Endocrinol Metab 265: E752–E760, 1993. [DOI] [PubMed] [Google Scholar]
- 33.Rossiter HB. Exercise: kinetic considerations for gas exchange. Compr Physiol 1: 203–244, 2011. [DOI] [PubMed] [Google Scholar]
- 34.Rossiter HB, Ward SA, Doyle VL, Howe FA, Griffiths JR, Whipp BJ. Inferences from pulmonary O2 uptake with respect to intramuscular [phosphocreatine] kinetics during moderate exercise in humans. J Physiol 518: 921–932, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rossiter HB, Ward SA, Howe FA, Wood DM, Kowalchuk JM, Griffiths JR, Whipp BJ. Effects of dichloroacetate on V̇o2 and intramuscular 31P metabolite kinetics during high-intensity exercise in humans. J Appl Physiol 95: 1105–1115, 2003. [DOI] [PubMed] [Google Scholar]
- 36.Scheuermann BW, Bell C, Paterson DH, Barstow TJ, Kowalchuk JM. Oxygen uptake kinetics for moderate exercise are speeded in older humans by prior heavy exercise. J Appl Physiol 92: 609–616, 2002. [DOI] [PubMed] [Google Scholar]
- 37.Spriet LL, Heigenhauser GJ. Regulation of pyruvate dehydrogenase (PDH) activity in human skeletal muscle during exercise. Exerc Sport Sci Rev 30: 91–95, 2002. [DOI] [PubMed] [Google Scholar]
- 38.Spriet LL, Peters SJ. Influence of diet on the metabolic responses to exercise. Proc Nutr Soc 57: 25–33, 1998. [DOI] [PubMed] [Google Scholar]
- 39.St Amand TA, Spriet LL, Jones NL, Heigenhauser GJ. Pyruvate overrides inhibition of PDH during exercise after a low-carbohydrate diet. Am J Physiol Endocrinol Metab 279: E275–E283, 2000. [DOI] [PubMed] [Google Scholar]
- 40.St-Pierre P, Keith LJ, Richards SM, Rattigan S, Keske MA. Microvascular blood flow responses to muscle contraction are not altered by high-fat feeding in rats. Diabetes Obes Metab 14: 753–761, 2012. [DOI] [PubMed] [Google Scholar]
- 41.Starling RD, Trappe TA, Parcell AC, Kerr CG, Find WJ, Costill DL. Effects of diet on muscle triglyceride and endurance performance. J Appl Physiol 82: 1185–1189, 1997. [DOI] [PubMed] [Google Scholar]
- 42.Staudacher HM, Carey AL, Cummings NK, Hawley JA, Burke LM. Short-term high-fat diet alters substrate utilization during exercise but not glucose tolerance in highly trained athletes. Int J Sport Nutr Exerc Metab 11: 273–286, 2001. [DOI] [PubMed] [Google Scholar]
- 43.Steer P, Sarabi DM, Karlstrom B, Basu S, Berne C, Vessby B, Lind L. The effect of a mixed meal on endothelium-dependent vasodilation is dependent on fat content in healthy humans. Clin Sci (Lond) 105: 81–87, 2003. [DOI] [PubMed] [Google Scholar]
- 44.Timmons JA, Gustafsson T, Sundberg CJ, Jansson E, Greenhaff PL. Muscle acetyl group availability is a major determinant of oxygen deficit in humans during submaximal exercise. Am J Physiol Endocrinol Metab 274: E377–E380, 1998. [DOI] [PubMed] [Google Scholar]
- 45.Tschakovsky ME, Hughson RL. Interaction of factors determining oxygen uptake at the onset of exercise. J Appl Physiol 86: 1101–1113, 1999. [DOI] [PubMed] [Google Scholar]
- 46.Turvey EA, Heigenhauser GJ, Parolin M, Peters SJ. Elevated n-3 fatty acids in a high-fat diet attenuate the increase in PDH kinase activity but not PDH activity in human skeletal muscle. J Appl Physiol 98: 350–355, 2005. [DOI] [PubMed] [Google Scholar]
- 47.Vogel RA, Corretti MC, Plotnick GD. Effect of a single high-fat meal on endothelial function in healthy subjects. Am J Cardiol 79: 350–354, 1997. [DOI] [PubMed] [Google Scholar]
- 48.Watt MJ, Heigenhauser GJ, Stellingwerff T, Hargreaves M, Spriet LL. Carbohydrate ingestion reduces skeletal muscle acetylcarnitine availability but has no effect on substrate phosphorylation at the onset of exercise in man. J Physiol 544: 949–956, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wheatley CM, Rattigan S, Richards SM, Barrett EJ, Clark MG. Skeletal muscle contraction stimulates capillary recruitment and glucose uptake in insulin-resistant obese Zucker rats. Am J Physiol Endocrinol Metab 287: E804–E809, 2004. [DOI] [PubMed] [Google Scholar]