An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people (original) (raw)
. Author manuscript; available in PMC: 2015 Feb 6.
Published in final edited form as: Science. 2012 May 17;337(6090):100–104. doi: 10.1126/science.1217876
Abstract
Rare genetic variants contribute to complex disease risk; however, the abundance of rare variants in human populations remains unknown. We explored this spectrum of variation by sequencing 202 genes encoding drug targets in 14,002 individuals. We find rare variants are abundant (one every 17 bases) and geographically localized, such that even with large sample sizes, rare variant catalogs will be largely incomplete. We used the observed patterns of variation to estimate population growth parameters, the proportion of variants in a given frequency class that are putatively deleterious, and mutation rates for each gene. Overall we conclude that, due to rapid population growth and weak purifying selection, human populations harbor an abundance of rare variants, many of which are deleterious and have relevance to understanding disease risk.
Understanding the genetic contribution to human disease requires knowledge of the abundance and distribution of functional genetic diversity within and among populations. The “common-disease rare-variant” hypothesis posits that variants affecting health are under purifying selection, and thus should be found only at low frequencies in human populations (1-3). This hypothesis has become increasingly credible, since very large genome-wide association studies of common variants have explained only a fraction of the known heritability of most traits (4, 5). Investigating the role of rare variants for complex trait mapping has led to tests that aggregate rare variants (6), and determine the abundance, distribution, and phenotypic effects of rare variants in human populations (7, 8).
Population genetic models predict that mutation rates, the strength of selection, and demography affect the abundance of rare variants, although the relative importance of each is a long-standing question (9-11). To understand rare variant diversity in humans, we sequenced 202 genes in a sample of 14,002 well-phenotyped individuals (table S1). These genes represent approximately 1% of the coding genome and approximately 7% of genes considered current or potential drug targets (12), enriched for cell signaling proteins and membrane-bound transporters (table S2). A total of 864 kb were targeted, including 351 kb of coding and 323 kb of untranslated (UTR) exon regions (database S1). Over 93% of target bases were successfully sequenced at a median depth of 27 reads per site (13). Because rare variant discovery can easily be confounded with sequencing errors, we performed numerous experiments to demonstrate high data quality (table S3, (13)). The sequenced subjects include two population samples (n=1,322 and 2,059) and 12 disease collections (n=125–1,125 cases, table S4). The self-reported ancestry of the sample was predominantly European (12,514), African American (594) and South Asian (567). Some of the following analyses focus on the European subset, which is well-powered to investigate rare variants. Based on our sample size we expect that 94% of variant alleles with minor allele frequency (MAF) 0.01% in Europeans were sampled at least once.
Sequencing revealed an abundance of rare (MAF<0.5%) single nucleotide variants (SNVs), compared to common variants (Fig. 1A, B). We observed on average one variant per 17 bp in the overall sample and one variant per 21 bp in the Europeans (table S5). Among all variants, more than 95% were rare (MAF≤0.5%), and more than 74% were observed in only one or two subjects. ~90% of rare variants were not previously reported, as opposed to ~5% of common variants (MAF>0.5%) (fig. S1). For the large European subset, Watterson’s θ_W_, a metric of genetic diversity (Table 1), was much larger (40.38×10-4) than in previous smaller scale studies, and an order of magnitude larger than the pairwise metric θπ (3.96×10-4). We observed a third allele at 2.0% of variable sites, and among those, 1.6% had a fourth allele. We found between 1.2 and 1.9 non-diallelic SNVs per kb of sequence (fig. S2), which tended to occur at sites under lower evolutionary conservation (fig. S3, (13)). The rate of variant discovery remained nearly constant with increasing sample size (Fig. 2A). We expect 111–153 variants per kb in a sample of 100,000 Europeans and 337–452 variants per kb in a sample of 1 million (Fig. 2A, B).
Fig. 1.
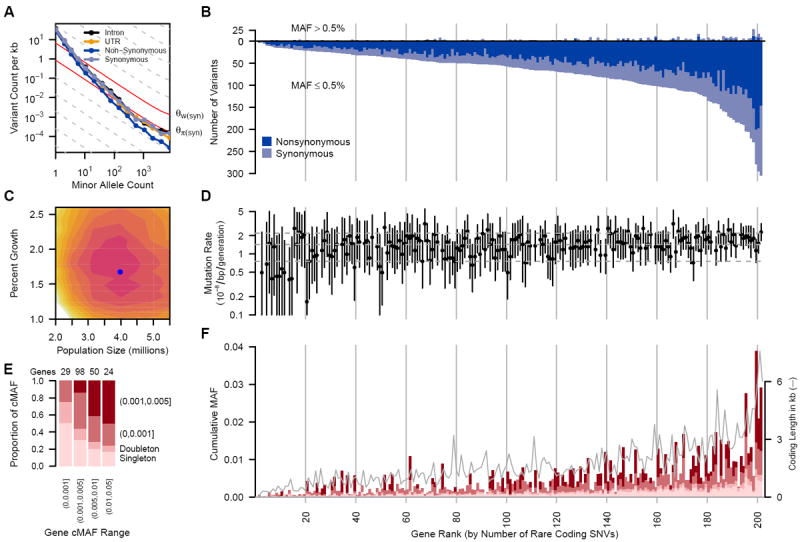
(A) Frequency spectrum of variants relating the number of variants per kb within minor allele counts. Solid gray lines provide expectations from nucleotide diversity (θπ) and the number of segregating sites (θ_W_). (B) The number of common (MAF>0.5%, above the origin) and rare (MAF≤ 0.5%, below the origin) coding variants observed in each gene are shown as stacked bars of NS and S variants. (C) Log-likelihood surface of European population growth (r) and population size (Ne) in a demographic model. Colored contours correspond to 2 log-likelihood intervals. The blue point is the maximum likelihood estimate of r and Ne. (D) Per-gene mutation rates with 2 log-likelihood intervals. Horizontal lines are 10th, 50th and 90th mutation rate percentiles. Seven genes on the X chromosome and four genes with low target coverage or yielding too few common variants for inference (ADRB3, CCR5, MIF and PTGER1) were excluded. (E) Proportion of rare cumulative MAF (cMAF) accounted for by SNVs of increasing frequency. (F) Proportion of rare variants in four cMAF ranges falling within the MAF categories shown in (E). The successfully sequenced coding length of each gene (in kb) is overlaid as a gray line. cMAFs in (E) and (F) are for amino acid-changing variants in each gene predicted to be damaging or are evolutionarily conserved (phyloP≥2). Genes in (B), (D) and (F) are ordered by number of rare coding variants per gene and vertical lines correspond to rank deciles.
Table 1.
Comparison of classical population genetic measures of sequence diversity across studies
| Study | No. of genesa | N | Sampleb | Length (Mb) | θπ (×10-4) | θ_W_ (×10-4) |
|---|---|---|---|---|---|---|
| Akey (27) | 132 | 23 | EU | 2.50 | 3.41 | 7.35 |
| SeattleSNPs (28) | 213 | 23 | EU | 7.26 | 6.81 | 6.36 |
| Ahituv (29) | 58 | 757 | EU | 0.13 | 4.32 | 10.11 |
| Current study | 202 | 500c | EU | 0.74 | 3.96 | 8.79 |
| 11,000 | EU | 0.74 | 3.96 | 40.38 | ||
| 500c | SA | 0.69 | 4.04 | 10.67 | ||
| Akey et al. | 132 | 24 | AA | 2.50 | 4.49 | 12.10 |
| Seattle SNPs | 213 | 24 | AA | 7.26 | 8.97 | 10.15 |
| Current study | 202 | 500c | AA | 0.70 | 4.89 | 13.78 |
Fig. 2.
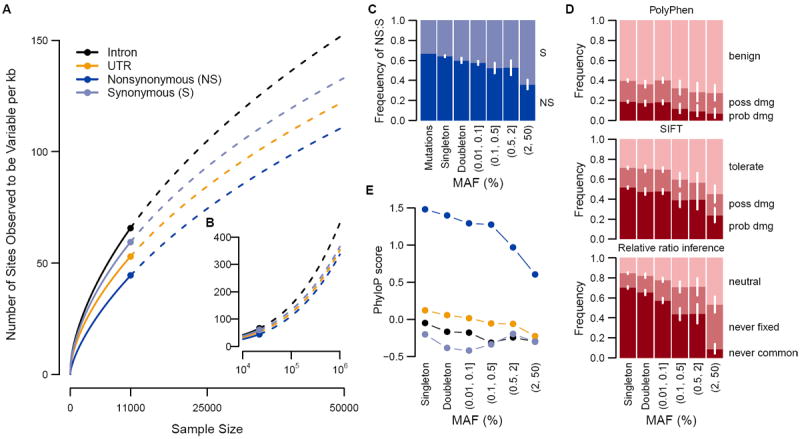
(A,B) Number of variants per kilobase of intronic, UTR, nonsynonymous (NS) or synonymous (S) sequence with sample size increasing to 50,000 (A) and one million (B) Europeans. Observed numbers are given as a dot, solid and dashed lines indicate hyper-geometric expectations and jack-knife projections, respectively. (C) Expected ratios of NS to S variants in the absence of selection and observed ratios for different minor allele frequency (MAF) bins. (D) The proportion of NS variants predicted to be benign, possibly damaging or probably damaging by PolyPhen or SIFT and the proportion of NS variants that is neutral, deleterious such that they will never become common (MAF >5%) or never be fixed in Europeans as predicted by the relative ratios of NS:S variant abundances observed at different MAF (2). (C,D) 95% confidence intervals are represented by white lines. (E) phyloP score for intronic, UTR, NS and S variants for different MAF bins.
These patterns are at odds with notions that human genetic diversity can be summarized by use of an effective population size (Ne) of 10,000 individuals (14). An Ne of 10,000 individuals is predictive of the average pairwise differences between human sequences (θπ, Table 1), and is reflective of our emergence from a small population in Africa (15). However, the excess of rare variants observed here (i.e.θW ≫ θπ) is a signature of the rapid growth and large population sizes that typify more recent human demographic history (8). When we fit a demographic model to the four-fold degenerate synonymous (S) variants in Europeans, we obtained a maximum-likelihood estimate for a recent growth rate of 1.7% (95% confidence interval [CI]=1.2%–2.3%), and a recent European effective population size of 4.0 million (95% CI=2.5–5.0 million; Fig. 1C).
Taking advantage of the unprecedented size of this study for population genetics inference (8, 16), we estimated mutation rates for each gene (Fig. 1D, (13)) and obtained a median estimate of 1.38×10−8 per bp per generation with 90% of estimates falling between 1.7×10−9 and 2.4×10−8. Incorporating singleton discovery false negative rates from 2–8% resulted in median estimates no greater than 1.45×10−8. These population-genetic-based rate estimates are similar to recent pedigree-based mutation rate estimates of 1.36×10−8 per bp per generation (17) and 1.17×10−8 per bp per generation (13, 18). Further, these data reject a model of uniform mutation rates across genes (p<2×10−8) and show synonymous mutation rates are correlated with the number of NS rare variants (p=0.04) and GC content (p<2.4×10−9) (13).
The excess of rare variants observed in coding regions is also due to an abundance of nonsynonymous (NS) variants segregating at low frequencies that are not seen at more common variant frequencies as a result of purifying selection. Summing across all frequencies of variant sites, S and intronic variants occurred more frequently (~70 variants per kb each) compared to UTR and nonsynonymous (NS) variant sites (~55 and ~45 per kb of UTR or NS sequence, respectively, Fig. 2A). Yet, examining the abundance of rare variants across functional categorizations of variant sites reveals little difference among classes when minor allele count is low (Fig. 1A). These patterns are likely due to an equal input of mutations for each category followed by purifying selection preventing deleterious NS and UTR variants from reaching higher frequencies (13, 19). The ratio of NS:S in singletons is close to that expected amongst new mutations and then decreases with increasing frequency (Fig. 2C). Using the approach of (2) we estimate that while ~70% of all NS singletons in our sample are sufficiently deleterious that they will never reach frequencies >5%, only 13% of new NS mutations appear so deleterious that they would not be observed even as singletons in a sample of this size (13), putting an upper bound on the frequency of dominant lethal mutations (15). The output of functional prediction algorithms (Fig. 2D, 2E) also suggest that rare variants are enriched for damaging variants.
On average, each subject carried a rare minor allele at 0.02% of all NS sites, of which ~56% are expected to be deleterious enough to never be fixed. Over 0.3% of sequenced subjects carried at least one mutation reported to be a dominant cause of disease (table S6, (13)). We also identified variants at 0.5%<MAF≤2%, the so-called goldilocks variants (20), in that they would be common enough to be detected in large population samples and rare enough to be enriched for variants under purifying selection (Fig. 2C-E). In the European sample, we observed 105 amino acid-changing variants in 73 genes falling within this frequency range. Half of these were predicted to be functionally damaging, relative to 31% of more common coding SNVs (>2%) and 65% of singletons. By comparison, we found 210 goldilocks variants in African Americans and 132 in South Asians, supporting the value of non-European samples for the genetic analysis of complex traits (21).
Rare variants can be tested in aggregate for an association with disease (6), where the power of the test is strongly correlated with the cumulative minor allele frequency (cMAF) of potentially deleterious SNVs within each gene (Figs. 1E, 1F, S4, S5). 37% of genes had cMAFs >0.5% of rare alleles predicted to be deleterious. We tested associations of common variants individually and rare coding variants in aggregate with the diseases represented in this study (13). When possible, we matched controls with cases using genome-wide genetic similarity. Nevertheless, type 1 error rate inflation consistent with effects of population stratification was observed (table S7 and fig. S6) and was worse for rare variant tests. There were no statistically significant rare variant associations, and thus no compelling evidence connecting any genes with the studied diseases. Of 13 more closely examined genes reported to be associated with six of the diseases investigated (table S8, (22)), only the association of rare variants in IL6 with multiple sclerosis was noteworthy (OR=12, p=0.007; table S9).
Because rare variants are typically the result of recent mutations, they are expected to be geographically clustered or even private to specific populations. Using a measure of variant sharing between two samples (7), we found that for common variants, any two European populations appear to be panmictic, while for rare variants, European populations show lower levels of sharing (fig. S7). In general, the level of sharing depends on geographic distance, with the dependence increasing substantially with decreasing allele frequency (fig. S8). The Finnish population shows substantially lower levels of sharing with other European populations than predicted by geographic distance, consistent with hypotheses of a historical Finnish demographic bottleneck (23). Levels of rare variant sharing are even lower when comparing populations from distinct continents. Thus catalogs of rare variants will need to be generated locally across the globe (7, 24).
We found substantial variation in the total abundance of variants across populations, even within Europe (Figs. 3, S7D), likely due to demographic history. In particular we observed a north-south gradient in the abundance of rare variants across Europe, with increased numbers of rare variants in Southern Europe and a very small number of variants among Finns, who had about one third as many variants as southern Europeans. The gradient is consistent with observed gradients in haplotype diversity (25) and a Finnish ancestral bottleneck (23). Association mapping approaches based on rare variant diversity levels will be more susceptible to subtle effects of population stratification (26) and more likely to result in false positive disease associations.
Fig. 3.
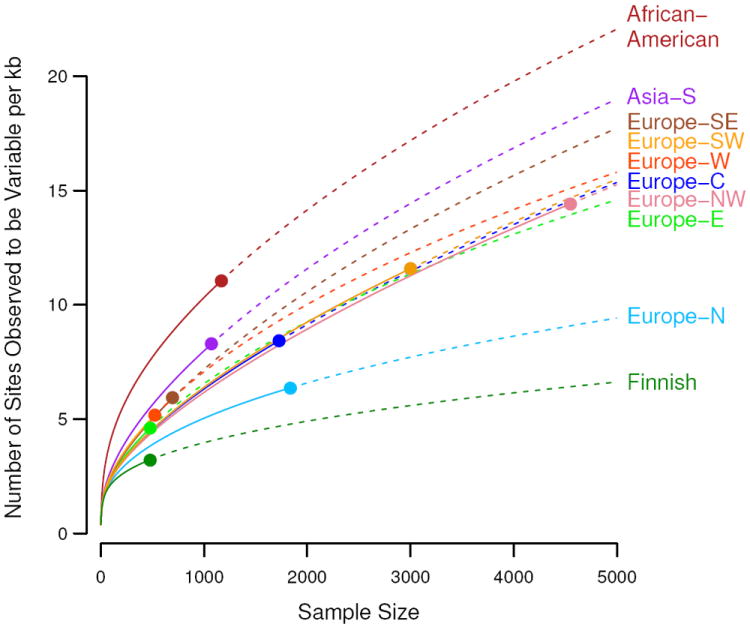
Number of variants per kilobase of sequence with sample sizes increasing to 5,000 people for multiple populations. Observed numbers are given as a dot, solid and dashed lines indicate hyper-geometric expectations and jack-knife projections, respectively.
To evaluate our conclusions relative to the rest of the genome, we compared the NS:S variant ratios of the sequenced genes to the entire coding genome within the low coverage CEU 1000 Genomes Project data. The average per subject NS:S ratio from our 202 genes was 0.54, while all other genes had an average ratio of 0.94 (p<10−15, fig. S9). By comparison, genes found in OMIM and the genome-wide association studies catalog (22) had average ratios of 0.75 and 0.78, respectively. This implies that the genes in this study are under stronger purifying selection, consistent with their choice as drug targets and importance to human health. Hence, our results cannot be simply extrapolated to the whole exome. Instead, it is likely that our results underestimate the average genetic diversity that will be found in more typical human gene coding regions, primarily regarding the amount of NS variation.
This large-scale resequencing study provides a unique description of variation for 202 drug target genes and insight into the very rare spectrum of variation. Although sequencing error might be a concern, we show that the error rates in this study are low (table S3). Another caveat is that our inference of demographic parameters and mutation rates ignores the effects of background selection on synonymous variants. Despite these caveats, the results show there is an abundance of rare variation in human populations, and that surveys of common variants are only observing a small fraction of the genetic diversity in any gene. Further, as we observe, much of the rare variation in coding regions appears to be functional and may be crucial for yielding insights into the genetic basis of human disease. Because the genes studied are related to drug discovery, development or repositioning efforts this work has potential to help investigate target biology and drug response.
Supplementary Material
Data table 1
Data table 2
Data table 3
Supplementary Materials
Acknowledgments
We thank GSK colleagues who advised on the selection of genes and collections especially W. Anderson, L. Condreay, P. Agarwal, A. Hughes, J. Rubio, C. Spraggs and D. Waterworth, the sample preparation team especially J. Charnecki, M. E. Volk, D. Duran., D. Briley and K. King for data preparation, A. Slater for subject selection and preparation of genome-wide genotype data, E. Woldu for capillary sequencing, A. Nelsen, S. Buhta-Halburnt, L. Amos, J. Forte for consent review, M. Lawson for assistance in running the association analyses, J. Brown for discussions about gene feature analyses and S. Ghosh for providing reviews of the manuscript and G. Tian, H. Jiang, Z. Su, X. Sun, L. Yang and X. Zhang at BGI for sequencing. We acknowledge the work of collaborating clinicians and researchers who contributed to recruiting and characterizing subjects (13). J.N. and D.W. were supported by a Searle Scholars Program award to J.N. D.K. is supported by an NIH Genome Analysis training grant. All variants described in this study have been submitted to dbSNP; accession numbers are included in database S2. Subject level sequence data for CoLaus and LOLIPOP studies are available in dbGaP. Additional subject level sequence data can be made available upon request from the authors under a Data Transfer Agreement for the purpose to understand, assess or extend the conclusions of this paper.
Reference List
- 1.Pritchard JK. Am J Hum Genet. 2001;69:124. doi: 10.1086/321272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kryukov GV, Pennacchio LA, Sunyaev SR. Am J Hum Genet. 2007;80:727. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marth GT, et al. Genome Biol. 2011;12:R84. doi: 10.1186/gb-2011-12-9-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manolio TA, et al. Nature. 2009;461:747. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eichler EE, et al. Nat Rev Genet. 2010;11:446. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asimit J, Zeggini E. Annu Rev Genet. 2010;44:293. doi: 10.1146/annurev-genet-102209-163421. [DOI] [PubMed] [Google Scholar]
- 7.Gravel S, et al. Proc Natl Acad Sci U S A. 2011;108:11983. doi: 10.1073/pnas.1019276108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coventry A, et al. Nat Commun. 2010;1:131. doi: 10.1038/ncomms1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohta T. Nature. 1973;246:96. doi: 10.1038/246096a0. [DOI] [PubMed] [Google Scholar]
- 10.Williamson SH, et al. Proc Natl Acad Sci U S A. 2005;102:7882. doi: 10.1073/pnas.0502300102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muller HJ. Am J Hum Genet. 1950;2:111. [PMC free article] [PubMed] [Google Scholar]
- 12.Russ AP, Lampel S. Drug Discov Today. 2005;10:1607. doi: 10.1016/S1359-6446(05)03666-4. [DOI] [PubMed] [Google Scholar]
- 13.See supporting material at Science online. 2012.
- 14.Jobling MA, Hurles M, Tyler-Smith C. Human Evolutionary Genetics: Origins, Peoples and Disease. Garland Science; 2003. [Google Scholar]
- 15.Livi-Bacci M. A concise history of world population. 2. Wiley-Blackwell; 2007. pp. 1–250. [Google Scholar]
- 16.Wakeley J, Takahashi T. Mol Biol Evol. 2003;20:208. doi: 10.1093/molbev/msg024. [DOI] [PubMed] [Google Scholar]
- 17.Awadalla P, et al. Am J Hum Genet. 2010;87:316. doi: 10.1016/j.ajhg.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conrad DF, et al. Nat Genet. 2011;43:712. doi: 10.1038/ng.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Messer PW. Genetics. 2009;182:1219. doi: 10.1534/genetics.109.105692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Price AL, et al. Am J Hum Genet. 2010;86:832. doi: 10.1016/j.ajhg.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kotowski IK, et al. Am J Hum Genet. 2006;78:410. doi: 10.1086/500615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hindorff LA, et al. Proc Natl Acad Sci U S A. 2009;106:9362. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salmela E, et al. PLoS One. 2008;3:e3519. doi: 10.1371/journal.pone.0003519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bustamante CD, Burchard EG, De la Vega FM. Nature. 2011;475:163. doi: 10.1038/475163a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lao O, et al. Curr Biol. 2008;18:1241. doi: 10.1016/j.cub.2008.07.049. [DOI] [PubMed] [Google Scholar]
- 26.Lander ES, Schork NJ. Science. 1994;265:2037. doi: 10.1126/science.8091226. [DOI] [PubMed] [Google Scholar]
- 27.Akey JM, et al. PLoS Biol. 2004;2:e286. doi: 10.1371/journal.pbio.0020286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.SeattleSNPs. 2012 http://pga.gs.washington.edu.
- 29.Ahituv N, et al. Am J Hum Genet. 2007;80:779. doi: 10.1086/513471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data table 1
Data table 2
Data table 3
Supplementary Materials