Distinct Conformational Spectrum of Homologous Multidrug ABC Transporters (original) (raw)
. Author manuscript; available in PMC: 2016 Mar 3.
Published in final edited form as: Structure. 2015 Feb 5;23(3):450–460. doi: 10.1016/j.str.2014.12.013
Abstract
ATP-binding cassette (ABC) exporters are ubiquitously found in all kingdoms of life and their members play significant roles in mediating drug pharmacokinetics and multidrug resistance in the clinic. Significant questions and controversies remain regarding the relevance of their conformations observed in X-ray structures, their structural dynamics, and mechanism of transport. Here we used single particle electron microscopy (EM) to delineate the entire conformational spectrum of two homologous ABC exporters (bacterial MsbA and mammalian P-glycoprotein) and the influence of nucleotide and substrate binding. Newly developed amphiphiles in complex with lipids that support high protein stability and activity enabled EM visualization of individual complexes in a membrane-mimicking environment. The data provide a comprehensive view of the conformational flexibility of these ABC exporters under various states and demonstrate not only similarities but striking differences between their mechanistic and energetic regulation of conformational changes.
INTRODUCTION
ATP-binding cassette (ABC) transporters constitute a large family of integral membrane proteins that utilize the energy of ATP hydrolysis to translocate ions, lipids, nutrients and drugs across lipid bilayers. Based on the directionality of transport, they are classified as either exporters or importers, with the former found in all living species and the latter only reported in prokaryotic systems (Dassa, 2011). Many ABC exporters are promiscuous and bind a wide array of structurally unrelated compounds, in contrast to most importers that are functionally dependent on peripheral binding proteins for specific substrate recognition (Locher et al., 2002; Oldham et al., 2007). ABC exporters are medically important since their members contribute to antibiotic or antifungal resistance of human pathogens, the development of multiple drug resistance (MDR), and several human genetic disorders due to protein dysfunctions. A prominent example is P-glycoprotein (P-gp) that affects the pharmacokinetics of numerous drugs and is implicated in MDR of many human cancers, HIV, and epileptic diseases (Eckford and Sharom, 2009; Giacomini et al., 2010).
ABC exporters share a common architecture including a minimum of two transmembrane domains (TMDs) and two highly conserved nucleotide binding domains (NBDs). The four core domains are commonly either coexpressed as a dimer of TMD-NBD halves, or fused into a single polypeptide chain (Figure S1). The TMDs form the translocation pathway and determine the substrate specificity, whereas the NBDs are thought to associate upon ATP binding and dissociate driven by ATP hydrolysis. The ATP binding and hydrolysis steps are coupled to significant conformational rearrangements of the TMDs opening towards the cytoplasm (also termed inward-facing: IF) or the periplasm (outward-facing: OF) (Higgins and Linton, 2004). The alternate access presentation of membrane openings of ABC transporters and other types of membrane pumps has long been used to explain the substrate translocation (Jardetzky, 1966). However, despite a wealth of biochemical and structural data obtained on these transporters from decades of research, many aspects of the translocation process such as the spectrum of conformational dynamics, the impact of substrate binding, and how the NBD and TMD movements are coupled remain to be fully elucidated.
Previous high resolution X-ray structural studies revealed large conformational variability within the group of ABC exporters, including prokaryotic MsbA (Ward et al., 2007), Sav1866 (Dawson and Locher, 2006), TM287/288 (Hohl et al., 2012; Hohl et al., 2014), and eukaryotic P-gp (Aller et al., 2009; Jin et al., 2012; Ward et al., 2013), ABCB10 (Shintre et al., 2013), and ABCB homologues (Kodan et al., 2014; Lee et al., 2014; Srinivasan et al., 2014) (Figure S1). Notably, most of these structures have been solved in IF states both in the absence and the presence of nucleotide, and a range of amplitudes of the NBD separation has been observed in different species. X-ray structures of OF states have only been obtained for two prokaryotic proteins with bound nucleotides (Sav1866 and MsbA) (Dawson and Locher, 2006; Ward et al., 2007). Most recently, a novel nucleotide-bound, occluded outward conformation has been reported for an antibacterial peptide ABC exporter (McjD) (Choudhury et al., 2014). This newly solved structure is proposed as a transition intermediate between previously reported inward-open and outward-open states (Figure S1), providing further steps along the conformational pathway of ABC exporters. The available structures are commonly used as a framework to describe the trajectory of a “universal ABC transporter”. As the data originates from multiple species it is not clear to what extent the findings can be generalized (Rees et al., 2009). Also, the large NBD separation observed in some IF X-ray structures and its physiological relevance are under much dispute, as this conformation could arise from crystallographic constraints, the use of detergents, or the absence of nucleotides and ligands (Gottesman et al., 2009; Jones and George, 2014). Adding to this complexity, measurements from different methods, including cross-linking (Loo et al., 2010; Loo and Clarke, 2014), fluorescence or luminescence resonance energy transfer (FRET or LRET) (Borbat et al., 2007; Cooper and Altenberg, 2013; Qu and Sharom, 2001; Verhalen et al., 2012), electron spin resonance (EPR) spectroscopy (Borbat et al., 2007; van Wonderen et al., 2014; Wen et al., 2013; Zou et al., 2009), or mass spectrometry (Marcoux et al., 2013), are not always in agreement, which has resulted in significant debate in the field.
Here we used single particle electron microscopy (EM) to directly visualize the conformational spectra of two homologous (~30% sequence identity) ABC exporters: bacterial MsbA (E. coli) and the mammalian P-gp (M. musculus) (Figure S2). MsbA is a homodimer of TMD-NBD halves and reported to function as a lipid A and phospholipid flippase (Doerrler et al., 2004; Zhou et al., 1998) and also to transport multiple drugs (Eckford and Sharom, 2008; Reuter et al., 2003). P-gp is a monomer with two pseudosymmetric halves of TMD-NBD fused together by a flexible linker of ~70 amino acids (Figures S1 and S2) (Ward et al., 2013). Newly developed amphiphiles (Tao et al., 2013) in complex with lipids that support high ATPase activity and improve protein stability enabled EM visualization of individual complexes of both transporters in a lipid-bilayer mimicking environment. EM imaging and analysis using an unbiased approach to 3D model construction was used to delineate the entire conformational spectrum of P-gp and MsbA and the influence of nucleotide and substrate binding. Our analysis reveals striking differences between the two transporters regarding the effect of binding nucleotides and substrates on their conformational changes and the range of NBD separation across the entire structural spectrum. Overall, the data provide a comprehensive view of the conformational flexibility of two homologous ABC transporters and add essential new insights into the mechanistic understanding of these machines.
RESULTS
Stabilization and EM imaging of ABC transporters in a lipid environment
We have recently developed novel β-sheet peptide (BP) assemblies for stabilizing integral membrane proteins and demonstrated their advantages in maintaining the function and providing well defined structures of MsbA for EM-based single particle analysis (Tao et al., 2013). For detailed conformational analysis in this report, we prepared MsbA and P-gp in BP solutions with exogenous lipids added to compose a bicelle (Zhang et al., 2011) or nanodisc (Nath et al., 2007) bilayer mimicking system. Thus the single protein molecules are surrounded by a thin layer of BP and lipid, more amenable to EM imaging than large micelles or vesicles resulting from commonly used detergent and lipid mixtures. For P-gp, the presence of lipid is a prerequisite for drug-stimulated ATPase activity and enhances its thermostability (Bai et al., 2011). The unprocessed EM micrographs and 2D class averages (see below) clearly show individual particles in multiple conformations for both MsbA and P-gp (Figures 1 and S3). Comparing samples prepared with and without added exogenous lipids, we note that upon inclusion of lipids the EM densities near the transmembrane region of MsbA and P-gp appear enlarged, suggesting that lipids preferentially associate with the hydrophobic TMDs as expected (Figure 1A vs. B, C).
Figure 1.

Unprocessed micrographs (top) and class averages (bottom) of MsbA (A, B) and P-gp (C). Addition of exogenous lipid to MsbA (B) and P-gp (C) in BP detergent solution enlarges the electron density in proximity to the TMD region (white arrows) compared to MsbA prepared without lipids (A). Various conformations of the transporters can be readily identified in the unprocessed micrographs shown in the top panels (OF: dotted circle, IF: double circle, and NBD wide open: full circle) and in the 2D class averages shown in the bottom panels. Scale bar corresponds to 50 nm for raw micrographs and 10 nm for class averages.
We further used specific protein labeling on MsbA, to unambiguously identify the location of the NBDs and the extracellular site in 2D images (Figure 2). Two single cysteine (Cys) mutants, one in the extracellular loop 2 (Q166C) and the other in the NBD near the cytoplasmic C-terminus (T561C), were selected for labeling with biotin C2 maleimide for subsequent binding of the 60 kDa tetrameric NeutrAvidin protein (Hiller et al., 1987). Using EM we observed NeutrAvidin binding to either one or two copies of the periplasmic Q166C, or the cytoplasmic T561C, respectively, both in the OF (left panels) or in different IF conformations (middle and right panels) (Figure 2). The data confirm assignments of the TMDs and NBDs in both IF and OF orientations of MsbA in the 2D images. Binding of NeutrAvidin to the T561C NBD greatly diminished ATPase activity, possibly due to impaired NBD mobility and/or steric hindrance of this large domain (Figure S4) and, consistent with the latter, no OF particles labeled with NeutrAvidin-labeled were observed for this mutation. For detailed conformational analysis we have therefore focused our studies on wild type and unlabeled transporters.
Figure 2.
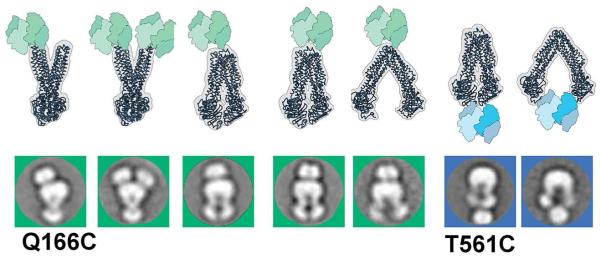
NeutrAvidine protein labeling of MsbA particles. Two single Cys mutants of MsbA (periplasmic Q166C and cytoplasmic T561C) were labeled with the tetrameric NeutrAvidine biotin binding protein. Shown are 2D class-averages of the complexes (Q166C images in green squares; T561C in blue squares) and corresponding 3D models for comparison. The NeutrAvidine labeling was observed for either one or two halves of MsbA, and in both IF and OF conformations. The position of NeutraAvidine densities further confirms the assignment of MsbA orientations.
Visualizing the conformational spectrum of MsbA and P-gp
To determine the relative population of individual conformations of MsbA and P-gp we developed a standardized single particle analysis routine, incorporating reference free alignment and classification protocols (Hohn et al., 2007; Lander et al., 2009). This approach sorts the isolated particles into homogenous class averages based on similarity. Averaging of aligned particles significantly increases the signal to noise ratio of each class compared to individual particles facilitating interpretation of the conformational state. To distinguish between different conformations vs. relative orientations, we also used a tilt pair approach (Random Conical Tilt, or RCT (Radermacher et al., 1987)) to reconstruct 3D maps. RCT is an established procedure that uses alignment and classification parameters of untilted particle images to determine the relative orientation of their respective partners in the paired tilted particle image and thus directly reconstruct a 3D model of an individual particle class, requiring no a priori information about the particle conformation. This method has been shown to be well suited to the characterization of small flexible proteins (Campbell et al., 2014; Chen et al., 2010; Lyumkis et al., 2013). From multiple 3D maps (25 to 40 Å resolution range) generated from the RCT analysis, we determined the inter NBD separation by comparison with a reference library of models created by linear interpolation of the atomic coordinates between the intermediate structures solved by crystallography (Ward et al., 2007) (Figures 3 and 4). The fit of the EM volume maps to the X-ray structure-derived models verifies that, to the resolution of interest in interpreting these structures, there are no significant structural artifacts introduced by the EM methods used here.
Figure 3.
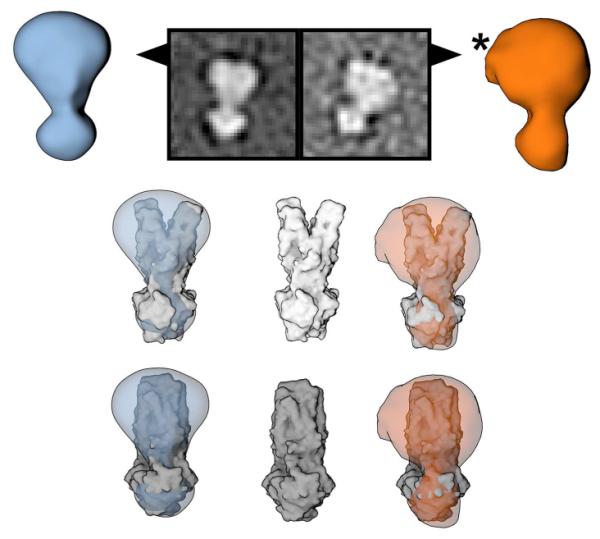
OF conformations of MsbA (blue) and P-gp (orange). Top row: 3D EM models derived from the RCT analysis and corresponding 2D class averages. The EM densities are consistent with the published OF X-ray structures of MsbA and SAV1866 (middle row), and the occluded OF structure of McjD (bottom row). In OF states the NBDs are associated creating a characteristic arrow head shape, which can be unambiguously identified in 2D averages and raw micrographs (Figure 1) and clearly distinguished from IF conformations. Superposition of EM densities and low pass filtered X-ray structures reveals additional densities in the transmembrane region. The asterisk marks an additional density attributed to tightly associated lipids, which is apparent in both 2D class averages and 3D reconstitutions.
Figure 4.

Conformational spectra of IF MsbA and P-gp. (A) X-ray structures of MsbA and P-gp previously solved in various IF conformations. The color scheme is the same as in Figure S1. Distances between T561 Cα positions of E. coli MsbA and its equivalent positions (marked by red dots) in V. cholerae MsbA and P-gp structures are shown above the PDB ID code. 3G61 included two P-gp molecules solved with slightly different NBD separations. (B) Surface representations of reference models created by linear interpolation of MsbA X-ray structures that are used to categorize the NBD spacings in the structures displayed in (C and D). (C) First row, shows experimental IF 3D-densities from APO MsbA arranged by ascending NBD separation using the distance between T561 as an internal standard. Second row shows superposition of EM density onto X-ray models to emphasize the correspondence between the two. Differences in densities surrounding the TMD come from the detergent/lipid. (D) Experimental maps of P-gp (first row) and superposition onto the same reference library (second row) shown in B. (E) Statistical occurrence of particles contributing to individual IF categories for nucleotide-free, APO MsbA (light blue), ATP-bound MsbA (dark blue), and nucleotide-free P-gp (orange). For MsbA the entire conformational spectrum is occupied, and no significant differences in individual IF distributions within the IF population are observed between the APO and ATP sample. The spectrum of P-gp conformations is clustered in the region representing NBD separation between 50 and 75 Å; no structures outside of this range are detected.
The OF particles assume a characteristic arrow head shape in the 2D class averages, readily distinguishable from dissociated NBD shapes of IF particles (Figure 1). The OF particles are confirmed by examination of the 3D density maps corresponding to the 2D class averages (Figure 3) and comparison of the 3D EM maps to the published X-ray structures (Choudhury et al., 2014; Dawson and Locher, 2006; Ward et al., 2007). Because a side view image of the ABC transporter in IF conformations, rotated by 90°, might have an appearance similar to the OF conformation, we cannot generally exclude the possibility of confusing different views and different conformers. However, our 3D analysis of arrow head shaped particles always corresponded to OF conformations and never to IF conformations of the transporters. Of note, our OF EM models cannot distinguish between the outward-open (shown in MsbA and Sav1866) and outward-occluded (McjD) conformations which have overall similar architecture with an almost identical NBD dimer shape (Figure 3, middle vs. bottom row). Therefore, we refer to both conformations as OF.
Nucleotide-dependent conformational changes in MsbA
We observed a continuum of IF conformations for nucleotide-free (APO) MsbA, flanked by conformations representing the two published IF crystal structures (PDB ID codes: 3B5W and 3B5X) (Figure 4A-C). The measured NBD distance, using the amino acid T561 located at the center tip of the NBDs as a reference, is 37 and 87 Å in 3B5X and 3B5W, respectively (Figure 4A). The majority of particles display NBD distances ranging between 45 and 60 Å (Figure 4E, light blue bar), suggesting that these are the preferred conformations in nucleotide-free solution. We also consistently observed a minor population, between 1 to 3%, of OF particles in nucleotide-free samples (Figure 5A and Table S1). This observation is unexpected since nucleotide binding is thought to be a prerequisite to the induction of OF conformation according to the prevalent ATP switch model (Dawson and Locher, 2006; Higgins and Linton, 2004; Ward et al., 2007). The possibility of co-purification of endogenous nucleotide and consequent trapping of some MsbA molecules in the OF conformation is low since the protein was purified through multiple columns and then dialyzed with nucleotide-free buffers for extended times. We used a highly sensitive luciferin-luciferase bioluminescence assay (Ito et al., 2004) to measure ATP in purified MsbA samples, and the calculated ATP to MsbA ratio is neglegible (< 1:20,000, within experimental error compared to blank controls).
Figure 5.
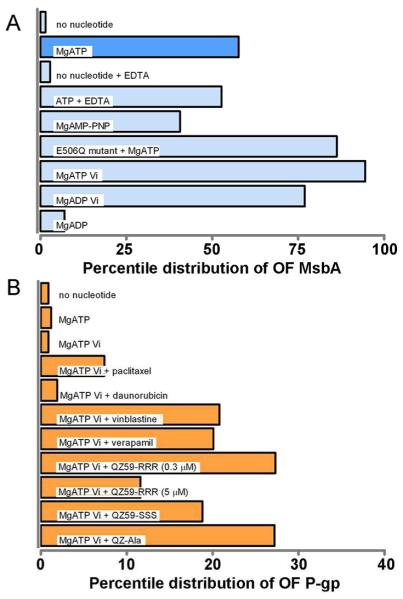
Statistical assessment of MsbA and P-gp conformations for different states of the ATP hydrolysis cycle. (A) Compared to the APO state, nucleotide binding to MsbA significantly increases the population of OF particles under conditions corresponding to the presence of ATP, or capturing the ATP-bound pre-hydrolysis state (AMP-PNP, ATP + EDTA in the absence of Mg2+, or the catalytically deficient E506Q mutant), the hydrolytic transition state (AT/DP plus Vi), and the post hydrolysis state (ADP). A small percentage of OF particles was observed even in the absence of nucleotide. (B) OF conformation of P-gp co-induced by ligands and ATP plus Vi. Ligands and concentrations are detailed in the text. A concentration-dependent stimulation of ATPase activity for each compound is shown in Figure S5. In the absence of Vi, a, residual percentage of OF conformation (< 3%) was detected in either the presence or absence of nucleotides and/or ligands (see Table S1).
To determine conformational effects of nucleotide binding we exposed MsbA to MgATP (1 mM) and repeated the 2D and 3D EM analyses. A brief incubation of MsbA with MgATP for one minute at room temperature (RT) increased the percentage of particles in the OF conformation to over 58% (Figure 5A). Within the 42% molecules remaining in the IF conformations, class averages similar to those seen in the nucleotide–free conformations (Figure 4C) were observed, and the relative percentile distribution of each IF conformer did not significantly differ compared to the nucleotide-free condition (Figure 4E, dark blue vs. light blue bars). Based on the measured ATPase activity of 1.3 ± 0.1 μmol ATP/min/mg of MsbA at RT and an MsbA concentration of 0.01 mg/mL used for the EM experiments, a one-minute incubation corresponds to an average of about 170 cycles of ATP hydrolysis per MsbA molecule and the consumption of less than 2% of total ATP. Thus, the occurrence of OF (58%) and IF (42%) MsbA conformers is interpreted as a representative snapshot of various states occurring during ATP binding and hydrolysis.
We subsequently determined MsbA conformational changes through discrete states of an ATP binding and hydrolysis cycle, including the ATP-bound pre-hydrolysis state, an intermediate transition state, and the ADP-bound post-hydrolysis state. Since the range of IF MsbA conformers did not change in response to nucleotide binding (Figure 4E), we present only the fraction of OF particles in subsequent studies to simplify data analysis.
First, to capture an ATP-bound, pre-hydrolysis state, we analyzed several conditions, including MsbA incubations with a non-hydrolyzable ATP analog AMP-PNP, depletion of Mg2+, an essential cofactor for ATP hydrolysis, and the use of a catalytically inactive MsbA mutant, E506Q, that permits ATP binding but abolishes ATP hydrolysis (Cooper and Altenberg, 2013; Schultz et al., 2011). The results show that all conditions mimicking the ATP-bound state resulted in an increased population of OF particles compared to the APO state (Figure 5A, and Table S1), indicating that the OF state is populated by nucleotide binding. Specifically, binding of MgAMP-PNP or ATP in the absence of Mg2+ (but inclusion of 10 mM EDTA) resulted in 41% and 53% of OF particles, respectively, somewhat lower as compared to the 58% OF population observed under MgATP hydrolysis conditions. One of the most pronounced effects was seen with the catalytic carboxylate mutant E506Q which resulted in a total of 86% OF particles in the presence of MgATP.
Next, to capture MsbA conformations in an intermediate transition state we pretreated MsbA with MgATP and orthovanadate (Vi). Under this condition, ATP is hydrolyzed and Vi replaces the dissociated phosphate (Pi) to form a tightly bound MgADP•Vi complex (Senior, 2011; Urbatsch et al., 2003). As a result, an almost saturated numbers of OF particles (95%) were observed (Figure 5A). High numbers of OF particles (77%) were also obtained by addition of MgADP plus Vi (Figure 5A), suggesting that MsbA can also assume the vanadate-induced MgADP•Vi transition state in the absence of hydrolysis. In comparison, binding of MgADP alone, which represents a post-hydrolysis state, only slightly increased the number of observed OF particles (7%) compared to the APO state (Figure 5A). Overall, our data indicate the presence of heterogeneous and inter-changeable conformations for MsbA and that nucleotide binding shifts the conformational distribution from the IF to the OF state, with about 50% in the OF state under steady state MgATP hydrolysis conditions, supporting previous data from FRET, LRET, and EPR spectroscopy studies that measure the average distances or the distance range between labeled residues (Borbat et al., 2007; Cooper and Altenberg, 2013; Zou et al., 2009).
Conformational spectrum of P-gp differs from MsbA
We applied the approach described above to conformational analysis of P-gp and observed significant differences from the results obtained for MsbA. Compared to MsbA, the NBD separation in IF P-gp conformations is much more constrained with NBD distances ranging between 50 and 75 Å (Figure 4D-E). The more limited conformational spectrum of P-gp compared to MsbA is also directly apparent from a comparison of 2D class averages (Figure 1C). For P-gp we did not detect extremely wide-open NBDs (up to 85 Å apart) or closed IF conformations (less than 40 Å apart) as observed in MsbA crystal structures and EM maps (Figures 4A and 4C). The measured range of NBD distance between the MsbA T561 analogous residues N607 and T1252 is very similar to the reported X-ray structures of P-gp that were solved in IF conformations (3G61, 46 and 52 Å; 4KSB, 66 Å; 4F4C, 72 Å) (Aller et al., 2009; Jin et al., 2012; Ward et al., 2013) (Figure 4A). Similar to MsbA, we also detected a minor population (< 3%) of OF particles for P-gp in the absence of nucleotide (with or without ligands, Table S1), suggesting a dynamic equilibrium of various conformers, including the OF state, and that this could be a common theme for the function of ABC transporters.
P-gp prevails in IF states in the presence of nucleotide
Contrary to observations for MsbA, where binding of nucleotides substantially increased the OF population, incubation of P-gp with either MgATP, or MgAMP-PNP, and even MgATP plus Vi, produced no apparent increase of OF particles compared to nucleotide-free conditions (Figure 5B and Table S1). A possible explanation is that a high energy barrier exists to prevent a change in conformational state from IF to OF, and that nucleotide binding is insufficient to induce this conformational change in P-gp compared to MsbA. A similar conclusion has been reached in a recent report using ion-mobility mass spectrometry to probe P-gp conformational changes (Marcoux et al., 2013), where induction of OF conformations required synergy between a substrate and nucleotides.
Compared to MsbA, P-gp displays low basal ATPase activity that can be substantially stimulated by transport substrates and ATPase modulators (Figure S5). Binding of these compounds to P-gp may therefore lower the activation energy barrier for NBD dimerization and transition to the OF state (Higgins and Linton, 2004). To test this hypothesis we conducted experiments on P-gp samples preincubated with anti-cancer drugs (paclitaxel, daunorubicin and vinblastine) that are known P-gp substrates, or with P-gp modulators, including verapamil, the small cyclic peptides QZ59-RRR and QZ59-SSS that were previously co-crystallized with P-gp (Aller et al., 2009), and a new QZ59-SSS derivative QZ-Ala (Figure 5B). As shown in Figure S5, all compounds showed stimulation of the basal ATPase activity (~0.05-0.10 μmol ATP/min/mg P-gp) in a concentration dependent manner. The maximal ATPase activity of P-gp in the presence of verapamil and QZ59-RRR, both at RT and 37 °C, was comparable or even higher than the activity of MsbA. Among these compounds, QZ59-RRR and vinblastine stimulated P-gp’s ATPase activity at low concentrations but inhibited at higher concentrations (Figures S5C and S5E), unlike others that enhanced the activity but did not inhibit with an increase of concentration.
All compounds yielded higher percentages of OF conformations but only when pre-incubated in combination with MgATP plus Vi. Figure 5B lists results for a parallel comparison of each sample after incubation for 15 minutes at RT. Each compound was tested at a selected concentration (daunorubicin, 100 μM; paclitaxel, 100 μM; vinblastine, 10 μM; verapamil, 100 μM; QZ59-RRR, 5 μM; QZ59-SSS, 5 μM; and QZ-Ala, 2 μM) that conferred the highest level of ATPase activity stimulation relative to basal activity (Figure S5). Notably, pre-incubation of P-gp with compound and MgATP alone had no effects (Table S1), suggesting that vanadate trapping stabilizes the OF state. The highest population of OF particles (up to 27%) was observed after preincubation with QZ59-RRR and QZ-Ala in combination with MgATP plus Vi (Figure 5B). This is far lower than observed for MsbA, which showed saturation of 95% particles in the OF conformation under conditions of vanadate trapping (after only one minute of preincubation with ATP plus Vi, Figure 5A). We note a correlation between the increased population of OF P-gp with ATPase activity by individual compounds that becomes evident when comparing results using QZ-RRR at two different concentrations; decreasing ATPase activity from 0.3 μM (44 fold) to 5 μM QZ-RRR (10 fold stimulation) (Figure S5) was accompanied with a drop in OF percentage of P-gp from 27% to 12% (Figure 5B). For daunorubicin and paclitaxel, which showed relatively small stimulatory effects for ATP hydrolysis, we detected the smallest percentage of OF P-gp (2% and 7%, respectively). Taken together, our data show that P-gp, unlike MsbA, prevails in IF states in the presence of ATP with or without transport substrates, and that the OF P-gp is only partially stabilized under artificial Vi trapping conditions, clearly demonstrating divergence in the energetic and mechanistic regulation of the two closely related ABC transporter homologs.
DISCUSSION
We have applied single particle EM and multi model analysis to determine the structural conformations of two homologous ABC exporters: the prokaryotic MsbA and eukaryotic P-gp. The development of BP for stabilizing single protein molecule assemblies of MsbA and Pgp in the presence of lipid underpinned our success in visualizing single particles by EM with low background, which allowed us to distinguish conformational states of individual ABC transporter molecules. We characterized the dynamic trajectories of MsbA and P-gp and monitored the conformational changes induced in response to nucleotides and/or substrates. Our data can be interpreted as providing “snapshots” of individual active ABC transporters within an ensemble of molecules, revealing information about conformational flexibility and heterogeneity of these highly dynamic proteins.
Contrary to our EM visualization of a broad spectrum of heterogeneous conformations, high resolution X-ray structural studies of individual ABC transporters have thus far been confined to snapshots of single conformational states. Similarly, recent breakthroughs in EM technologies have culminated in a higher resolution (8 Å) single particle EM structure for a heterodimeric ABC exporter TmrAB (T. thermophilus), which was solved in a nucleotide-free, IF conformation (Kim et al., 2014). In this case, labeling of TmrAB with fragment antigen-binding (Fab) domains helped to overcome the protein size limitation, and possibly constrained the flexible conformations for high resolution imaging. Instead, our studies herein have focused on the changes in conformations of ABC transporters subjected to nucleotide and/or substrate binding using wild-type proteins, avoiding possible interferences by protein engineering and labeling.
Both MsbA and P-gp display a dynamic range of interchangeable IF and OF conformations but substantial differences are revealed between the two closely related ABC transporter homologs (Figure 6). MsbA was captured in both OF and a continuum of IF conformations from wide open to closed NBD states under steady state hydrolysis conditions. Surprisingly P-gp remained predominantly in IF conformations even in the presence of mM concentrations of MgATP, and only accumulated appreciable levels of up to 27% OF when locked in the Vi transition state conformation after exposure to ATP and hydrolysis stimulating substrates. The data indicate a distinct energy landscape of conformations for the two transporters, and imply that the OF state of P-gp, unlike MsbA, is transient (a likely high energy state) even under continuous MgATP hydrolysis conditions when turnover through IF and OF states is fast based on the ATPase activity measurements (Figure S5). There is currently no X-ray structure of P-gp in the OF conformation, and we note not of any eukaryotic ABC transporter to date, albeit several high resolution structures of ABCB10 in IF conformations were solved with bound nucleotides (Shintre et al., 2013), which may indicate a trend of mammalian ABC transporters to preferentially reside in IF conformations even in presence of nucleotide.
Figure 6.

Dynamic conformational changes of ABC transporters under steady-state ATP hydrolysis conditions. (A) Illustration of the prevailing ATP switch model by which ATP binding causes NBD dimerization leading to a conformational switch from IF to OF. Statistical distribution of conformational changes observed by EM for (B) MsbA and for (C) P-gp. In the nucleotide-free state (left) MsbA and P-gp are predominantly in IF conformations sampling a range of conformations with various degrees of NBD separation. Under steady-state MgATP hydrolysis conditions, when the NBDs continuously associate and dissociate, MsbA (B, right) was captured in a nearly equal distribution of IF and OF conformations (~60% to 40%). In contrast, P-gp prevailed in IF conformations (C, right) suggesting that the OF conformation is short-lived, and likely reflects a high energy state that rapidly resets back to IF. In both cases, the OF conformation can be enriched under stabilizing conditions by Vi-induced nucleotide trapping (see Figure 5).
Our observation of IF conformations with wide open NBDs for both MsbA and P-gp substantiates the relevance of previously published X-ray structures (Aller et al., 2009; Jin et al., 2012; Ward et al., 2007; Ward et al., 2013), as our EM structures are not influenced by crystal packing, and were obtained in a bilayer-mimicking environment under a broad range of conditions such as the presence of nucleotides and/or substrates. Our EM data cannot easily reconcile with proposed constant contact models, in which the NBDs remain associated during the catalytic cycle (Gottesman et al., 2009; Jones and George, 2014). Since both MsbA and P-gp can recognize large sizes of substrates (e.g. 1.8 kDa lipid A for MsbA; 4 kDa β-amyloid peptides for P-gp), the wide opening of NBDs and the TMD pocket may be necessary to better accommodate substrate binding prior to translocation.
Interestingly, we found that the amplitude of NBD separation in the IF states is more constrained in P-gp than MsbA at both ends of its conformational spectrum (Figure 3D). A plausible explanation for this difference may be related to the presence of a short flexible linker connecting the two homologous halves in the single polypeptide chain of P-gp (Figures S1 and S2). This linker region contains phosphorylation sites with no defined role in function of P-gp (Germann et al., 1995; Goodfellow et al., 1996; Szabo et al., 1997) but previous studies have shown that cleavage of this linker increased the basal and drug-stimulated ATPase activity (Nuti et al., 2000; Sato et al., 2009). The short linker may constrain the degree of separation of the NBDs and also create a barrier preventing their close association for ATP binding and hydrolysis. The presence of a flexible linker region in several eukaryotic ABC transporters, including P-gp, the multidrug resistance associated proteins (MRPs) and the cystic fibrosis transmembrane conductance regulator (CFTR), but not in prokaryotic MDR homologues, may indicate that it is an integrated structural component in regulating the transporter’s conformational changes. Notably, CFTR has a much larger linker region (R-region) than P-gp which has been shown to regulate through phosphorylation and dephosphorylation NBD dimerization and channel gating. As a unique ion-channel type ABC transporter CFTR supposedly has a much more restrained range of conformations (Riordan, 2008).
Previous EM images of negatively stained 2D crystals of P-gp grown in detergent or in lipid monolayers gave clues that the NBDs undergo nucleotide/substrate dependent structural changes albeit the projection images might represent an average of the ensemble of all molecules in a particular state (Lee et al., 2008; Rosenberg et al., 2003). More recently, FRET between fluorescent probes attached to single cysteines in each of the two NBDs of P-gp reported that the ensemble-averaged ratios of donor/acceptor fluorescence did not change much for the APO, nucleotide-bound, substrate-stimulated MgATP continuous hydrolysis, or the vanadate-trapped states (Verhalen et al., 2012). Also consistent with our results, an EPR study of P-gp using the same cysteine pairs showed a rather broad distance distribution of the NBDs in the absence of nucleotide (Wen et al., 2013). Accompanying molecular dynamics simulations of the APO and MgATP docked states resulted in only marginal changes of the distance distribution with an increased propensity of closed NBDs after MgATP binding, and overall underlined the highly mobile nature of P-gp. The broad distribution of NBD distances accounting for at least four and possibly up to nine conformational states was also described in single-molecule FRET studies (Verhalen et al., 2012; Zarrabi et al., 2014), which further showed that conformations with closely spaced NBDs are short lived during active MgATP hydrolysis conditions. Overall, although these studies mainly focused on the separation of the NBDs, the data are in line with our whole molecule, single particle EM observations.
Taken together, our studies demonstrate a divergence in the conformational pathway of closely related homologous ABC exporters in the context of a common conformational cycle. Sequence evolution within the family of ABC transporters may account for the mechanistic divergence to fulfill their distinct biological functions. MsbA and P-gp are prototypical ABC exporters with the core NBD and TMD domains assembled in the forms of homodimer and a linker-fused pseudo heterodimer, respectively. Nevertheless, these are only two examples and certainly underrepresent the large variations in sequences and structures among the superfamily of ABC transporters. Particularly, some ABC exporters including the MRPs and CFTR, contain asymmetric NBDs with one active and one degenerate nucleotide binding site, unlike MsbA and P-gp of which both sites are catalytically active. Several recent structural studies of such heterodimeric, asymmetric ABC exporters have shown that the two NBDs remain partially or fully engaged during a catalytic cycle, i.e. the degenerate site composed of residues from both NBDs tends to stay associated (Hohl et al., 2012; Hohl et al., 2014; Mishra et al., 2014). These studies illustrate that a universal transport model that is applicable to all ABC transporters may not exist. Thus, detailed analyses must be undertaken in order to elucidate the conformational pathways for different subclasses of ABC transporters.
Materials and Methods
Preparation of MsbA
MsbA was prepared as described previously with slight modification (Lee et al., 2013). E506Q mutant was engineered by site-directed mutagenesis of wild type MsbA from E. Coli in the pET19b vector using QuickChange site-directed mutagenesis (Agilent Technologies). Single Cys mutants Q166C and T561C were generated using the Cys-less MsbA as a template in which two wild-type Cys residues (C88 and C315) were replaced with Ala (Cooper and Altenberg, 2013). All constructs were confirmed by DNA sequencing (Genewiz Inc.). Briefly, bacterial cell pellets containing MsbA were directly solubilized in 20 mM Tris (pH 8.0), 20 mM NaCl, 1% (wt/vol) β-D-undecyl maltopyranoside, 10% (vol/vol) glycerol, 0.1 mg/ml DNase I and a cocktail of proteinase inhibitors (Roche). After centrifugation at 38,000 × g for 45 min, MsbA was purified from the supernatant using an immobilized metal ion affinity chromatography column followed by size exclusion chromatography using a Superdex 200 16/60 column. Endogenous E. Coli lipids have been detected in purified MsbA samples (lipid:MsbA ratio ~ 0.03:1 wt/wt) (Tao et al., 2013). The purity of MsbA was analyzed using SDS-PAGE gels stained with Coomassie Brilliant Blue R-250. 0.025% (wt/vol) of BP-1 and 0.02 mg/mL E. coli polar lipids (Avanti Polar Lipids, Inc.) were mixed with 0.2 mg/ml of purified MsbA and then dialyzed against detergent-free buffer (6 kDa cutoff membrane, 4 °C, 1-2 days). For the EM study, the samples were diluted 20 fold with dialysis buffer containing reagents described in Figure 5A. The samples contain 1 mM nucleotides (ATP, AMP-PNP, or ADP), 2 mM Mg2+ and/or 1 mM Vi, unless noted otherwise.
NeutrAvidine labeling of MsbA
The purified single Cys mutants of MsbA were labeled with biotin C2 maleimide (Anaspec) for overnight in a buffer containing 20 mM Tris (pH7.5), 20 mM NaCl, 0.1% β-D-undecyl maltopyranoside, and 0.04% FA-3 (Lee et al., 2013). Excess biotin C2 maleimide was removed by immobilizing MsbA onto Ni-NTA agarose resin. The biotin labeled MsbA was then incubated with NeutrAvidin protein (Thermo Pierce) overnight on the resin at 4°C. The complexes were eluted by 2 column volumes of buffer containing 300 mM imidazole. The labeled MsbA samples were analyzed for ATPase activity using a standard linked enzyme ATPase assay described below. Detergent exchange to BP-1 by dialysis was conducted before EM imaging.
Preparation of P-gp
Mouse P-gp was prepared as described previously with slight modification (Aller et al., 2009; Bai et al., 2011). P-gp harboring microsomes were resuspended in 20 mM Tris (pH 8.0), 10 μg/ml Leupeptin, 10 μg/ml Pepstatin A, 2.5 μg/ml Chymostatin, 0.2 mM TECP, 1 mM β-mercaptoethanol, 10 mM Imidazole, 20% glycerol and 500 mM NaCl and solubilized with 1.2% β-D-dodecyl maltopyranoside (DDM) for 10 min at 4°C. After centrifugation at 38,000 × g for 30 min, P-gp was purified from the supernatant using an immobilized metal ion affinity chromatography column in the presence of 0.067% DDM, 0.04% sodium cholate and 0.1 mg/ml 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE). The elute were further purified by size exclusion chromatography using a Superdex 200 column in the presence of 0.067% DDM, 0.04% sodium cholate and 0.1 mg/ml of (POPE). The purity of P-gp was analyzed using SDS-PAGE gels stained with Coomassie Brilliant Blue R-250. 0.025% (wt/vol) of BP-1 was mixed with 0.3 mg/ml of purified P-gp for ATPase assays, and no dialysis procedure was performed. For the EM studies, samples were diluted ~30 fold with buffer containing reagents described in Figure 5B. The samples contain 1 mM ATP, 2 mM Mg2+, and/or 1 mM Vi, unless noted otherwise.
ATPase activity
The ATPase activity of MsbA and P-gp was measured at RT or 37 °C using the ATP-regenerating system as described previously (Lee et al., 2013). Briefly, 0.5–1 μg MsbA or P-gp were added to 100 μL of 50 mM Tris (pH 7.5) buffer containing 10 mM ATP, 12 mM MgCl2, 6 mM phosphoenolpyruvate, 1 mM NADH, 10 units lactate dehydrogenase, 10 units pyruvate kinase, and compounds indicated in the study. The rate of ATP hydrolysis was determined by the decrease in NADH absorbance at 340 nm using a Filtermax F5 Multiplate Spectrophotometer. ATPase activity was calculated using the following equation: ΔOD/(ε × [protein] × time), where ΔOD is the change in absorbance and ε is the extinction coefficient of NADH. The concentration of purified MsbA and P-gp was measured by comparing the SDS/PAGE intensity of Coomassie-stained protein bands with known amounts of BSA.
Electron Microscopy
All samples were applied onto freshly made carbon-copper grids within 3 minutes of sample preparation, unless stated otherwise. Sample dilution was adjusted to achieve a homogenous separation of particles. Samples were stained as previously described (Moeller et al., 2012) (Tao et al., 2013) using a 2% Uranyl formate solution. EM micrographs were acquired using a Tecnai F20 Twin transmission electron microscope operating at 200 kV, using a dose of ~45 e−/A°2 and nominal underfocus ranging from 0.7 μm to 1.7 μm. Utilizing the Leginon software package (Suloway et al., 2005) images were automatically collected onto a Tietz F415 4,000 × 4,000 pixel charge-coupled device (CCD) camera (15 μm pixel) at a nominal magnification of 64,000x. Images were collected binned by a factor of two, with a pixel size of 0.328 nm.
EM data processing and image analysis
Experimental data were processed using Sparx (Hohn et al., 2007; Yang et al., 2012) and the Appion software package (Lander et al., 2009) interfaced with the Leginon database infrastructure. Particles were automatically selected using a difference of Gaussian (DoG) algorithm and extracted with a box size of 112 pixels (Voss et al., 2009). The defoci of the raw micrographs were determined using CTFFIND3 (Mindell and Grigorieff, 2003) as implemented in the Appion package and phases were corrected on the full micrograph prior to particle extraction.
To determine the percentage of OF particles we used two different standardized protocols. In both cases we selected exclusively side views, which allowed a straightforward conformational characterization from 2D images. Putative top or bottom views were seldom observed presumably due to a strong preferred orientation of the transporters on the carbon surface.
Protocol 1: False positives are first removed from the particle stack using routines in the Sparx software package. The particles in the stack were initially aligned without a reference and averaged into a single class, which was then used for a subsequent reference based alignment. The aligned particles were then clustered into groups using kmeans classification as implemented in Sparx; the number of groups varied based on the stack size (~ 80 particles/group on average). Particles contributing to groups that represented noise or false positives were manually excluded from the stack. The procedure was repeated until the number of particles not contributing to identifiable class averages was less than 2%.
Protocol 2: To minimize user bias we used the iterative stable alignment and classification method (ISAC) (Yang et al., 2012) that automatically identifies homogeneous subsets of images.
Because ISAC is very computationally expensive we started with a stack composed of particles after one round of manual sorting (as per Protocol 1). Extensive testing confirmed that the ISAC results were not boased by this pre-sorting step.
The number of particles in groups representing OF particles were determined by manually examining the final class averages and counting the particles contributing to classes in the OF conformation. The results for Protocol 1 and 2 did not differ by more than 5% for any of the experiments. To further evaluate the reproducibility of the methods used we also collected and analyzed data from two independent protein purifications prepared under identical conditions, and these results were also within a 5% of each other.
Random conical tilt reconstructions were calculated using the Appion pipeline on 2D class averages calculated using multiple iterations of reference based Spider (Frank et al., 1996) alignment and kmeans classification with intermittent deselection of false positives. References were generated by XMIPP maximum likelihood 2D classification (Scheres et al., 2005). Resolutions were ~25-40 Å. The 3D structures were compared to a 3D reference set derived from a linear interpolation of X-ray structures (Ward et al., 2007) calculated using the Chimera software package(Pettersen et al., 2004_ENREF_44). The number of particles contributing to the individual volumes were used to determine the percentages provided.
Supplementary Material
HIGHLIGHTS.
- New tools enabled EM visualization of dynamic transporter conformations in lipids.
- Distinct conformational spectra of two homologous ABC transporters were revealed.
- P-gp prevails in inward-facing conformations under ATP hydrolysis conditions.
- MsbA with ATP displays both outward and a continuum of inward-facing conformations.
ACKNOWLEDGMENT
This work was supported by the National Institute of Health grant P50 GM073197 (C.S.P., B.C. and Q.Z.), R01 GM098538 (Q.Z.), P41 GM103310 (C.S.P. and B.C.), RGM102928 (I.L.U.), U54 GM94610 (G.C. and I.L.U.), and R01 GM94367 (G.C.). We thank Drs. Peter Wright, Andrew Ward, Takanori Otomo, and Guillermo Altenberg for discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
COMPETING INTERESTS
The authors declare that no competing interests exist.
DATA DEPOSITION
The 3D EM structures have been deposited in the Electron Microscopy Data Bank (accession nos. 6033, 6032, 6031, 6030, 6029, 6028, 6027, 6026, 6025, 6024, 6023, 6022, 6021, 6020, 6019, 6018, 6007).
REFERENCES
- Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J, Swartz DJ, Protasevich II, Brouillette CG, Harrell PM, Hildebrandt E, Gasser B, Mattanovich D, Ward A, Chang G, et al. A gene optimization strategy that enhances production of fully functional P-glycoprotein in Pichia pastoris. PloS One. 2011;6:e22577. doi: 10.1371/journal.pone.0022577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbat PP, Surendhran K, Bortolus M, Zou P, Freed JH, McHaourab HS. Conformational motion of the ABC transporter MsbA induced by ATP hydrolysis. PLoS Biol. 2007;5:e271. doi: 10.1371/journal.pbio.0050271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MG, Underbakke ES, Potter CS, Carragher B, Marletta MA. Single-particle EM reveals the higher-order domain architecture of soluble guanylate cyclase. Proc. Natl. Acad. Sci. U. S. A. 2014;111:2960–2965. doi: 10.1073/pnas.1400711111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xie C, Nishida N, Li Z, Walz T, Springer TA. Requirement of open headpiece conformation for activation of leukocyte integrin alphaXbeta2. Proc. Natl. Acad. Sci. U. S. A. 2010;107:14727–14732. doi: 10.1073/pnas.1008663107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury HG, Tong Z, Mathavan I, Li Y, Iwata S, Zirah S, Rebuffat S, van Veen HW, Beis K. Structure of an antibacterial peptide ATP-binding cassette transporter in a novel outward occluded state. Proc. Natl. Acad. Sci. U. S. A. 2014;111:9145–9150. doi: 10.1073/pnas.1320506111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper RS, Altenberg GA. Association/dissociation of the nucleotide-binding domains of the ATP-binding cassette protein MsbA measured during continuous hydrolysis. J. Biol. Chem. 2013;288:20785–20796. doi: 10.1074/jbc.M113.477976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dassa E. Natural history of ABC systems: not only transporters. Essays Biochem. 2011;50:19–42. doi: 10.1042/bse0500019. [DOI] [PubMed] [Google Scholar]
- Dawson RJ, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- Doerrler WT, Gibbons HS, Raetz CR. MsbA-dependent translocation of lipids across the inner membrane of Escherichia coli. J. Biol. Chem. 2004;279:45102–45109. doi: 10.1074/jbc.M408106200. [DOI] [PubMed] [Google Scholar]
- Eckford PD, Sharom FJ. Functional characterization of Escherichia coli MsbA: interaction with nucleotides and substrates. J. Biol. Chem. 2008;283:12840–12850. doi: 10.1074/jbc.M708274200. [DOI] [PubMed] [Google Scholar]
- Eckford PD, Sharom FJ. ABC efflux pump-based resistance to chemotherapy drugs. Chem. Rev. 2009;109:2989–3011. doi: 10.1021/cr9000226. [DOI] [PubMed] [Google Scholar]
- Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- Germann UA, Chambers TC, Ambudkar SV, Pastan I, Gottesman MM. Effects of phosphorylation of P-glycoprotein on multidrug resistance. J. Bioenerg. Biomembr. 1995;27:53–61. doi: 10.1007/BF02110331. [DOI] [PubMed] [Google Scholar]
- Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, et al. Membrane transporters in drug development. Nat. Rev. Drug Disc. 2010;9:215–236. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodfellow HR, Sardini A, Ruetz S, Callaghan R, Gros P, McNaughton PA, Higgins CF. Protein kinase C-mediated phosphorylation does not regulate drug transport by the human multidrug resistance P-glycoprotein. J. Biol. Chem. 1996;271:13668–13674. doi: 10.1074/jbc.271.23.13668. [DOI] [PubMed] [Google Scholar]
- Gottesman MM, Ambudkar SV, Xia D. Structure of a multidrug transporter. Nat. Biotechnol. 2009;27:546–547. doi: 10.1038/nbt0609-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins CF, Linton KJ. The ATP switch model for ABC transporters. Nat. Struct. Mol. Biol. 2004;11:918–926. doi: 10.1038/nsmb836. [DOI] [PubMed] [Google Scholar]
- Hiller Y, Gershoni JM, Bayer EA, Wilchek M. Biotin binding to avidin. Oligosaccharide side chain not required for ligand association. Biochem. J. 1987;248:167–171. doi: 10.1042/bj2480167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohl M, Briand C, Grutter MG, Seeger MA. Crystal structure of a heterodimeric ABC transporter in its inward-facing conformation. Nat. Struct. Mol. Biol. 2012;19:395–402. doi: 10.1038/nsmb.2267. [DOI] [PubMed] [Google Scholar]
- Hohl M, Hurlimann LM, Bohm S, Schoppe J, Grutter MG, Bordignon E, Seeger MA. Structural basis for allosteric cross-talk between the asymmetric nucleotide binding sites of a heterodimeric ABC exporter. Proc. Natl. Acad. Sci. U. S. A. 2014;111:11025–11030. doi: 10.1073/pnas.1400485111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohn M, Tang G, Goodyear G, Baldwin PR, Huang Z, Penczek PA, Yang C, Glaeser RM, Adams PD, Ludtke SJ. SPARX, a new environment for Cryo-EM image processing. J. Struct. Biol. 2007;157:47–55. doi: 10.1016/j.jsb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Ito Y, Son M, Sato S, Ishikawa T, Kondo M, Nakayama S, Shimokata K, Kume H. ATP release triggered by activation of the Ca2+-activated K+ channel in human airway Calu-3 cells. Am. J. Respir. Cell Mol. Biol. 2004;30:388–395. doi: 10.1165/rcmb.2003-0184OC. [DOI] [PubMed] [Google Scholar]
- Jardetzky O. Simple allosteric model for membrane pumps. Nature. 1966;211:969–970. doi: 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- Jin MS, Oldham ML, Zhang Q, Chen J. Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature. 2012;490:566–569. doi: 10.1038/nature11448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PM, George AM. A reciprocating twin-channel model for ABC transporters. Q. Rev. Biophys. 2014;47:189–220. doi: 10.1017/S0033583514000031. [DOI] [PubMed] [Google Scholar]
- Kim J, Wu S, Tomasiak TM, Mergel C, Winter MB, Stiller SB, Robles-Colmanares Y, Stroud RM, Tampe R, Craik CS, et al. Subnanometre-resolution electron cryomicroscopy structure of a heterodimeric ABC exporter. Nature. 2014 doi: 10.1038/nature13872. doi: 10.1038/nature13872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodan A, Yamaguchi T, Nakatsu T, Sakiyama K, Hipolito CJ, Fujioka A, Hirokane R, Ikeguchi K, Watanabe B, Hiratake J, et al. Structural basis for gating mechanisms of a eukaryotic P-glycoprotein homolog. Proc. Natl. Acad. Sci. U. S. A. 2014;111:4049–54. doi: 10.1073/pnas.1321562111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander GC, Stagg SM, Voss NR, Cheng A, Fellmann D, Pulokas J, Yoshioka C, Irving C, Mulder A, Lau PW, et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J. Struct. Biol. 2009;166:95–102. doi: 10.1016/j.jsb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Urbatsch IL, Senior AE, Wilkens S. Nucleotide-induced structural changes in P-glycoprotein observed by electron microscopy. J. Biol. Chem. 2008;283:5769–5779. doi: 10.1074/jbc.M707028200. [DOI] [PubMed] [Google Scholar]
- Lee JY, Yang JG, Zhitnitsky D, Lewinson O, Rees DC. Structural basis for heavy metal detoxification by an Atm1-type ABC exporter. Science. 2014;343:1133–1136. doi: 10.1126/science.1246489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Bennett BC, Hong WX, Fu Y, Baker KA, Marcoux J, Robinson CV, Ward AB, Halpert JR, Stevens RC, et al. Steroid-based facial amphiphiles for stabilization and crystallization of membrane proteins. Proc. Natl. Acad. Sci. U. S. A. 2013;110:E1203–1211. doi: 10.1073/pnas.1221442110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locher KP, Lee AT, Rees DC. The E. coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science. 2002;296:1091–1098. doi: 10.1126/science.1071142. [DOI] [PubMed] [Google Scholar]
- Loo TW, Bartlett MC, Clarke DM. Human P-glycoprotein is active when the two halves are clamped together in the closed conformation. Biochem. Biophys. Res. Commun. 2010;395:436–440. doi: 10.1016/j.bbrc.2010.04.057. [DOI] [PubMed] [Google Scholar]
- Loo TW, Clarke DM. Identification of the distance between the homologous halves of P-glycoprotein that triggers the high/low ATPase activity switch. J. Biol. Chem. 2014;289:8484–8492. doi: 10.1074/jbc.M114.552075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyumkis D, Doamekpor SK, Bengtson MH, Lee JW, Toro TB, Petroski MD, Lima CD, Potter CS, Carragher B, Joazeiro CA. Single-particle EM reveals extensive conformational variability of the Ltn1 E3 ligase. Proc. Natl. Acad. Sci. U. S. A. 2013;110:1702–1707. doi: 10.1073/pnas.1210041110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcoux J, Wang SC, Politis A, Reading E, Ma J, Biggin PC, Zhou M, Tao H, Zhang Q, Chang G, et al. Mass spectrometry reveals synergistic effects of nucleotides, lipids, and drugs binding to a multidrug resistance efflux pump. Proc. Natl. Acad. Sci. U. S. A. 2013;110:9704–9709. doi: 10.1073/pnas.1303888110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindell JA, Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 2003;142:334–347. doi: 10.1016/s1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- Mishra S, Verhalen B, Stein RA, Wen PC, Tajkhorshid E, McHaourab HS. Conformational dynamics of the nucleotide binding domains and the power stroke of a heterodimeric ABC transporter. eLife. 2014;3:e02740. doi: 10.7554/eLife.02740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller A, Kirchdoerfer RN, Potter CS, Carragher B, Wilson IA. Organization of the influenza virus replication machinery. Science. 2012;338:1631–1634. doi: 10.1126/science.1227270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath A, Atkins WM, Sligar SG. Applications of phospholipid bilayer nanodiscs in the study of membranes and membrane proteins. Biochemistry. 2007;46:2059–2069. doi: 10.1021/bi602371n. [DOI] [PubMed] [Google Scholar]
- Nuti SL, Mehdi A, Rao US. Activation of the human P-glycoprotein ATPase by trypsin. Biochemistry. 2000;39:3424–3432. doi: 10.1021/bi992392w. [DOI] [PubMed] [Google Scholar]
- Oldham ML, Khare D, Quiocho FA, Davidson AL, Chen J. Crystal structure of a catalytic intermediate of the maltose transporter. Nature. 2007;450:515–521. doi: 10.1038/nature06264. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Qu Q, Sharom FJ. FRET analysis indicates that the two ATPase active sites of the P-glycoprotein multidrug transporter are closely associated. Biochemistry. 2001;40:1413–1422. doi: 10.1021/bi002035h. [DOI] [PubMed] [Google Scholar]
- Radermacher M, Wagenknecht T, Verschoor A, Frank J. Three-dimensional reconstruction from a single-exposure, random conical tilt series applied to the 50S ribosomal subunit of Escherichia coli. J. Microsc. 1987;146:113–136. doi: 10.1111/j.1365-2818.1987.tb01333.x. [DOI] [PubMed] [Google Scholar]
- Rees DC, Johnson E, Lewinson O. ABC transporters: the power to change. Nat. Rev. Mol. Cell Biol. 2009;10:218–227. doi: 10.1038/nrm2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter G, Janvilisri T, Venter H, Shahi S, Balakrishnan L, van Veen HW. The ATP binding cassette multidrug transporter LmrA and lipid transporter MsbA have overlapping substrate specificities. J. Biol. Chem. 2003;278:35193–35198. doi: 10.1074/jbc.M306226200. [DOI] [PubMed] [Google Scholar]
- Riordan JR. CFTR function and prospects for therapy. Annu. Rev. Biochem. 2008;77:701–726. doi: 10.1146/annurev.biochem.75.103004.142532. [DOI] [PubMed] [Google Scholar]
- Rosenberg MF, Kamis AB, Callaghan R, Higgins CF, Ford RC. Three-dimensional structures of the mammalian multidrug resistance P-glycoprotein demonstrate major conformational changes in the transmembrane domains upon nucleotide binding. J. Biol. Chem. 2003;278:8294–8299. doi: 10.1074/jbc.M211758200. [DOI] [PubMed] [Google Scholar]
- Sato T, Kodan A, Kimura Y, Ueda K, Nakatsu T, Kato H. Functional role of the linker region in purified human P-glycoprotein. FEBS J. 2009;276:3504–3516. doi: 10.1111/j.1742-4658.2009.07072.x. [DOI] [PubMed] [Google Scholar]
- Scheres SH, Valle M, Carazo JM. Fast maximum-likelihood refinement of electron microscopy images. Bioinformatics. 2005;2(21 Suppl):ii243–244. doi: 10.1093/bioinformatics/bti1140. [DOI] [PubMed] [Google Scholar]
- Schultz KM, Merten JA, Klug CS. Characterization of the E506Q and H537A dysfunctional mutants in the E. coli ABC transporter MsbA. Biochemistry. 2011;50:3599–3608. doi: 10.1021/bi101666p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior AE. Reaction chemistry ABC-style. Proc. Natl. Acad. Sci. U. S. A. 2011;108:15015–15016. doi: 10.1073/pnas.1111863108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintre CA, Pike AC, Li Q, Kim JI, Barr AJ, Goubin S, Shrestha L, Yang J, Berridge G, Ross J, et al. Structures of ABCB10, a human ATP-binding cassette transporter in apo- and nucleotide-bound states. Proc. Natl. Acad. Sci. U. S. A. 2013;110:9710–9715. doi: 10.1073/pnas.1217042110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan V, Pierik AJ, Lill R. Crystal structures of nucleotide-free and glutathione-bound mitochondrial ABC transporter Atm1. Science. 2014;343:1137–1140. doi: 10.1126/science.1246729. [DOI] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, Carragher B. Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Szabo K, Bakos E, Welker E, Muller M, Goodfellow HR, Higgins CF, Varadi A, Sarkadi B. Phosphorylation site mutations in the human multidrug transporter modulate its drug-stimulated ATPase activity. J. Biol. Chem. 1997;272:23165–23171. doi: 10.1074/jbc.272.37.23165. [DOI] [PubMed] [Google Scholar]
- Tao H, Lee SC, Moeller A, Roy RS, Siu FY, Zimmermann J, Stevens RC, Potter CS, Carragher B, Zhang Q. Engineered nanostructured beta-sheet peptides protect membrane proteins. Nat. Methods. 2013;10:759–761. doi: 10.1038/nmeth.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbatsch IL, Tyndall GA, Tombline G, Senior AE. P-glycoprotein catalytic mechanism: studies of the ADP-vanadate inhibited state. J. Biol. Chem. 2003;278:23171–23179. doi: 10.1074/jbc.M301957200. [DOI] [PubMed] [Google Scholar]
- van Wonderen JH, McMahon RM, O'Mara ML, McDevitt CA, Thomson AJ, Kerr ID, Macmillan F, Callaghan R. The central cavity of ABCB1 undergoes alternating access during ATP hydrolysis. FEBS J. 2014;281:2190–2201. doi: 10.1111/febs.12773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhalen B, Ernst S, Borsch M, Wilkens S. Dynamic ligand-induced conformational rearrangements in P-glycoprotein as probed by fluorescence resonance energy transfer spectroscopy. J. Biol. Chem. 2012;287:1112–1127. doi: 10.1074/jbc.M111.301192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss NR, Yoshioka CK, Radermacher M, Potter CS, Carragher B. DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J. Struct. Biol. 2009;166:205–213. doi: 10.1016/j.jsb.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward A, Reyes CL, Yu J, Roth CB, Chang G. Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc. Natl. Acad. Sci. U. S. A. 2007;104:19005–19010. doi: 10.1073/pnas.0709388104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward AB, Szewczyk P, Grimard V, Lee CW, Martinez L, Doshi R, Caya A, Villaluz M, Pardon E, Cregger C, et al. Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc. Natl. Acad. Sci. U. S. A. 2013;110:13386–13391. doi: 10.1073/pnas.1309275110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen PC, Verhalen B, Wilkens S, McHaourab HS, Tajkhorshid E. On the origin of large flexibility of P-glycoprotein in the inward-facing state. J. Biol. Chem. 2013;288:19211–19220. doi: 10.1074/jbc.M113.450114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Fang J, Chittuluru J, Asturias FJ, Penczek PA. Iterative stable alignment and clustering of 2D transmission electron microscope images. Structure. 2012;20:237–247. doi: 10.1016/j.str.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrabi N, Ernst S, Verhalen B, Wilkens S, Borsch M. Analyzing conformational dynamics of single P-glycoprotein transporters by Forster resonance energy transfer using hidden Markov models. Methods. 2014;66:168–179. doi: 10.1016/j.ymeth.2013.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Tao H, Hong WX. New amphiphiles for membrane protein structural biology. Methods. 2011;55:318–323. doi: 10.1016/j.ymeth.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, White KA, Polissi A, Georgopoulos C, Raetz CR. Function of Escherichia coli MsbA, an essential ABC family transporter, in lipid A and phospholipid biosynthesis. J. Biol. Chem. 1998;273:12466–12475. doi: 10.1074/jbc.273.20.12466. [DOI] [PubMed] [Google Scholar]
- Zou P, Bortolus M, McHaourab HS. Conformational cycle of the ABC transporter MsbA in liposomes: detailed analysis using double electron-electron resonance spectroscopy. J. Mol. Biol. 2009;393:586–597. doi: 10.1016/j.jmb.2009.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.