RNA quaternary structure and global symmetry (original) (raw)
. Author manuscript; available in PMC: 2016 Apr 1.
Published in final edited form as: Trends Biochem Sci. 2015 Mar 13;40(4):211–220. doi: 10.1016/j.tibs.2015.02.004
Abstract
Many proteins associate into symmetric multisubunit complexes. Structural analyses suggested that, in contrast, virtually all RNAs with complex three-dimensional structures function as asymmetric monomers. Recent crystal structures revealed that several biological RNAs exhibit global symmetry at the level of their tertiary and quaternary structures. Here, we survey known examples of global RNA symmetry, including the true quaternary symmetry of the bacteriophage ϕ29 prohead RNA (pRNA), and the internal pseudosymmetry of the single-chain flavin mononucleotide (FMN), glycine, and cyclic diadenosine monophosphate (c-di-AMP) riboswitches. For these RNAs, global symmetry stabilizes the RNA fold, coordinates ligand-RNA interactions, and facilitates association with symmetric binding partners.
Keywords: X-ray crystallography, cooperativity, c-di-AMP riboswitch, glycine riboswitch, FMN riboswitch, bacteriophage ϕ29 pRNA
Quaternary structure in proteins and RNA
A striking feature of the three-dimensional structures of many proteins is the presence of symmetric quaternary structure. Quaternary structure arises from the association of multiple identical or different polypeptide chains, resulting in homo- or heterooligomers, respectively. Most homooligomers, and many heterooligomers, exhibit symmetry, which can give rise to finite bounded structures with point-group symmetry (see Glossary) (ranging from small peptide homodimers such as gramicidin to icosahedral viral capsids), or extended filaments, helices, and tubes that combine point-group and translational symmetry (Box 1). By oligomerizing, proteins can form larger structures while maintaining efficient genomic coding, increase their stability by reducing solvent-exposed surface area, and acquire sophisticated behaviors such as cooperativity and allostery (reviewed in [1]).
Box 1. Symmetry, pseudosymmetry and quasisymmetry.
Since proteins are chiral, the only allowed symmetry operations are rotations and translations (reviewed in [1, 42]). Many oligomers exhibit closed (point-group) symmetry, which results in finite globally symmetric quaternary structure. If a translation component is present, filaments or tubes in an open symmetry can be formed (e.g., hemoglobin fibrils characteristic of sickle-cell disease), leading to a semi-infinite quaternary structure. Protein quaternary structure was discovered in the 1920s by Svedberg, who determined via ultracentrifugation that hemoglobin and hemocyanin sedimented as multisubunit complexes [89]. The crystal structure of hemoglobin first elucidated the symmetry involved in quaternary protein structure [90] and exemplifies both macromolecular symmetry and pseudosymmetry (Figure I). Specifically, pseudosymmetry is defined by approximate symmetry formed by two different molecules which adopt similar tertiary structures despite having differences in primary sequence (reviewed in [1]). In contrast, quasisymmetry describes the approximate symmetry between identical subunits which form similar overall tertiary structures existing in different conformations or forming different subunit-subunit interactions. Hemoglobin contains two α and two β subunits, which associate to form a symmetrical (αβ)2 dimer of two αβ pseudosymmetrical dimers (Fig. I). Alternatively, hemoglobin may be considered a tetramer with D2 (dihedral) pseudosymmetry.
Figure I.
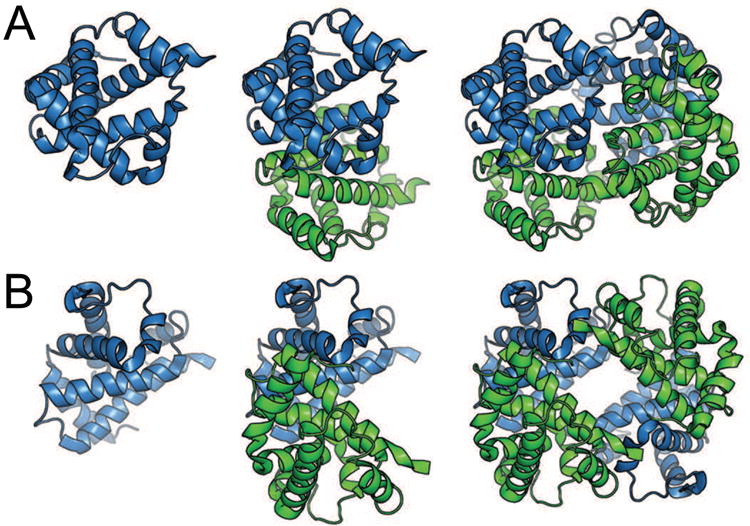
Symmetry and pseudosymmetry of tetrameric horse hemoglobin (PDB 2DHB) [91]. (a) Cartoon representation of the α subunit (blue) of hemoglobin (left), the αβ pseudo-dimer with β subunit colored green (middle), and the (αβ)2 tetramer (right). This view emphasizes the symmetry between α subunits and between β subunits. (b) Rotated view of hemoglobin, emphasizing pseudosymmetry between α and β subunits within the tetramer. Heme ligands are omitted for clarity.
The basic building block of nucleic acid structure is the antiparallel double-stranded helix, whose beautiful symmetry was revealed by X-ray fiber diffraction and modeling in 1953 [2]. However, as structures of RNAs were reported subsequently by X-ray crystallography, a puzzling lack of quaternary structure became apparent. The structures of tRNA (1974), the hammerhead ribozyme (1993), the P4-P6 domain of the group I intron (1996), the HDV ribozyme (1998) and the hairpin ribozyme (2001) successively revealed diverse RNA tertiary architectures, but none of these molecules were found to oligomerize to give rise to symmetric quaternary structures [3-7]. Indeed, even the ribosome, which is formally an RNA heterotrimer (comprising 5S, 16S and 23S rRNAs in addition to numerous ribosomal proteins), exhibits no point-group symmetry ([8-11]) though the peptidyl transferase center exhibits local pseudosymmetry [12]. Thus, it appeared that RNAs could readily adopt complex three-dimensional structures, but very rarely did they form symmetric oligomers. Prior to the discovery of pseudosymmetry in riboswitches discussed below, the only known exceptions were the kissing-loop dimerization elements of retroviruses and plasmid-encoded RNAs [13, 14] and the homo-oligomer formed by a component of the packaging motor of bacteriophage ϕ29 [15].
In this review, we survey examples of naturally occurring global RNA symmetry, focusing on symmetry and pseudosymmetry (Box 1) at the level of tertiary and quaternary structures. Thus, examples of local symmetry arising from pairing, G quadruplexes (reviewed in [16]), RA motifs [17], and engineered RNAs possessing pseudosymmetry by design [18, 19] are not discussed.
Kissing-loops and ϕ29 pRNA: quaternary structure through Watson-Crick base pairing
The first example of symmetric RNA oligomerization (other than helix formation) characterized structurally was the kissing-loop complex of the RNAs encoded by certain bacterial plasmids [14]. These structures arise from the association of the loops of two stem-loops through Watson-Crick complementarity. In the case of the ColE1 RNAs (Figure 1), the seven complementary loop nucleotides of each RNA form a double helix, which stacks coaxially with the two stems of the protomer RNAs. Kissing-loop dimerization elements are essential for packaging retroviral RNA genomes [20], and structures of dimers of the human immunodeficiency virus (HIV) dimerization element have also been reported [13, 21]. In the latter, the loop sequences are palindromic (e.g., GCGCGC), allowing homo-dimerization.
Figure 1.
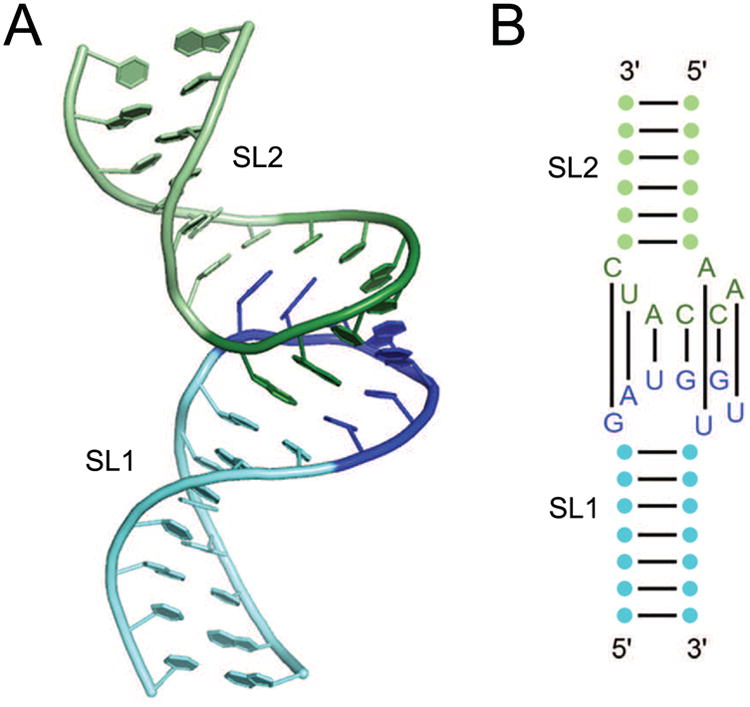
The ColE1 RNA kissing dimer. (A) Cartoon representation two RNAs resembling the ColE1 RNAs based on the NMR solution structure [14, 86]. Stemloops SL1 (green) and SL2 (blue) interact via a loop-loop kissing between SL1 loop residues (dark green) and SL2 residues (dark blue). (B) Secondary structure of the two RNAs with the seven base pairs between the two loops indicated.
A similar mode of association underlies oligomerization of the ϕ29 prohead RNA (pRNA) (Fig. 2). Bacteriophage ϕ29 encapsulates its ∼20 kB genomic DNA in its capsid using an ATP-dependent DNA packaging motor. The pRNA ring interacts with other elements of the packaging motor, including the ATPases that drive DNA into the capsid, and the head-tail connector that attaches the packaging motor to the capsid. Individual 174-nucleotide pRNA protomers oligomerize to form a closed 5- or 6-membered planar ring through the association of two different sequence motifs that make kissing-loop interactions (Fig. 2) [15, 22, 23]. Bulge B23 (i.e., the bulge between helices 2 and 3) of one RNA interacts with loop L4 (i.e., the loop that caps helix 4) of a neighboring RNA, an interaction scheme that is continued cyclically until B23 of the last RNA interacts with L4 of the first RNA (Fig. 2B). While each interaction only produces 4 intermolecular base pairs, stacking interactions on both sides of the resulting 4-bp duplex create a continuous helical stack from the P4 of one pRNA to the P3 of the neighbor [24]. As with ColE1 RNAs, pRNAs oligomerize essentially via Watson-Crick pairing in loop-loop interactions, which are one of the many types of interactions that guide riboswitch folding, as described below.
Figure 2.
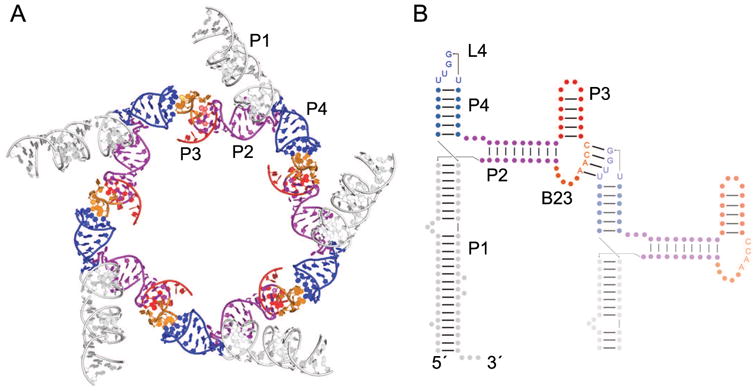
The homooligomeric ϕ29 bacteriophage pRNA. (A) Cartoon representation of a partial model of the pentameric pRNA based on cryo-electron microscopy reconstruction of the bacteriophage prohead [22]. A single monomer consists of helix P1 (white), helix P2 (purple), bulge B23 (orange), helix P3 (red), and helix P4 (blue), which contains loop L4. (B) Secondary structure of two units of the pRNA (nts 1-119) showing the intramolecular kissing-loop interaction between bulge B23 of one RNA and L4 of a neighboring pRNA (faded). Helix P1 has been shortened in the neighboring RNA for clarity. The second domain of the pRNA (nts 120-172) is not shown. Arrows indicate chain connectivities in the 5′ to 3′ direction.
Riboswitches: pseudosymmetry in the structure of ligand-binding RNAs
In the past decade, riboswitches have proved a rich vein of RNA structural information. Riboswitches are mRNA domains that directly sense the intracellular concentrations of small molecule ligands and regulate gene expression in response to ligand binding. By the end of 2014, the 3D folds of 23 structurally distinct classes of riboswitches had been elucidated by X-ray crystallography (reviewed in [25-27]). The cognate ligands of these riboswitches range in size from simple ions (F-, Mg2+), to small molecules such as purines, amino acids and vitamins, to macromolecules such as full-length tRNA [28-33]. Consistent with the chemical diversity of their ligands, riboswitch classes exhibit diverse and structurally distinct 3D folds. Within this trove of RNA structural riches, the structures of three riboswitch classes (which respond to c-di-AMP, FMN and glycine) were found to exhibit clear internal symmetry [34-39]. Unlike pRNA, none of these RNAs, at least in their biological context, are oligomers. Instead, their symmetry arises from the presence of structurally homologous domains within one RNA chain. We refer to such structures as “pseudo-quaternary” as they exhibit pseudosymmetry (Box 1).
RNA folding, in general, proceeds hierarchically (for an introduction, see [40]). Watson-Crick paired RNA helices (the secondary structure) are thermodynamically stable in isolation, and fold first. Tertiary structure, which to a first approximation is the packing of preformed helices against each other, can be mediated by (1) the connectivity of the RNA chain itself, (2) by stereochemical constraints imposed by multi-helical junctions, (3) by interactions between non-helical elements (such as bulges and loops) with other non-helical elements as well as helices, and (4) by ligands, such as divalent cations and small molecules. As in the case of proteins [41, 42], the quaternary and pseudo-quaternary structures of RNA molecules are mediated by the same interaction types that stabilize their tertiary structure. Thus, kissing-loops and pRNA associate through Watson-Crick base pairing. The pseudo-quaternary structure of the three riboswitches necessarily requires RNA chain connectivity, but in addition, the interfaces between the homologous subdomains form through the interaction of loops (FMN), helical and other non-helical elements (glycine), and small molecule ligands (c-di-AMP).
The FMN riboswitch: RNA pseudosymmetry with an asymmetrically bound ligand
Flavin mononucleotide (FMN) riboswitches reside upstream of genes important to flavin synthesis and transport, such as the ribHDE(B/A) operon in Fusobacterium nucleatum [33, 43], and were found to be the target of the antibiotic roseoflavin [44-46]. The structure of the FMN riboswitch (Fig. 3A) is organized into two pseudosymmetric domains that associate to form the pocket that binds a single FMN molecule [37]. Each of the two domains contains a 5-nucleotide T-loop from loop P2 or P5 that, with an intercalated adenine from loop P3 or P6, forms a GAA(A) tetraloop-like motif to create one “wing” of the butterfly-shaped RNA. These longdistance loop-loop interactions are mediated by an adenine at each wingtip (A38 and A90, Fig. 3B), in a manner reminiscent of other T-loop interactions [47, 48]. Adding to the overall pseudosymmetry, GU dinucleotides G41-U42 from P6 and G93-U94 from P3 base pair with A81-C82 and A29-C30, respectively, within the stems P2 and P5, and form the center of the RNA. This intricate pairing arrangement occurs within the RNA such that P2 interdigitates with P6 and P3 with P5, rather than with their neighbors in primary structure (i.e., P2 with P3 and P5 with P6), a folding scheme reminiscent of a T-loop PK domain [49].
Figure 3.
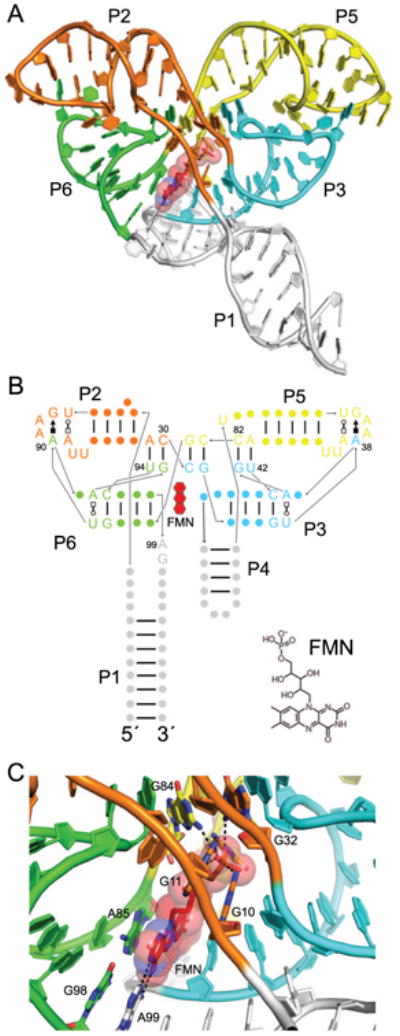
The FMN riboswitch. (A) Cartoon representation of the F. nucleatum flavin mononucleotide (FMN) riboswitch bound to FMN (red) [37]. Symmetry elements are colored, and helices are labeled except for P4, which is obscured by P1 and P3. (B) Secondary structure of the FMN riboswitch with the approximate position of FMN indicated by a red, three-ringed shape. Noncanonical base pairs are shown with Leontis-Westhof symbols [87]. As is common to T-loop motifs, a noncanonical base pair (U•A Trans Watson-Crick/Hoogsteen) formed by T-loop residues 1 and 5 precedes the GAA(A) motifs in loops P2 and P5. The chemical structure of FMN is shown (bottom right). (C) View of the FMN binding site with coloring the same as in A. Hydrogen bonds are indicated by dotted black lines. FMN riboswitch residues interacting with the FMN ligand (red) are labeled, including G10, G11, G32, and G84, which hydrogen bond to the FMN phosphate.
A single FMN molecule is bound asymmetrically by nucleotides in the linker regions between the helical elements which comprise the two domains (Fig. 3B). On its more hydrophilic side, the isoalloxazine ring contacts the Watson-Crick face of A99 (Fig. 3C) while on its more apolar side, its two methyl groups interact with the ribose of U61. In addition, the flavin ring stacks on the G98•A85 base pair, and the phosphate of FMN is bound through numerous hydrogen bonds to the RNA (Fig. 3C) [37]. Riboflavin, which lacks the phosphate group, binds the FMN riboswitch ∼1000-fold more weakly than FMN [37].
Pseudosymmetry in glycine riboswitches
In many riboswitches, it is possible to identify two separable (although typically overlapping) RNA segments that function, respectively, in ligand binding and in the downstream control of gene expression by interfacing with the transcriptional or translational machinery. By analogy with ligand-binding RNAs selected in vitro [50], the former has been referred to as the “aptamer” domain, and the latter as the “expression platform” [51]. Tandem riboswitches are genetic control elements composed of two or more riboswitches, each comprised of its own aptamer and expression platform domains. Tandem riboswitches are present in a number of bacterial genes, allowing for multi-input control of gene expression, such as by the intracellular concentrations of two different metabolites [52] or by two molecules of the same metabolite [30, 53, 54]. The glycine riboswitch is noteworthy because, in the majority of cases, it consists of two homologous aptamer domains (each capable of binding to glycine) that are upstream of a single expression platform. Thus, even at the level of sequence, it was immediately apparent that this riboswitch class may exhibit pseudo-quaternary structure [30].
Crystal structures have been determined of the isolated aptamer domain 2 of the glycine riboswitch from Vibrio cholerae [39] and of the two tandem aptamer domains of the glycine riboswitch from F. nucleatum [38]. When compared side by side, the overall similarity of the two domains of the F. nucleatum riboswitch is readily apparent (Fig. 4). The three-helix junction P1-P2-P3 of aptamer 1 (Fig. 4A) is recapitulated by the P4-P5-P6 three-helix junction of aptamer 2 (Fig. 4B), except that P1 is longer than P4 and P6 is longer than P3. Each aptamer binds to one glycine molecule using A-rich bulges within P3 and P6, for aptamer 1 and 2, respectively (Fig. 4A, B). Within each aptamer domain, the Watson-Crick face of a conserved U residue (U50 or U141) contacts the bound glycine, which is sandwiched between an AAG base triple and a Watson-Crick base pair (either A•U or G•C) [38, 39]. Each individual glycine-binding motif contains a looped-out A residue (A36 or A111) which stacks between A residues from the A-rich junctions connecting P1 to P2/P3 and P4 to P5/P6, respectively. These interactions mediate intradomain packing of helices within each aptamer domain, bringing P1 next to P3 and P4 next to P6.
Figure 4.
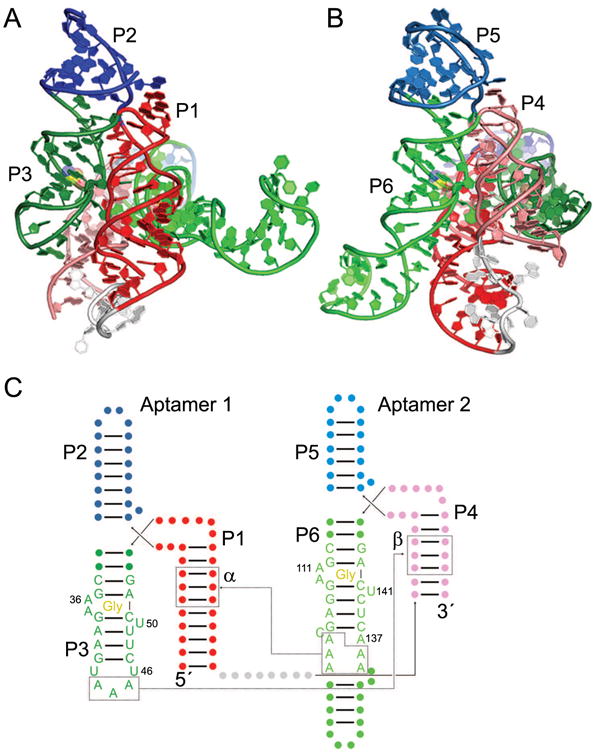
The two-domain glycine riboswitch. Cartoon representation of the F. nucleatum glycine riboswitch bound to two molecules of glycine (yellow) [38], with views of Aptamer 1 (A) and Aptamer 2 (B). The U1A binding site and U1A protein used for crystallization have been omitted for clarity. (C) Secondary structure of the glycine riboswitch with approximate ligand locations indicated by “Gly”. The α and β interactions are indicated by dotted lines, and the γ interaction occurs between U46 and A137. The kink-turn motif connecting the two aptamer domains [67] has been omitted for clarity.
Interdomain, or pseudo-quaternary, contacts occur at three sites in both reported crystal structures (between two separate identical chains within the crystal, in the case of the V. cholerae structure) [38, 39]. Two of these sites (termed α and β [38]) involve similar A-minor [55] interactions between A residues from helix P3 or P6 from one domain with the minor groove of helix P1 or P4 of the other domain, respectively. The third contact (termed γ) occurs in the center of the riboswitch in which looped-out residue U47 from domain 1 forms a Hoogsteen base pair with looped-out A137 from domain 2 (Fig. 4C). Despite the relatively modest conservation at these sites, they covary to preserve pairing [38], and disruption of pairing greatly affects riboswitch dimerization [56].
Symmetrical ligands within the pseudosymmetrical c-di-AMP riboswitches
C-di-AMP is an essential bacterial second messenger involved in cell wall homeostasis, growth, and sporulation (reviewed in [57]). A widespread class of riboswitches, typified by that in the 5′ UTR of the ydaO/yuaA operon in Bacillus subtilis, binds to c-di-AMP to control gene expression of downstream genes [58]. Recent crystal structures [34-36] of c-di-AMP riboswitches revealed that they bind two copies of c-di-AMP using an RNA fold that is strikingly symmetric (Fig. 5A). The structure of the RNA-ligand complex results from pseudo-quaternary association of two three-helix junctions (helices P1-P2-P3 and P4-P5-P6) (Fig. 5B). P2 forms a continuous helical stack with P3, and similarly P5 stacks on P6. Helices P3 and P4 are connected by a linker region, and helices P1 and P6 are connected via a pseudoknot between the loop of P6 and the sequence 3′ of helix P1 [34, 35, 58, 59]. Each three-helix junction is organized around an interaction that closely mimics the non-junctional structure resulting from association of a GNRA tetraloop with its receptor sequence [4, 60]. The NAA(A) tetraloop-like motif of each junction also interacts with c-di-AMP [36]. The tetraloop is completed by a fourth residue from a distal part of the RNA.
Figure 5.
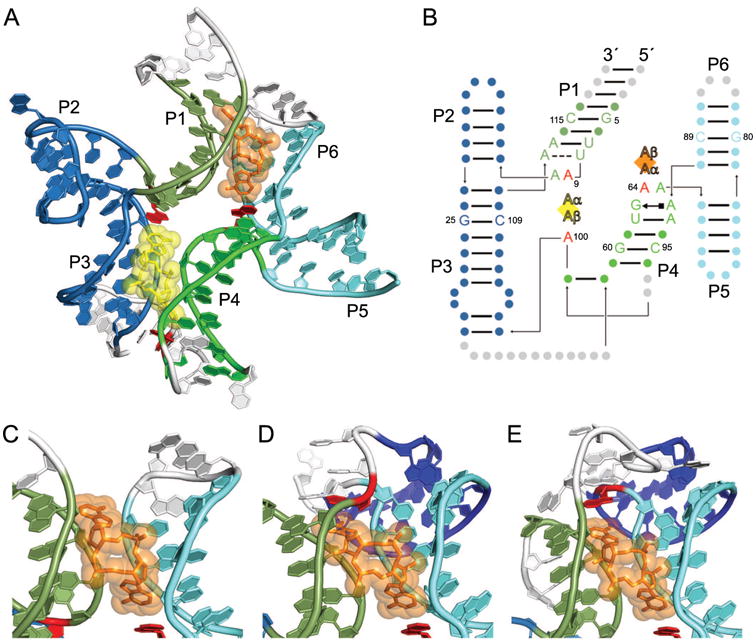
C-di-AMP riboswitches bind two c-di-AMP molecules with a pseudosymmetric fold. (A) The cartoon representation of the B. subtilis c-di-AMP riboswitch [36] bound to two molecules of c-di-AMP, CDA1 (yellow) and CDA2 (orange). Portions of the riboswitch mutated for crystallography are shown in white. The U1A binding site and U1A protein used for crystallization have been omitted for clarity. (B) Secondary structure of the B. subtilis c-di-AMP riboswitch with approximate ligand locations indicated by diamonds and helices P1-P6 labeled as such. Individual A nucleobases of each c-di-AMP ligand are indicated by Aα and Aβ. The identities of symmetric nucleotides between the two binding pockets are shown, with other residues signified by circles. Residues stacking on the c-di-AMP ligands are colored red. (C) View of CDA2 in the B. subtilis c-di-AMP riboswitch, emphasizing the lack of the interaction at CDA2 Aβ. Coloring is the same as in A. (D) View of CDA2 in the T. tengcongensis c-di-AMP riboswitch with coloring of helices the same as in C, except for helix P7 (dark blue), which is formed by base pairing of the pseudoknot. In this riboswitch, residue U112 (red) stacks on CDA2 Aβ. (E) View of CDA2 in the T. pseudothanolicus c-di-AMP riboswitch with coloring the same as in D, indicating helix P7 (dark blue) formed by pseudoknot base pairs. In this riboswitch, residue A72 (red) stacks on CDA2 Aβ.
Each c-di-AMP binding pocket itself is twofold pseudosymmetric as both AMP moieties of each c-di-AMP (CDA1 and CDA2) are recognized similarly [34-36]. The pseudo symmetry within the c-di-AMP riboswitch largely occurs at the level of helices and gross structural features. The sugar face of each adenine nucleobase of c-di-AMP makes A-minor contacts with neighboring G residues, which are G25 and G60 for CDA1 and G5 and G80 for CDA2 (Fig. 5B). The Hoogsteen face of each adenine nucleobase forms hydrogen bonds between N2 and neighboring bridging phosphate oxygens. Additionally, each c-di-AMP adenine stacks with a critical riboswitch residue, which is A for three of the four stacking interactions (A9, A65, and A100, Fig. 5B, red). The fourth stacking interaction, which would stack on Aβ of CDA2, was not present in the crystallized version of the B. subtilis riboswitch (Fig. 5C) [36]. In other c-di-AMP riboswitches, this residue is present as the P1 loop residue U112 in Thermoanaerobacter tengcongensis (Fig. 5D) [34] and P6 loop residue A72 in Thermoanaerobacter pseudothanolicus (Fig. 5E) [35]. Thus, the topology of this region varies among c-di-AMP riboswitch structures known to date, which could reflect different mechanisms of communication between the aptamer domain and the expression platform. Despite this variation, overall pseudosymmetry of the riboswitch is preserved in this region, as the pseudoknot helix is mimicked by the linker region connecting helices P3 and P4 in the T. tengcongensis and T. pseudothanolicus structures [34, 35].
The three classes of riboswitches known to exhibit global symmetry embody three different ligand-interaction modes. In FMN riboswitches, the pseudosymmetrical fold surrounds FMN such that the repeated structural region is one-half of a single, asymmetric ligand binding site. In glycine riboswitches, two individual aptamer domains each bind a single glycine molecule. In c-di-AMP riboswitches, each three-helix junction of the riboswitch binds to both ligands so that the repeated domain constitutes half of each of the two ligand sites. Consequently, the contribution of global symmetry to function should reflect the differences in the RNA architectures.
Functional consequences of RNA quaternary and pseudo-quaternary structure
To date, the ϕ29 pRNA is the only biological RNA that has been shown to assemble into a true symmetrical quaternary structure. The number of protomers in the oligomeric complex is dictated by the packing of each monomer, a mutated version of which crystallized as a tetramer [24] even though the physiologically relevant form has been proposed to be either a pentamer or a hexamer [15, 22]. A symmetric, cyclical homopentamer (or any oligomer with a prime number of protomers) would have the built-in geometric requirement of being planar. Symmetry mismatch between bacteriophage tails and capsids reduces the overall interactions between the two domains in motion, thus allowing for relatively free rotation (i.e., no large potential energy wells) and reducing the amount of ATP required for rotation [61]. As the ϕ29 connector is 6-fold symmetric, a symmetry mismatch occurs between the it and the 5-fold symmetric capsid; therefore, it has been proposed that a 6-fold symmetric pRNA would interact with connector [15]. However, if the pRNA is 5-fold symmetric, symmetry mismatch would occur between pRNA and the 6-fold symmetric connector, such that the stationary part of the packaging motor would include the capsid, pRNA, and ATPase [22]. In either case, the pRNA's symmetry promotes its interaction with one part of the viral machinery that shares symmetry while minimizing interaction with other viral components through symmetry mismatch.
The mechanistic consequences of protein quaternary structure have been classically associated with allostery and cooperativity based on the model of Monod, Wyman, and Changeux [62]. Thus, one might hypothesize by analogy that pseudosymmetrical riboswitches display cooperative ligand binding. For the FMN riboswitch, which binds a single FMN molecule, the overall stability of the RNA fold is more likely the explanation, possibly due to the inherent stability of symmetric structures [63]. Small-angle X-ray scattering (SAXS) experiments, which report on the overall shape of the riboswitch in the presence or absence of ligand, have suggested that the ligand binding site is preformed for this RNA [64] in contrast to glycine and c-di-AMP riboswitches, which compact greatly upon ligand binding (compaction being a proxy for stabilization) [36, 65]. This finding is consistent with biochemical probing experiments on the FMN riboswitch [29, 37] that suggested that ligand binding-induced changes are limited to residues surrounding the ligand phosphate and to helix P1 (Fig. 3B).
The early report [30] that the glycine riboswitch binds its ligands cooperatively appears to be an artifact resulting from analysis of truncated riboswitch constructs lacking a functionally important K-turn motif [38, 39, 56, 65-68]. This K-turn is formed by a long-distance interaction between the 5′ end of the RNA and the linker region connecting the two aptamer domains [67, 68]. Inclusion of the K-turn enhanced glycine binding by ∼10-fold [68] and allowed for detection of glycine binding via isothermal titration calorimetry (ITC) at near-physiological Mg2+ concentrations, whereas no binding was detected for the RNAs lacking a K-turn under similar solution conditions [65]. SAXS experiments indicated that the K-turn-containing riboswitch is more compact than the K-turn-lacking riboswitch even in the absence of glycine, consistent with partial folding of the riboswitch [65]. Recently, mutational analysis of each glycine-binding site has suggested that, although independent and not cooperative, the two ligand sites communicate via pseudo-quaternary contacts α, β, and γ [56] (Fig. 4). In the case of the V. cholerae riboswitch, mutational analysis suggests that while binding of glycine to both domains is required for domain-domain interaction, aptamer 1 is more critical for dimerization and aptamer 2 relays glycine binding to gene expression [56].
Like the glycine riboswitch, the c-di-AMP riboswitch binds two ligands, raising the possibility of cooperativity. However, the functional consequence of binding two c-di-AMP molecules is not entirely clear. Biochemical probing experiments suggested that the c-di-AMP riboswitch binds c-di-AMP with 100 pM to 1 nM dissociation constant [58], but these experiments were performed in relatively high Mg2+ concentrations (10 or 20 mM), which likely stabilizes ligand binding by shielding the negatively charged phosphate backbones from the two halves of the riboswitch brought together by the anionic c-di-AMP molecules. ITC experiments indicate the affinity to be ∼10-300 nM across a variety of species [34-36]. Mutations eliminating CDA binding to site 1 (Fig. 5 A, yellow) affect binding to both sites and thus are more detrimental than mutations to the less conserved site 2 (Fig. 5A, orange) [35], but how each site contributes to gene expression remains to be established. The cellular concentration of c-di-AMP has been estimated to be ∼1-5 μM in B. subtilis depending on whether the cells are growing or sporulating [69], and other c-di-AMP-binding proteins have dissociation constants in the mid-nM to low-μM range [70-72]. Thus, cooperative binding may allow for a more exquisitely tuned response to fluctuating c-di-AMP levels, or for rapid binding in kinetically controlled systems [73]. Based on its proximity to the pseudoknot, binding of CDA2 may facilitate pseudoknot folding while CDA1 facilitates overall folding, which is consistent with the observation that mutation of site 1 also affects binding to site 2 [35]. Alternatively, as for the other RNAs above, symmetry may largely play a stabilizing role, contributing to RNA folding rather than ligand binding cooperativity.
While binding a twofold symmetric ligand like c-di-AMP using a twofold pseudosymmetric RNA is an elegant solution, this is clearly not the only way. There are two known classes of riboswitches that recognize the closely-related bacterial second messenger cyclic diguanosine monophosphate (c-di-GMP) [74, 75]. These riboswitches recognize their cognate ligand asymmetrically as monomers, and their structures do not exhibit any pseudosymmetry [36, 76-78]. C-di-GMP riboswitches bind their ligands in a C-shape conformation, in which a single helical stack continues through the G-bases of the ligand. In contrast, c-di-AMP riboswitches bind their cognate second messenger in an extended conformation, in which the Watson-Crick faces of the adenine moieties point in opposite directions (Fig. 5A). As for the c-di-GMP riboswitch [76], SAXS experiments have suggested that the c-di-AMP riboswitch greatly compacts in the presence of ligand [36], consistent with a large conformational change likely tied to regulation of gene expression. The numerous interactions between c-di-AMP and the riboswitch bridge the two halves of the RNA, contributing to ∼2/3 of the overall dimerization interface [36].
Global symmetry in RNA quaternary structure results in diverse, if not idiosyncratic, functional implications. As the role of each ligand to c-di-AMP riboswitch function remains to be established, the example of domain-domain communication in the glycine riboswitch serves as a guide for how multiple ligands inform function in the absence of cooperativity. For the FMN riboswitch, stability appears to be a consequence of its pseudosymmetrical fold. And bacteriophage ϕ29 pRNAs use symmetry and symmetry mismatch to enhance and diminish interactions with other viral components, respectively.
Evolution of RNA quaternary structure
The general function and evolution of quaternary structure and symmetry is a complex topic and has been discussed in depth for proteins [1, 62, 79]. For example, viral structural proteins possess symmetry for the sake of genomic thrift, as in the cases of icosahedrally symmetric capsids, which associate to form megadalton complexes that were ultimately translated from a relatively meager sequence of 500-1000 nucleotides. Similarly, it is advantageous for the ϕ29 bacteriophage to utilize a 5-fold symmetric pRNA as part of the motor that packages viral DNA [22] because the monomeric pRNA sequence requires 5-fold less sequence space in the phage genome. For this reason, examples of symmetry within viral proteins and nucleic acids abound, as genomic space is limited physically by the amount of RNA or DNA that can fit within the virus particle. Evolution of symmetry is believed to occur in a stepwise fashion, from no symmetry to cyclic symmetry to dihedral symmetry (e.g., C1 to C2 to D2), with evolution between cyclic symmetries forbidden (e.g., from C2 to C3) and higher symmetries observed less often than lower symmetries [80]. For pRNA, the evolution of C5 or C6 symmetry was likely influenced by the viral capsid's symmetry through protein-RNA interaction, followed by the evolution of the RNA-RNA interface.
For riboswitches, which by definition fold into one state or another depending on cellular conditions, the evolution of symmetry may solve the need for folding rapidly and stably. Folding of symmetric complexes has been predicted to take place on a smoother folding landscape than asymmetric complexes due to the preservation of symmetry during folding [63]. This feature may be particularly relevant to riboswitches, which fold cotranscriptionally and rapidly decide the fate of gene expression [25]. As only 3 of the 23 characterized riboswitch classes display pseudo-quaternary structure, one might speculate that true quaternary structure requires order on a timescale too slow for riboswitch activation. In principle, evolutionary sequence space can be explored more rapidly in a dimeric structure as mutations to each amino acid or nucleotide affect two residues at the dimer interface instead of one [1, 42]. Thus, once weak dimerization has been achieved, the pathway to a more stable dimeric interaction, if advantageous, would be relatively rapid. After a gene duplication event, the evolution of each monomer would proceed independently, suggesting that the fastest route toward pseudo-dimer evolution would be through the evolution of at least weak dimerization prior to gene duplication. This type of two-stage evolution may be particularly important for RNA structures, which are thought to evolve on rugged fitness landscapes [81, 82].
The evolution of symmetry within the FMN riboswitch is puzzling as the riboswitch only binds a single ligand which breaks the symmetry imposed by the repeated RNA fold. One might speculate that the riboswitch had originated from a gene duplication event in which the proto-riboswitch bound flavins pseudosymmetrically by virtue of the approximately symmetrical isoalloxazine ring of the FMN molecule (Fig. 3B). Subsequently, evolution of asymmetry within the ligand binding site resulted in a better functioning riboswitch that maintained overall symmetry due to the stability of the fold. Evolution of the c-di-AMP riboswitch is also unclear due to the long-range interactions involved in forming the c-di-AMP binding sites (Fig. 5). For these riboswitch classes, a monomeric “half-riboswitch” with weaker ligand binding may very well have been a step in their evolutionary history.
Unlike the FMN and c-di-AMP riboswitches, which appear to be obligate dimers, the evolution of tandem glycine riboswitches can be easily rationalized as a gene duplication event from the single-domain riboswitches observed in ∼12% of organisms [83]. As glycine is a small molecule, initial duplication of the glycine riboswitch may have allowed for higher specificity to glycine by requiring two bound glycine molecules for gene activation. Evolution of the tandem riboswitch configuration then resulted in the loss of the first expression domain and the creation of domain-domain interactions for a more nuanced response to glycine. Other tandem riboswitch configurations respond to the same ligand or different ligands [52], but they are rare [83] and appear to operate independently, which does not necessitate interactions between the tandem domains. An exception is the glutamine riboswitch, which exists in tandem configurations of two or three aptamer domains with a single expression domain [53].
In summary, the evolution of symmetric RNA quaternary structures is a consequence of the interaction with symmetric binding partners, as in the case of pRNA, or of gene duplication followed by domain-domain association (glycine riboswitch). In the case of the FMN and c-di-AMP riboswitches, the construction of monomeric versions of these riboswitches in vitro would at least demonstrate the plausibility of an evolutionary path from asymmetric monomer to their symmetric counterparts.
RNA symmetry at large: future prospects for new quaternary structures
Common features of the RNA topologies and interfaces described above are well-known themes that govern RNA tertiary structure. Kissing-loop interactions bring together pRNAs to form an integral part of the bacteriophage DNA packaging motor. Both the c-di-AMP and glycine riboswitches contain extensive A-minor interactions between the two domains, with the ligand mediating those interactions for c-di-AMP riboswitches. A-minor interactions are common through RNA-RNA interfaces [55, 84] and stitch together the three-helix junctions of the c-di-AMP riboswitches and even play a role in bringing P1 and P3 together in the FMN riboswitch [37]. Both the c-di-AMP and FMN riboswitches utilize NAA(A) tetraloop-like motifs, although the contexts are different.
One might have predicted symmetry within two-domain glycine riboswitches based on sequence alone because the two aptamer domains are so similar. However, discovering the pseudosymmetry within c-di-AMP and FMN riboswitches was unexpected. Thus, unless RNA structure prediction improves markedly, the search for global symmetry within known RNA motifs will continue to rely heavily on structural techniques like cryo-EM [22] and crystallography [34-39]. Viruses have been an ample source of macromolecular symmetry in the past (reviewed in [85]), so we may yet uncover new symmetry or pseudosymmetry in viral RNA structures, either as binders to symmetric viral proteins or as independently functioning motifs. Likewise, global pseudosymmetry is a theme in some riboswitch structures, and the future investigation of other riboswitches like tandem glutamine aptamers [53] may deepen our knowledge of how structured RNA molecules achieve gene regulation.
Highlights.
- Quaternary structure arises from the association of multiple macromolecular chains
- In contrast to proteins, few RNAs with symmetric quaternary structures are known
- Some riboswitches are now known to have intramolecular, pseudo-quaternary symmetry
- Pseudo-quaternary structure stabilizes riboswitches and tunes their ligand binding response
Acknowledgments
This work was supported in part by the Intramural Program of the National Heart, Lung and Blood Institute, NIH, and a Lenfant Biomedical Postdoctoral Fellowship awarded to C. P. J.
Glossary Box
A-minor motif
A widespread class of RNA interactions between the minor groove edge of A nucleobases and the minor groove of a neighboring helix
Allostery
A conformational change within a macromolecule at one site induced by binding of a small molecule or ligand to a second distinct binding site (see original description in [88])
Aptamer domain
The domain of the riboswitch that primarily interacts with and senses the ligand
Bulge
A single-stranded RNA sequence interrupting an otherwise continuous helix
Cyclic diadenosine monophosphate (c-di-AMP)
C-di-AMP consists of two adenosine monophosphate molecules linked via 3′-to-5′ linkages and serves as a bacterial second messenger regulating cell wall homeostasis
Cyclic diguanosine monophosphate (c-di-GMP)
C-di-GMP is a cyclic dinucleotide made of two guanosine monophosphates linked via 3′-to-5′ linkages and serves as a bacterial second messenger regulating biofilm formation
Cooperativity
Cooperativity describes the observed change in ligand binding affinity upon binding of another identical ligand, exemplified by O2 binding to hemoglobin
Expression platform domain
The domain of the riboswitch that alters gene expression, typically by changing base pairing that affects transcription or translation
Flavin mononucleotide (FMN)
FMN is an enzyme cofactor used to catalyze one- and two-electron transfer reactions for a wide variety of biological processes.
Helix
Also referred to as a duplex, or a paired region in an RNA structure
Kissing-loop interaction
This interaction refers to Watson-Crick pairing between the loop nucleotides of two RNA stemloops to form a continuous helical stack from one RNA stem through the kissing loop to the second RNA stem. See example in Figure 2
Kink turn (K-turn)
A K-turn is an RNA structural motif consisting of two helices, one of them containing two noncanonical G•A sheared pairs, separated by a three-nucleotide bulge that results in a ∼120° turn in the direction of the strands
Loop
An unpaired region in an RNA structure, either at the end of RNA stemloop or connecting two RNA helices
Point-group symmetry
Symmetry about a fixed origin such that all symmetry operations intersect at the origin
Prohead RNA (pRNA)
pRNA is a homooligomeric RNA that functions as a structural element connecting the head and tail of the ϕ29 bacteriophage
Pseudoknot
Often associated with translation regulation, a pseudoknot is an RNA motif in which the loop of the stemloop binds through Watson-Crick pairing to a distant sequence
Riboswitch
Often found in noncoding regions of bacterial mRNAs, riboswitches are a class of structured noncoding RNAs that coordinate highly specific binding of small molecules or metabolites with the regulation of gene expression
Small-angle X-ray scattering (SAXS)
SAXS is a widely used technique for examining the overall size and shape of a macromolecule in solution, potentially yielding structural information at a resolution of ∼20 Å
Tetraloop
A tetraloop consists of a Watson-Crick paired helix ending in a 4-nucleotide loop (e.g., GAAA), which is highly stable due to the loop forming noncanonical base pairs and stacking interactions
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Goodsell DS, Olson AJ. Structural symmetry and protein function. Annual Review of Biophysics and Biomolecular Structure. 2000;29:105–153. doi: 10.1146/annurev.biophys.29.1.105. [DOI] [PubMed] [Google Scholar]
- 2.Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171:737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- 3.Robertus JD, et al. Structure of yeast phenylalanine tRNA at 3 Å resolution. Nature. 1974;250:546–551. doi: 10.1038/250546a0. [DOI] [PubMed] [Google Scholar]
- 4.Cate JH, et al. Crystal structure of a group I ribozyme domain: principles of RNA packing. Science. 1996;273:1678–1685. doi: 10.1126/science.273.5282.1678. [DOI] [PubMed] [Google Scholar]
- 5.Ferré-D'Amaré AR, et al. Crystal structure of a hepatitis delta virus ribozyme. Nature. 1998;395:567–574. doi: 10.1038/26912. [DOI] [PubMed] [Google Scholar]
- 6.Rupert PB, Ferré-D'Amaré A R. Crystal structure of a hairpin ribozyme-inhibitor complex with implications for catalysis. Nature. 2001;410:780–786. doi: 10.1038/35071009. [DOI] [PubMed] [Google Scholar]
- 7.Pley HW, et al. Three-dimensional structure of a hammerhead ribozyme. Nature. 1994;372:68–74. doi: 10.1038/372068a0. [DOI] [PubMed] [Google Scholar]
- 8.Ban N, et al. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 9.Schluenzen F, et al. Structure of functionally activated small ribosomal subunit at 3.3 ångströms resolution. Cell. 2000;102:615–623. doi: 10.1016/s0092-8674(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 10.Yusupov MM, et al. Crystal structure of the ribosome at 5.5 Å resolution. Science. 2001;292:883–896. doi: 10.1126/science.1060089. [DOI] [PubMed] [Google Scholar]
- 11.Wimberly BT, et al. Structure of the 30S ribosomal subunit. Nature. 2000;407:327–339. doi: 10.1038/35030006. [DOI] [PubMed] [Google Scholar]
- 12.Bashan A, et al. Structural basis of the ribosomal machinery for peptide bond formation, translocation, and nascent chain progression. Molecular Cell. 2003;11:91–102. doi: 10.1016/s1097-2765(03)00009-1. [DOI] [PubMed] [Google Scholar]
- 13.Mujeeb A, et al. Structure of the dimer initiation complex of HIV-1 genomic RNA. Nature Structural Biology. 1998;5:432–436. doi: 10.1038/nsb0698-432. [DOI] [PubMed] [Google Scholar]
- 14.Marino JP, et al. Bent helix formation between RNA hairpins with complementary loops. Science. 1995;268:1448–1454. doi: 10.1126/science.7539549. [DOI] [PubMed] [Google Scholar]
- 15.Guo P, et al. Inter-RNA interaction of phage phi29 pRNA to form a hexameric complex for viral DNA transportation. Molecular Cell. 1998;2:149–155. doi: 10.1016/s1097-2765(00)80124-0. [DOI] [PubMed] [Google Scholar]
- 16.Neidle S, Balasubramanian S. Quadruplex Nucleic Acids. RSC Pub; 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grabow WW, et al. The right angle (RA) motif: a prevalent ribosomal RNA structural pattern found in group I introns. Journal of Molecular Biology. 2012;424:54–67. doi: 10.1016/j.jmb.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chworos A, et al. Building programmable jigsaw puzzles with RNA. Science. 2004;306:2068–2072. doi: 10.1126/science.1104686. [DOI] [PubMed] [Google Scholar]
- 19.Geary C, et al. RNA nanostructures. A single-stranded architecture for cotranscriptional folding of RNA nanostructures. Science. 2014;345:799–804. doi: 10.1126/science.1253920. [DOI] [PubMed] [Google Scholar]
- 20.Moore MD, Hu WS. HIV-1 RNA dimerization: It takes two to tango. AIDS Reviews. 2009;11:91–102. [PMC free article] [PubMed] [Google Scholar]
- 21.Ennifar E, et al. Crystal structures of coaxially stacked kissing complexes of the HIV-1 RNA dimerization initiation site. Nature Structural Biology. 2001;8:1064–1068. doi: 10.1038/nsb727. [DOI] [PubMed] [Google Scholar]
- 22.Simpson AA, et al. Structure of the bacteriophage phi29 DNA packaging motor. Nature. 2000;408:745–750. doi: 10.1038/35047129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo PX, et al. A small viral RNA is required for in vitro packaging of bacteriophage phi 29 DNA. Science. 1987;236:690–694. doi: 10.1126/science.3107124. [DOI] [PubMed] [Google Scholar]
- 24.Ding F, et al. Structure and assembly of the essential RNA ring component of a viral DNA packaging motor. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7357–7362. doi: 10.1073/pnas.1016690108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang J, et al. Ribozymes and riboswitches: modulation of RNA function by small molecules. Biochemistry. 2010;49:9123–9131. doi: 10.1021/bi1012645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Serganov A. The long and the short of riboswitches. Current Opinion in Structural Biology. 2009;19:251–259. doi: 10.1016/j.sbi.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Breaker RR. Riboswitches and the RNA world. Cold Spring Harbor Perspectives in Biology. 2012;4 doi: 10.1101/cshperspect.a003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dann CE, 3rd, et al. Structure and mechanism of a metal-sensing regulatory RNA. Cell. 2007;130:878–892. doi: 10.1016/j.cell.2007.06.051. [DOI] [PubMed] [Google Scholar]
- 29.Winkler WC, et al. An mRNA structure that controls gene expression by binding FMN. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:15908–15913. doi: 10.1073/pnas.212628899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mandal M, et al. A glycine-dependent riboswitch that uses cooperative binding to control gene expression. Science. 2004;306:275–279. doi: 10.1126/science.1100829. [DOI] [PubMed] [Google Scholar]
- 31.Baker JL, et al. Widespread genetic switches and toxicity resistance proteins for fluoride. Science. 2012;335:233–235. doi: 10.1126/science.1215063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grundy FJ, Henkin TM. tRNA as a positive regulator of transcription antitermination in B. subtilis. Cell. 1993;74:475–482. doi: 10.1016/0092-8674(93)80049-k. [DOI] [PubMed] [Google Scholar]
- 33.Mironov AS, et al. Sensing small molecules by nascent RNA: a mechanism to control transcription in bacteria. Cell. 2002;111:747–756. doi: 10.1016/s0092-8674(02)01134-0. [DOI] [PubMed] [Google Scholar]
- 34.Ren A, Patel DJ. c-di-AMP binds the ydaO riboswitch in two pseudo-symmetry-related pockets. Nature Chemical Biology. 2014;10:780–786. doi: 10.1038/nchembio.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao A, Serganov A. Structural insights into recognition of c-di-AMP by the ydaO riboswitch. Nature Chemical Biology. 2014;10:787–792. doi: 10.1038/nchembio.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones CP, Ferré-D'Amaré AR. Crystal structure of a c-di-AMP riboswitch reveals an internally pseudo-dimeric RNA. The EMBO Journal. 2014 doi: 10.15252/embj.201489209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Serganov A, et al. Coenzyme recognition and gene regulation by a flavin mononucleotide riboswitch. Nature. 2009;458:233–237. doi: 10.1038/nature07642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Butler EB, et al. Structural basis of cooperative ligand binding by the glycine riboswitch. Chemistry & Biology. 2011;18:293–298. doi: 10.1016/j.chembiol.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang L, et al. Structural insights into ligand recognition by a sensing domain of the cooperative glycine riboswitch. Molecular Cell. 2010;40:774–786. doi: 10.1016/j.molcel.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bloomfield VA, et al. Nucleic Acids: Structures, Properties, and Functions. University Science Books; 2000. [Google Scholar]
- 41.Janin J, Chothia C. The structure of protein-protein recognition sites. The Journal of Biological Chemistry. 1990;265:16027–16030. [PubMed] [Google Scholar]
- 42.Levy ED, Teichmann S. Structural, evolutionary, and assembly principles of protein oligomerization. Progress in Molecular Biology and Translational Science. 2013;117:25–51. doi: 10.1016/B978-0-12-386931-9.00002-7. [DOI] [PubMed] [Google Scholar]
- 43.Vitreschak AG, et al. Regulation of riboflavin biosynthesis and transport genes in bacteria by transcriptional and translational attenuation. Nucleic Acids Research. 2002;30:3141–3151. doi: 10.1093/nar/gkf433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deigan KE, Ferré-D'Amaré AR. Riboswitches: discovery of drugs that target bacterial gene-regulatory RNAs. Accounts of Chemical Research. 2011;44:1329–1338. doi: 10.1021/ar200039b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ott E, et al. The RFN riboswitch of Bacillus subtilis is a target for the antibiotic roseoflavin produced by Streptomyces davawensis. RNA Biology. 2009;6:276–280. doi: 10.4161/rna.6.3.8342. [DOI] [PubMed] [Google Scholar]
- 46.Lee ER, et al. Roseoflavin is a natural antibacterial compound that binds to FMN riboswitches and regulates gene expression. RNA Biology. 2009;6:187–194. doi: 10.4161/rna.6.2.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chan CW, et al. Structure and function of the T-loop structural motif in noncoding RNAs. Wiley Interdisciplinary Reviews RNA. 2013;4:507–522. doi: 10.1002/wrna.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang J, Ferré-D'Amaré AR. Co-crystal structure of a T-box riboswitch stem I domain in complex with its cognate tRNA. Nature. 2013;500:363–366. doi: 10.1038/nature12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jaeger L, et al. The UA_handle: a versatile submotif in stable RNA architectures. Nucleic Acids Research. 2009;37:215–230. doi: 10.1093/nar/gkn911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 51.Sudarsan N, et al. Thiamine pyrophosphate riboswitches are targets for the antimicrobial compound pyrithiamine. Chemistry & Biology. 2005;12:1325–1335. doi: 10.1016/j.chembiol.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 52.Sudarsan N, et al. Tandem riboswitch architectures exhibit complex gene control functions. Science. 2006;314:300–304. doi: 10.1126/science.1130716. [DOI] [PubMed] [Google Scholar]
- 53.Ames TD, Breaker RR. Bacterial aptamers that selectively bind glutamine. RNA Biology. 2011;8:82–89. doi: 10.4161/rna.8.1.13864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trausch JJ, et al. The structure of a tetrahydrofolate-sensing riboswitch reveals two ligand binding sites in a single aptamer. Structure. 2011;19:1413–1423. doi: 10.1016/j.str.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nissen P, et al. RNA tertiary interactions in the large ribosomal subunit: the A-minor motif. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:4899–4903. doi: 10.1073/pnas.081082398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ruff KM, Strobel SA. Ligand binding by the tandem glycine riboswitch depends on aptamer dimerization but not double ligand occupancy. RNA. 2014;20:1775–1788. doi: 10.1261/rna.047266.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Corrigan RM, Grundling A. Cyclic di-AMP: another second messenger enters the fray. Nature Reviews Microbiology. 2013;11:513–524. doi: 10.1038/nrmicro3069. [DOI] [PubMed] [Google Scholar]
- 58.Nelson JW, et al. Riboswitches in eubacteria sense the second messenger c-di-AMP. Nature Chemical Biology. 2013;9:834–839. doi: 10.1038/nchembio.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Block KF, et al. Evidence for widespread gene control function by the ydaO riboswitch candidate. Journal of Bacteriology. 2010;192:3983–3989. doi: 10.1128/JB.00450-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ferré-D'Amaré AR, et al. A general module for RNA crystallization. Journal of Molecular Biology. 1998;279:621–631. doi: 10.1006/jmbi.1998.1789. [DOI] [PubMed] [Google Scholar]
- 61.Hendrix RW. Symmetry mismatch and DNA packaging in large bacteriophages. Proceedings of the National Academy of Sciences of the United States of America. 1978;75:4779–4783. doi: 10.1073/pnas.75.10.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Monod J, et al. On the Nature of Allosteric Transitions: A Plausible Model. Journal of Molecular Biology. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 63.Wolynes PG. Symmetry and the energy landscapes of biomolecules. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:14249–14255. doi: 10.1073/pnas.93.25.14249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baird NJ, Ferré-D'Amaré AR. Idiosyncratically tuned switching behavior of riboswitch aptamer domains revealed by comparative small-angle X-ray scattering analysis. RNA. 2010;16:598–609. doi: 10.1261/rna.1852310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baird NJ, Ferré-D'Amaré AR. Modulation of quaternary structure and enhancement of ligand binding by the K-turn of tandem glycine riboswitches. RNA. 2013;19:167–176. doi: 10.1261/rna.036269.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Klein DJ, et al. The kink-turn: a new RNA secondary structure motif. The EMBO Journal. 2001;20:4214–4221. doi: 10.1093/emboj/20.15.4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kladwang W, et al. Automated RNA structure prediction uncovers a kink-turn linker in double glycine riboswitches. Journal of the American Chemical Society. 2012;134:1404–1407. doi: 10.1021/ja2093508. [DOI] [PubMed] [Google Scholar]
- 68.Sherman EM, et al. An energetically beneficial leader-linker interaction abolishes ligand-binding cooperativity in glycine riboswitches. RNA. 2012;18:496–507. doi: 10.1261/rna.031286.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oppenheimer-Shaanan Y, et al. c-di-AMP reports DNA integrity during sporulation in Bacillus subtilis. EMBO Reports. 2011;12:594–601. doi: 10.1038/embor.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Corrigan RM, et al. Systematic identification of conserved bacterial c-di-AMP receptor proteins. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:9084–9089. doi: 10.1073/pnas.1300595110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sureka K, et al. The cyclic dinucleotide c-di-AMP is an allosteric regulator of metabolic enzyme function. Cell. 2014;158:1389–1401. doi: 10.1016/j.cell.2014.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huynh TN, et al. An HD-domain phosphodiesterase mediates cooperative hydrolysis of c-di-AMP to affect bacterial growth and virulence. Proceedings of the National Academy of Sciences of the United States of America. 2015 doi: 10.1073/pnas.1416485112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang J, et al. Global analysis of riboswitches by small-angle X-ray scattering and calorimetry. Biochimica et Biophysica Acta. 2014 doi: 10.1016/j.bbagrm.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sudarsan N, et al. Riboswitches in eubacteria sense the second messenger cyclic di-GMP. Science. 2008;321:411–413. doi: 10.1126/science.1159519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee ER, et al. An allosteric self-splicing ribozyme triggered by a bacterial second messenger. Science. 2010;329:845–848. doi: 10.1126/science.1190713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kulshina N, et al. Recognition of the bacterial second messenger cyclic diguanylate by its cognate riboswitch. Nature Structural & Molecular Biology. 2009;16:1212–1217. doi: 10.1038/nsmb.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Smith KD, et al. Structural basis of ligand binding by a c-di-GMP riboswitch. Nature Structural & Molecular Biology. 2009;16:1218–1223. doi: 10.1038/nsmb.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Smith KD, et al. Structural basis of differential ligand recognition by two classes of bis-(3′-5′)-cyclic dimeric guanosine monophosphate-binding riboswitches. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7757–7762. doi: 10.1073/pnas.1018857108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Janin J, et al. Protein-protein interaction and quaternary structure. Quarterly Reviews of Biophysics. 2008;41:133–180. doi: 10.1017/S0033583508004708. [DOI] [PubMed] [Google Scholar]
- 80.Levy ED, et al. Assembly reflects evolution of protein complexes. Nature. 2008;453:1262–1265. doi: 10.1038/nature06942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pitt JN, Ferré-D'Amaré AR. Rapid construction of empirical RNA fitness landscapes. Science. 2010;330:376–379. doi: 10.1126/science.1192001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lau MW, Ferré-D'Amaré AR. An in vitro evolved glmS ribozyme has the wild-type fold but loses coenzyme dependence. Nature Chemical Biology. 2013;9:805–810. doi: 10.1038/nchembio.1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Barrick JE, Breaker RR. The distributions, mechanisms, and structures of metabolite-binding riboswitches. Genome Biology. 2007;8:R239. doi: 10.1186/gb-2007-8-11-r239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Geary C, et al. Promoting RNA helical stacking via A-minor junctions. Nucleic Acids Research. 2011;39:1066–1080. doi: 10.1093/nar/gkq748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rossmann MG. Structure of viruses: a short history. Quarterly Reviews of Biophysics. 2013;46:133–180. doi: 10.1017/S0033583513000012. [DOI] [PubMed] [Google Scholar]
- 86.Lee AJ, Crothers DM. The solution structure of an RNA loop-loop complex: the ColE1 inverted loop sequence. Structure. 1998;6:993–1005. doi: 10.1016/s0969-2126(98)00101-4. [DOI] [PubMed] [Google Scholar]
- 87.Leontis NB, Westhof E. Geometric nomenclature and classification of RNA base pairs. RNA. 2001;7:499–512. doi: 10.1017/s1355838201002515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Monod J, et al. Allosteric proteins and cellular control systems. Journal of Molecular Biology. 1963;6:306–329. doi: 10.1016/s0022-2836(63)80091-1. [DOI] [PubMed] [Google Scholar]
- 89.Svedberg T. Mass and size of protein molecules. Nature. 1929;123:871. [Google Scholar]
- 90.Perutz MF, et al. Three-dimensional Fourier synthesis of horse oxyhaemoglobin at 2.8 Å resolution: (1) x-ray analysis. Nature. 1968;219:29–32. doi: 10.1038/219029a0. [DOI] [PubMed] [Google Scholar]
- 91.Bolton W, Perutz MF. Three dimensional fourier synthesis of horse deoxyhaemoglobin at 2.8 Ångström units resolution. Nature. 1970;228:551–552. doi: 10.1038/228551a0. [DOI] [PubMed] [Google Scholar]