Inhibition of Myocardin-Related Transcription Factor/Serum Response Factor Signaling Decreases Lung Fibrosis and Promotes Mesenchymal Cell Apoptosis (original) (raw)
Abstract
Myofibroblasts are crucial to the pathogenesis of tissue fibrosis. Their formation of stress fibers results in the release of myocardin-related transcription factor (MRTF), a transcriptional coactivator of serum response factor (SRF). MRTF-A (Mkl1)-deficient mice are protected from lung fibrosis. We hypothesized that the SRF/MRTF pathway inhibitor CCG-203971 would modulate myofibroblast function in vitro and limit lung fibrosis in vivo. Normal and idiopathic pulmonary fibrosis lung fibroblasts were treated with/without CCG-203971 (N-[4-chlorophenyl]-1-[3-(2-furanyl)benzoyl]-3-piperidine carboxamide) and/or Fas-activating antibody in the presence/absence of transforming growth factor (TGF)-β1, and apoptosis was assessed. In vivo studies examined the effect of therapeutically administered CCG-203971 on lung fibrosis in two distinct murine models of fibrosis induced by bleomycin or targeted type II alveolar epithelial injury. In vitro, CCG-203971 prevented nuclear localization of MRTF-A; increased the apoptotic susceptibility of normal and idiopathic pulmonary fibrosis fibroblasts; blocked TGF-β1–induced myofibroblast differentiation; and inhibited TGF-β1–induced expression of fibronectin, X-linked inhibitor of apoptosis, and plasminogen activator inhibitor-1. TGF-β1 did not protect fibroblasts or myofibroblasts from apoptosis in the presence of CCG-203971. In vivo, CCG-203971 significantly reduced lung collagen content in both murine models while decreasing alveolar plasminogen activator inhibitor-1 and promoting myofibroblast apoptosis. These data support a central role of the SRF/MRTF pathway in the pathobiology of lung fibrosis and suggest that its inhibition can help resolve lung fibrosis by promoting fibroblast apoptosis.
Organ fibrosis is characterized by the accumulation of a collagen-rich extracellular matrix and the distortion of normal tissue architecture. This process typically impairs organ function, and in the lung, derangement of the delicate alveolar structure results in significant morbidity and mortality.1,2 Although many aspects of the pathogenesis of tissue fibrosis remain undefined, it is now well accepted that fibroblasts are important contributors to the production of the excessive extracellular matrix.3 Furthermore, in the context of organ fibrosis, fibroblasts consistently differentiate into a myofibroblast phenotype as defined by the formation of F-actin–containing stress fibers and the increased expression of α-smooth muscle actin (SMA).3,4 In addition to the enhanced production of α-SMA, myofibroblasts elaborate extracellular matrix proteins, including fibronectin and collagen, and exhibit an increased resistance to apoptosis.4 In conjunction with their stress fiber formation, myofibroblasts exert contractile forces on their environment, thereby accentuating the architectural distortion that is characteristic of tissue fibrosis. Myofibroblast contractile forces also contribute to the liberation of the profibrotic mediator transforming growth factor (TGF)-β1, thereby exacerbating the fibrotic process.5–8
Because of the crucial role that myofibroblasts play in tissue fibrosis, substantial effort has been directed toward understanding the mechanisms by which these cells undergo differentiation from fibroblasts. Through these investigations, multiple soluble mediators, including TGF-β1, endothelin, connective tissue growth factor, and lysophosphatidic acid, have been found to promote the transition.4,9 Beyond soluble mediators, the presence of a mechanically stiff extracellular substrate, as measured by resistance to mechanical deformation, can also stimulate fibroblasts to acquire a myofibroblast phenotype. Although the induction of fibroblast activation by a stiff environment is in part due to enhanced TGF-β activation, recent evidence also indicates that their phenotype is modified in response to the rigidity of the substrate to which the cell is attached via a process of mechanotransduction that is independent of enhanced TGF-β activation.8,10
The induction of fibroblast-to-myofibroblast transition, by either a soluble profibrotic mediator or a stiff extracellular matrix, depends on the activation of intracellular signaling pathways. The Rho family of GTPases and their target kinase, Rho-associated kinase (ROCK), were hypothesized to participate in myofibroblast differentiation and contraction based on the observation that these signaling molecules are important for actin polymerization, a crucial element of stress fiber formation.9,11–14 Importantly, recent studies have substantiated a key role for Rho/ROCK in both soluble mediator and stiff matrix-induced fibroblast-to-myofibroblast transition. Specifically, treatment of human lung fibroblasts with an inhibitor of ROCK significantly impeded the TGF-β–mediated acquisition of a myofibroblast phenotype.11,15 Furthermore, the inhibition of Rho/ROCK signaling disrupted myofibroblast differentiation that was induced when cells were cultured on a stiff extracellular substrate, and targeting the Rho/ROCK signaling pathway reduced the severity of fibrosis in an animal model of lung scarring.13
Besides impairing myofibroblast differentiation through the disruption of stress fiber formation, the targeting of Rho/ROCK alters the gene expression profile of (myo)fibroblasts.9,14 The concurrent inhibition of stress fiber formation and altered gene expression implicates a connection between these processes during myofibroblast differentiation, and myocardin-related transcription factor (MRTF) has been identified as a crucial link.14 MRTF functions as a transcriptional coactivator that, along with serum response factor (SRF), regulates the expression of myofibroblast genes, including α-SMA and collagen.3,9,14 SRF/MRTF signaling is intricately regulated by actin polymerization; when actin is depolymerized, MRTF is bound to the globular form and sequestered in the cytoplasm. During stress fiber formation, globular actin polymerizes into F-actin and releases MRTF, enabling it to translocate into the nucleus. Both soluble and mechanical stimuli associated with fibrosis, including TGF-β1 and exposure to stiff substrates, have been found to promote MRTF nuclear transport in fibroblasts.9,14,16–19 Furthermore, recent studies showed that the interruption of SRF/MRTF signaling attenuates myofibroblast differentiation and extracellular matrix synthesis in colonic fibroblasts and human skin fibroblasts from patients with scleroderma.16,20
Given the central role of SRF/MRTF signaling in myofibroblast differentiation, we hypothesized that inhibiting this pathway would prevent myofibroblast activation and reduce the severity of fibrosis in animal models of lung injury. To target this signaling pathway, we used a specific inhibitor, CCG-203971, a chemically modified derivative of CCG-1423, a compound which is known to block the nuclear localization of MRTF.21 CCG-1423 was initially identified by a high-throughput screen to identify small-molecule inhibitors of the RhoA signaling pathways.22 A recent report indicates that CCG-1423 binds to the N-terminal basic domain of MRTF, blocks its nuclear localization signal, prevents its interaction with importin α/β1, and induces sequestration in the cytoplasm.23 The precise interactions by which CCG-203971 inhibits SRF/MRTF have yet to be elucidated, although CCG-100602, another derivative of CCG-1423, has recently been shown to prevent the nuclear accumulation of MRTF in colon fibroblasts exposed to TGF-β1.16,21
Herein, we demonstrate that CCG-203971 inhibits nuclear accumulation of MRTF-A in lung fibroblasts, prevents myofibroblast differentiation, and reduces fibrosis in two distinct murine models. Moreover, we demonstrate that blockade of SRF/MRTF signaling affects several profibrotic mechanisms, including up-regulation of X-linked inhibitor of apoptosis (XIAP), fibroblast and myofibroblast resistance to apoptosis, and expression of plasminogen activator inhibitor (PAI)-1.
Materials and Methods
Cell Lines and Culture
CCL-210 (normal adult primary human lung) and IMR-90 (normal primary human fetal lung) fibroblasts were obtained from ATCC (Manassas, VA). Cells between passages 8 and 16 were cultured in Dulbecco’s modified Eagle's medium supplemented with 5% fetal bovine serum, and growth arrested for 16 to 24 hours before treatment as previously described.24 Deidentified normal and idiopathic pulmonary fibrosis (IPF) lung fibroblasts were provided by E.S.W. and cultured with the same protocol as the CCL-210 and IMR-90 fibroblasts, but using 10% fetal bovine serum. Mkl1 (MRTF-A) knockout murine lung fibroblasts were provided by Y.Z. (University of Alabama-Birmingham, Birmingham, AL),13 These cells were cultured in minimal essential medium supplemented with 20% fetal bovine serum.13 Because of substantial death within 2 to 4 hours of serum deprivation, treatments were given in media containing 5% fetal bovine serum. Additionally, these murine cells demonstrated increased sensitivity to CCG-203971, necessitating a reduction in concentration from 30 to 10 μmol/L.
Antibodies and Reagents
N-(4-chlorophenyl)-1-[3-(2-furanyl)benzoyl]-3-piperidinecarboxamide (CCG-203971) was synthesized by the Vahlteich Medicinal Chemistry Core(University of Michigan, Ann Arbor, MI) and provided by S.D.L..21 Porcine TGF-β1 was from R&D Systems (Minneapolis, MN). The activating anti-Fas antibody (clone CH11, designated as Fas-Ab) was purchased from Millipore (Billerica, MA). Antibodies to α-SMA and total fibronectin and fluorescein isothiocyanate–conjugated anti–α-SMA antibody were purchased from Sigma-Aldrich (St. Louis, MO). Antibodies to XIAP, glyceraldehyde-3-phosphate dehydrogenase, poly-(ADP-ribose) polymerase (PARP), phosphorylated Smad3, and total Smad3 were purchased from Cell Signaling (Danvers, MA). The antibody to MRTF-A was purchased from Santa Cruz Biotechnology (Dallas, TX). Horseradish peroxidase–conjugated secondary antibodies were purchased from Pierce (Rockford, IL). The In Situ Cell Death Detection Kit, TMR red, was purchased from Roche Life Science (Indianapolis, IN).
Immunofluorescence Staining
IMR-90 cells were cultured and treated in dishes containing sterilized glass coverslips (Fisher Scientific, Pittsburgh, PA), and immunofluorescence staining was performed as previously described25 using rabbit anti–MRTF-A primary antibody (Santa Cruz Biotechnology, Dallas, TX) at 1:50 dilution and AlexaFluor 555-conjugated goat anti-rabbit secondary antibody (Molecular Probes, Eugene, OR) (1:500 dilution). Images were acquired using an Olympus BX60 microscope with DP72 camera and CellSens Standard imaging software version 1.11 (Olympus America, Center Valley, PA). To quantify the nuclear-to-cytoplasmic ratio, images were imported into ImageJ software version 1.45s (NIH, Bethesda, MD). Using the CellMask stain, individual cells were outlined and the optical density of MRTF-A staining was measured and adjusted for the area of the cell. Next, the DAPI stain was used to similarly outline the nucleus and calculate the density of MRTF-A staining within the nucleus. The cytoplasmic fraction was determined by subtracting the nuclear fraction from the total cell calculation, and the nuclear-to-cytoplasmic ratio was determined by dividing the nuclear signal by the cytoplasmic signal.
Bleomycin Model of Lung Fibrosis
Weight- and age-matched (18 to 22 g at 6 to 8 weeks of age) C57BL/6 mice were anesthetized with ketamine and xylazine. A 0.5-cm incision was made in the neck to expose the trachea. Sterile bleomycin [1.2 U/kg in 50 μL of sterile phosphate-buffered saline (PBS)] was administered intratracheally with a 1.0-mL tuberculin syringe, and the incision was closed with surgical glue.
Targeted Type II Alveolar Epithelial Cell Injury Model of Lung Fibrosis
C57BL/6 mice aged 6 to 8 weeks and expressing the human diphtheria toxin (DT) receptor (DTR) in an alveolar epithelial cell (AEC)-restricted manner downstream of the surfactant protein C promoter (SPC-DTR+) and DTR– (wild-type) mice were injected with DT 10.0 μg/kg i.p. once daily for 14 days as previously described.26 Control mice were injected for the same duration with 100 μL of PBS alone.
In Vivo CCG-203971 Treatment
For both the bleomycin and targeted type II AEC injury models, 100 mg/kg of CCG-203971 dissolved in 50 μL of dimethyl sulfoxide (DMSO) was administered b.i.d. by i.p. injection20 beginning on day 11 of each model. Control mice received 50 μL of DMSO vehicle b.i.d. beginning at the same time point.
TUNEL Staining
Lungs were perfused with PBS, inflated with intratracheal OCT, removed, and immediately frozen in a dry-ice alcohol bath and stored at −80°C. Lung sections (7 μm) were fixed, mounted with ProLong Gold Antifade Mountant with DAPI (Life Technologies, Carlsbad, CA), permeabilized, and immunostained as previously described.27 Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed with the In Situ Cell Death Detection Kit, TMR Red, per the manufacturer's instruction manual. Fluorescein isothiocyanate–conjugated α-SMA staining was performed with a 1:200 dilution. Sections were visualized on an Olympus BX-51 fluorescence microscope and images were captured with an Olympus DP-70 camera and analyzed using DP controller software version 3.1.1.267. Numbers of TUNEL-positive cells were quantified from ten 400× fields for each mouse by an observer blinded to the treatment group (K.K.K.).
Lung Histological Examination and Picrosirius Red Stain
After perfusion of the pulmonary vasculature with PBS, the left lung was inflation-fixed at 25 cm H2O pressure with 10% neutral-buffered formalin, removed en bloc, further fixed in 10% neutral-buffered formalin overnight, and then paraffin-embedded. Five-micron sections were then stained using hematoxylin and eosin or picrosirius red.28
Hydroxyproline Assay
The hydroxyproline concentration was measured in both lungs as previously described.28
Western Immunoblot Analysis and Densitometry
Whole-cell lysates were collected and subjected to SDS-PAGE and Western blot analysis as previously described.24 All Western blots were stripped and reprobed for glyceraldehyde-3-phosphate dehydrogenase or, in the case of phosphorylated Smad3, for total Smad3. Densitometry and quantification of Western blots were performed with ImageJ software version 1.45s as previously detailed.24
Assessment of Apoptosis
Cleaved PARP was assessed via Western blot analysis. Histone-associated DNA fragments were detected using the Cell Death Detection enzyme-linked immunosorbent assay (ELISA) kit (Roche Applied Science, Indianapolis, IN) according to the manufacturer's instructions. Kinetic assessments of apoptosis were made by the analysis of caspase-3/7 activation using fluorescence imaging. For this assay, lung fibroblasts were seeded into a 6- or 24-well plate and cultured to approximately 50% to 60% confluence. After 16 hours of serum deprivation, cells were washed with PBS and placed in serum-free media containing 500 μL of CellPlayer 96-well kinetic caspase-3/7 reagent (Essen BioScience, Ann Arbor, MI) at a final concentration of 0.5 μmol/L. The activation of caspase-3/7, a marker of apoptosis, resulted in green fluorescence staining of nuclear DNA. Using an IncuCyte system (Essen BioScience), time-lapsed images were obtained with ×20 magnification from nine spots within each well of a 24-well plate or 16 spots per well in a 6-well plate every 2 hours.29 IncuCyte software version 2011A (Essen Bioscience, Ann Arbor MI) was used to quantify the fluorescent signal, which is represented as the object count per image.
PAI-1 Measurement
Endogenous total and/or active murine PAI-1 concentrations were measured in bronchoalveolar lavage fluid and cell culture supernatants using a carboxylated microsphere–based ELISA as previously described.30
Statistical Analysis
Comparisons were made with either analysis of variance with multiple comparisons post-test or, when comparing multiple variables over time, 2-way analysis of variance with multiple comparisons using GraphPad Prism version 6.01 (GraphPad Software, Inc., San Diego, CA).
Results
CCG-203971 Inhibits MRTF-A Nuclear Localization and Impairs TGF-β1–Induced Myofibroblast Differentiation and Fibronectin Deposition
Prior studies have implicated Rho and SRF/MRTF-mediated signaling pathways in fibroblast differentiation and collagen production.13,15,16,20,31 To examine the effect of a novel specific inhibitor of this pathway, CCG-203971, on TGF-β1–induced normal lung fibroblast differentiation and activation, we examined nuclear localization of MRTF-A in normal lung fibroblasts (Figure 1). IMR-90 fibroblasts cultured on glass coverslips were treated with/without TGF-β1 and/or CCG-203971 for 24 hours. The distribution of MRTF-A was assessed by immunofluorescence staining (Figure 1A), and the nuclear-to-cytoplasmic ratio was quantified (Figure 1B). CCG-203971, in the presence or absence of TGF-β1, led to a significant reduction in MRTF-A nuclear localization.
Figure 1.

CCG-203971 (CCG) inhibits myocardin-related transcription factor A (MRTF-A) nuclear localization. IMR-90 fibroblasts were cultured on glass coverslips and treated with/without 2 ng/mL transforming growth factor (TGF)-β1 and/or 30 μmol/L CCG-203971 for 24 hours. A: The distribution of MRTF-A was assessed by immunofluorescence staining. B: The nuclear-to-cytoplasmic ratio of MRTF-A was determined for each condition as described in Materials and Methods. Data are expressed as means ± SEM. ∗∗P < 0.01, ∗∗∗P < 0.001 versus untreated controls; †††P < 0.001 versus TGF-β1treated fibroblasts. Original magnification, ×20 (A).
Because nuclear localization of MRTF-A has been shown to be crucial for myofibroblast differentiation, we did a concentration–response analysis of α-SMA expression after TGF-β1 treatment in the presence/absence of CCG-203971. CCL-210 fibroblasts were exposed to 0.3, 3.0, or 30 μmol/L CCG-203971 30 minutes before TGF-β1 exposure, and α-SMA was assessed at 24 hours via Western blot analysis (Figure 2, A and B). The reported IC50 for CCG-203971 ranges from 4.2 to 6.2 μmol/L,16,21 and we observed significant suppression of TGF-β1–mediated α-SMA expression at the 30 μmol/L concentration (approximately sixfold the IC50). No effect was seen with the 0.3 or 3.0 μmol/L concentrations. These findings are concordant with recent findings in human colonic fibroblasts and in normal and scleroderma-associated skin fibroblasts.16,20 To ensure that the inhibition of myofibroblast differentiation was not due to nonspecific blockade of proximal TGF-β1 signaling, we treated fibroblasts with/without CCG-203971 followed by TGF-β1 and assessed for Smad activation (Figure 2C). Pretreatment with CCG-203971 had no impact on the rapid and robust phosphorylation of Smad3 by TGF-β1.
Figure 2.

Inhibition of serum response factor/myocardin-related transcription factor with CCG-203971 prevents myofibroblast differentiation but not Smad phosphorylation after treatment with transforming growth factor (TGF)-β1. Normal adult human lung fibroblasts (CCL-210) were cultured to 60% confluence in Dulbecco’s modified Eagle's medium (DMEM) supplemented with 5% fetal bovine serum and growth arrested in serum-free DMEM for 24 hours. Cells were treated with 0, 0.3, 3.0, or 30 μmol/L CCG-203971 for 30 minutes before treatment with/without 2 ng/mL TGF-β1 or with 30 μmol/L CCG-203970 in the absence of TGF-β1. A: Whole-cell lysates were obtained after 24 hours, and α-smooth muscle actin (SMA) expression was determined by Western blot analysis. Blots were stripped and probed for glyceraldehyde-3-phosphate dehydrogenase (GAPDH). B: For each Western blot, the density of the α-SMA band and the corresponding GAPDH band were measured with ImageJ version 1.45s (NIH, Bethesda, MD). The ratio of α-SMA to GAPDH was determined for each condition, and then normalized so that the ratio in untreated cells was equivalent to 1.0. Pooled analysis from at least three independent experiments with each concentration of CCG-203971 is shown. C: CCL-210 fibroblasts were treated with or without 2 ng/mL TGF-β1 in the presence or absence of 30 μmol/L CCG-203971 for 30 and 60 minutes. Whole-cell lysates were assessed for Smad3 phosphorylation by Western blot analysis, and blots were stripped and reprobed for total Smad3. Data are expressed as means ± SEM. ∗∗P < 0.01 versus untreated controls; †P < 0.05 versus TGF- β1 treatment in the absence of CCG-203971.
Rho- and SRF/MRTF-mediated signaling regulates collagen synthesis by fibroblasts.16,20 Aside from collagen, the extracellular matrix component fibronectin is a crucial mediator of fibroblast phenotype and function, including differentiation and survival.14,32 Accordingly, we sought to determine the impact of inhibiting this pathway on TGF-β1–mediated fibronectin accumulation. As with α-SMA expression, we found that 30 μmol/L of CCG-203971 completely abrogated TGF-β1–induced fibronectin deposition at 24 hours in CCL-210 fibroblasts (Figure 3). In contrast to α-SMA, basal expression of total fibronectin (in the absence of TGF-β1) was also markedly suppressed by CCG-203971. The inhibitory effects of CCG-203971 on the TGF-β1 induction of α-SMA and fibronectin expression were maintained at 48 hours (data not shown), and similar results were obtained in a second line of normal (fetal) human lung fibroblasts (IMR-90) (data not shown).
Figure 3.

CCG-203971 blocks basal and transforming growth factor (TGF)-β1 induced fibronectin expression with or without 2 ng/mL TGF-β1 in the presence or absence of 30 μmol/L CCG-203971 for 30 and 60 minutes. A: Total fibronectin expression was determined by Western blot analysis. Each blot was stripped and probed for glyceraldehyde-3-phosphate dehydrogenase (GAPDH). B: Fibronectin-to-GAPDH ratios were determined and normalized to the ratio observed in untreated cells. Data presented are a pooled analysis that includes a minimum of three experiments with each concentration of CCG-203971. Data are expressed as means ± SEM. ∗∗∗P < 0.001 versus untreated controls; †††P < 0.001 versus cells treated with TGF-β1 alone.
Postinflammatory Administration of CCG-203971 Attenuates Lung Fibrosis
Several studies have shown that strategies targeting myofibroblast phenotypes can decrease lung fibrosis.33–35 To determine whether pharmacological therapy directly targeting SRF/MRTF signaling also reduces lung fibrosis, we treated wild-type C57BL/6 mice with either CCG-203971 or vehicle control between days 11 and 21 after the administration of intratracheal bleomycin and assessed the impact of this compound during the postinflammatory phase of this model of lung injury and fibrosis (Figure 4).36,37 Mice receiving intratracheal bleomycin exhibited weight loss that was evident at day 3 and statistically significant by day 6, whereas the uninjured control mice receiving CCG-203971 did not demonstrate weight loss (Figure 4A). After day 11, the cohort of bleomycin-treated mice that received CCG-203971 had a significant reversal of weight loss, whereas the mice that received vehicle control continued to lose weight. By day 14, there was a statistically significant difference in weight between the bleomycin/CCG-203971 and bleomycin/DMSO groups. By day 20, there were no significant differences between the bleomycin/CCG-203971 cohort and the uninjured/DMSO and uninjured/CCG-203971 groups. Importantly, mice receiving CCG-203971 in the absence of bleomycin-induced lung injury did not exhibit any weight loss, and their weights over the course of the experiment were no different from those in the uninjured mice receiving vehicle control.
Figure 4.

CCG-203971 (CCG) reduces lung fibrosis after intratracheal bleomycin (bleo). Wild-type C57BL/6 mice were treated with 1.2 U/kg intratracheal bleomycin or phosphate-buffered saline (PBS) as previously described.30 Starting on day 11 after bleomycin administration, the mice received CCG-203971 (100 mg/kg b.i.d. via i.p. injection) or an equal volume of vehicle control (dimethyl sulfoxide; DMSO). A: Weight changes over time. Weights were obtained on days 4, 6, and 8 before the administration of CCG-203971 and then daily through day 20 after the initiation of CCG-203971 administration. B: Hydroxyproline concentrations of both lungs at day 21 after bleomycin administration. Two independent experiments were completed with similar results, and the figure shown represents one of the experimental replicates. C: Representative histological examination. D: Representative picrosirius red staining. Data are expressed as means ± SEM. n = 7 mice per group (A and B). ∗P < 0.05, ∗∗P < 0.01, bleomycin/CCG-203971 and bleomycin/DMSO versus PBS/CCG-203971 and PBS/DMSO; ††P < 0.01, PBS/CCG-203971 and PBS/DMSO versus bleomycin/DMSO; ‡P < 0.05, bleomycin/CCG-203971 versus bleomycin/DMSO; §§§§P < 0.0001 versus untreated controls and CCG-203971 alone; ¶¶¶¶P < 0.0001 versus bleomycin-treated mice.
Lung hydroxyproline was quantified at day 21 as a measure of lung collagen content.36 In mice that received intratracheal bleomycin with vehicle control, there was a significant increase in lung hydroxyproline content, which was markedly attenuated in the bleomycin-injured cohort that received CCG-203971 between days 11 and 21 (Figure 4B). Evaluation of hematoxylin and eosin stained lung sections from this same time point demonstrated extensive zones of lung fibrosis, including increased cellularity and matrix accumulation in the bleomycin-injured mice, which were markedly reduced in mice treated with CCG-203971 (Figure 4C). Consistently, CCG-203971 significantly reduced collagen accumulation as observed with picrosirius red staining (Figure 4D).
We used the targeted type II AEC injury model to investigate the efficacy of CCG-203971 in a second model of fibrosis that is not dependent on acute lung injury–induced inflammation (Figure 5).26,28,36 Therapeutic administration of CCG-203971 between days 11 and 21 was associated with an observable, but not statistically significant, improvement in the weight of SPC-DTR+ mice (Figure 5A). Control mice (SPC-DTR+ mice that did not receive DT) treated with CCG-203971 did not experience weight loss compared with untreated controls (data not shown). The therapeutic administration of CCG-203971 to DT-treated SPC-DTR+ mice was associated with a significant reduction in lung fibrosis as assessed by collagen accumulation (hydroxyproline quantification and picrosirius red staining) (Figure 5, B and C) and histological examination (Figure 5C). Thus, in complementary models of lung fibrosis, we found that the therapeutic administration of CCG-203971 significantly limited the extent of lung fibrosis.
Figure 5.

CCG-203971 (CCG) attenuates lung fibrosis after type II alveolar epithelial injury. Surfactant protein C promoter–diphtheria toxin receptor (SPC-DTR+) and DTR− transgenic mice were injected with/without 10.0 mg/kg diphtheria toxin (DT) i.p. once daily for 14 days. CCG-203971 100 mg/kg i.p. b.i.d. was administered from day 11 to 21 in the indicated groups. A: Weights were obtained at the indicated time points. CCG-203971 treatment alone did not lead to any weight loss in the DTR+ mice that did not receive DT (data not shown). B: At day 21, both lungs from at least five mice per group were assessed for hydroxyproline. The experiment was performed twice, with similar results. C: Representative histological examination and picrosirius red staining from additional mice at day 21. Insets represent digital enlargements of the indicated areas of the corresponding 40× image. Data are expressed as means ± SEM (A and B). n = 5 to 7 per group (A and B). ∗P < 0.05, ∗∗P < 0.01 versus DTR mice treated with DT alone; †P < 0.05 versus DTR controls without DT treatment; ‡‡P < 0.01 versus DTR mice treated with DT and CCG-203971. H&E, hematoxylin and eosin. DMSO, dimethyl sulfoxide.
Inhibition of SRF/MRTF Signaling with CCG-203971 Enhances Fibroblast Susceptibility to Apoptosis
Myofibroblast apoptosis is a crucial component of the resolution phase of normal wound repair, and our recent studies have shown that blockade of antiapoptotic signaling pathways can enhance myofibroblast susceptibility to apoptosis.3,24,36–41 To determine how blockade of SRF/MRTF-mediated signaling by CCG-203971 affected myofibroblast susceptibility to apoptosis, we first measured apoptosis induction over time in IMR-90 fibroblasts treated with/without Fas-Ab in the presence or absence of different concentrations of CCG-203971, using image-based quantification of caspase-3/7 activation (Figure 6A). Over the initial 9 hours, there was no significant apoptosis observed in the untreated IMR-90 fibroblasts, whereas cells treated with Fas-Ab showed a mild increase in apoptosis that did not reach statistical significance (means ± SEM, 11.4 ± 0.7 counts per image at baseline compared with 23.2 ± 0.7 counts per image at 9 hours). IMR-90 fibroblasts treated with the Fas-Ab in the presence of 30 μmol/L CCG-203971, in contrast, showed a significantly increased rate of apoptosis that was evident at 5 hours (2.3-fold increase, from 11.5 ± 0.7 to 26.6 ± 1.4 counts per image; P < 0.01) and progressed to a 4.7-fold increase by 9 hours (53.6 ± 9.7 counts per image; P < 0.0001).
Figure 6.

Fas-mediated apoptosis is enhanced by CCG-203971 (CCG) in IMR-90 fibroblasts. IMR-90 fibroblasts were treated with 0.3, 3.0, or 30 μmol/L CCG-203971 alone or in combination with 250 ng/mL Fas-activating antibody (Fas-Ab). A: Apoptosis was assessed by the detection of a green-fluorescent signal indicating cleavage by caspase-3/7 and quantified at the indicated time points using IncuCyte software version 2011A as described in Materials and Methods. B: Cells were treated with 250 ng/mL Fas-Ab, 30 μmol/L CCG-203971, or the combination, and apoptosis was similarly assessed at baseline and 16 hours. C and D: IMR-90 fibroblasts were treated with 250 ng/mL Fas-Ab and/or 0, 3.0, or 30 μmol/L CCG-203971 for 8 hours (C) or 16 hours (D). Apoptosis was assessed by Western blot analysis for cleaved poly-(ADP-ribose) polymerase (PARP); blots were stripped and probed for glyceraldehyde-3-phosphate dehydrogenase (GAPDH). E: Similarly, IMR-90 fibroblasts were treated with Fas-Ab and/or 30 μmol/L CCG-203971 for 8 or 16 hours, and apoptosis was assessed by enzyme-linked immunosorbent assay detection of histone-associated DNA fragments. Data are expressed as means ± SEM. Each data point represents three replicates, with each replicate comprised of nine different images from each well at each time point (A and B). n = 3 at each time point for each condition (E). ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, and ∗∗∗∗P < 0.0001 versus control; †P < 0.05 versus baseline controls; ‡‡‡P < 0.001 versus Fas-Ab alone and CCG-203971 at 16 hours; §§P < 0.01 versus Fas-Ab + CCG-203971 at 8 hours.
We have previously shown that Fas-Ab induce a modest, but significant, increase in the apoptosis of IMR-90 fibroblasts after 16 hours of treatment.36 Consistent with these previous results, we observed a 3.2-fold increase in apoptosis at 16 hours in these cells after treatment with the Fas-Ab (P < 0.01) (Figure 6B). Notably, at the 16-hour time point, treatment with 30 μmol/L of CCG-203971 alone induced a threefold increase in apoptosis compared with baseline (P < 0.05), which was not significantly different from that with the Fas-Ab alone. Compared with either CCG-203971 or Fas-Ab exposure alone, the combination of Fas-Ab with 30 μmol/L CCG-203971 induced an additional 2.5-fold increase in apoptosis at the 16-hour time point (P < 0.001 for each comparison).
To confirm the results of the automated quantification of caspase-3/7 activation method, two additional apoptosis assessments were performed. First, IMR-90 fibroblasts were treated with/without the Fas-Ab in the presence or absence of CCG-203971 at concentrations of 3.0 or 30.0 μmol/L for 8 hours (Figure 6C) or 16 hours (Figure 6D), and apoptosis was assessed by Western blot for cleaved PARP. Consistent with the results from the fluorescent imaging assay, at 8 hours, cells treated with the Fas-Ab combined with 30 μmol/L CCG-203971 exhibited a clear increase in the cleaved PARP band, indicative of apoptosis. At this early time point, treatment with the Fas-Ab alone or with either 3.0 or 30 μmol/L of CCG-203971 alone did not induce an increase in cleaved PARP, and these results were consistent with those from the imaging assay. A slight increase in cleaved PARP was seen in cells treated with the combination of 3.0 μmol/L CCG-203971 and the Fas-Ab. At 16 hours, there was a significant increase in the cleaved PARP band from cells treated with the Fas-Ab alone and from cells treated with the Fas-Ab combined with 30 μmol/L CCG-203971. Along with the increase in cleaved PARP, there was a marked reduction in the intact PARP band in cells treated with the combination of Fas-Ab and 30 μmol/L CCG-203971, indicative of the advanced stage of apoptosis.42,43
Because PARP cleavage is dependent on activated caspase-3, we used an additional non–caspase-based technique to further assess apoptosis. With an ELISA-based assay to detect histone-associated DNA fragments, we again found that the 30 μmol/L concentration of CCG-203971 in combination with a Fas-Ab induced a significant increase in apoptosis at 8 hours, which was further accentuated at 16 hours (Figure 6E).
To demonstrate that the increased susceptibility to Fas-mediated apoptosis induced by CCG-203971 was not limited to normal fetal lung fibroblasts, we performed a complementary set of experiments in CCL-210 (normal human adult) lung fibroblasts (Figure 7, A–C). As in the IMR-90 fibroblasts, treatment of CCL-210 fibroblasts with the combination of Fas-Ab and 30 μmol/L CCG-203971 induced apoptosis at 8 hours, as assessed by Western blot analysis for cleaved PARP (Figure 7, A and B) and by ELISA detection of histone-associated DNA fragments (Figure 7C). At 16 hours of treatment, there was extensive death of CCL-210 fibroblasts after treatment with the combination of Fas-Ab and 30 μmol/L CCG-203971, such that no cleaved or intact PARP was detected by Western blot analysis, precluding quantification (data not shown).
Figure 7.

Inhibition of serum response factor/myocardin-related transcription factor A (MRTF-A) signaling by CCG-203971 and MRTF-A deficiency promotes Fas-mediated apoptosis in fibroblasts. CCL-210 fibroblasts were exposed to 250 ng/mL Fas-activating antibody (Fas-Ab) with or without 0, 3.0, or 30 μmol/L CCG-203971 for 8 hours. A: Apoptosis was assessed by Western blot analysis for cleaved poly-(ADP-ribose) polymerase (PARP). Blots were stripped and probed for glyceraldehyde-3-phosphate dehydrogenase (GAPDH). B: Relative densitometry was based on the ratio of cleaved PARP to GAPDH for each condition. C: Apoptosis in CCL-210 fibroblasts was assessed by enzyme-linked immunosorbent assay (ELISA) detection of histone-associated DNA fragments. D: Lung fibroblasts from wild-type (WT) mice and transgenic mice deficient in MRTF-A (Mkl1) were treated with 250 ng/mL Fas-activating antibody (Fas-Ab), and apoptosis was assessed at 8 hours by detection of histone-associated DNA fragments. E: Normal primary adult lung fibroblasts (left graph) and fibroblasts from the lungs of patients with idiopathic pulmonary fibrosis (IPF; right graph) were treated with/without 250 ng/mL Fas-Ab and/or 30 μmol/L CCG-203971, and apoptosis was assessed after 8 hours by ELISA detection of histone-associated DNA fragments. Data are expressed as means ± SEM. n = 3 or more replicates per condition (B); n = 3 per group (C); n = 3, WT, and n = 6, Mkl_−/− (D); n = 4 normal and 4 IPF cell lines (E). ∗∗_P < 0.01 versus untreated control; †P < 0.05 versus Fas-Ab alone, CCG-203971 alone, and CCG-203971 3 μmol/L + Fas-Ab; ‡P < 0.05, ‡‡P < 0.01 versus untreated control; §P < 0.05 versus WT and _Mkl1_−/− control and WT fibroblasts treated with Fas-Ab.
Next, we examined the apoptotic susceptibility of lung fibroblasts from transgenic Mkl1 (the mouse orthologue of MRTF-A) knockout mice and found that, in contrast to normal lung fibroblasts, the cells lacking MRTF-A had a robust apoptotic response to treatment with Fas-Ab (Figure 7D), providing further evidence that MRTF-A signaling participates in the apoptosis-resistant phenotype of lung fibroblasts. Combined treatment with Fas-Ab and CCG-203971 did not lead to a further increase in apoptosis in the MRTF-A–deficient fibroblasts (data not shown), suggesting that the CCG-203971–mediated increase in fibroblast susceptibility to Fas-mediated apoptosis is unlikely to represent an off-target effect of the drug.
Previous studies from our laboratory have shown that inhibition of survival signaling pathways can restore the apoptotic susceptibility of fibrotic lung fibroblasts.36,41 To determine whether CCG-203971 similarly sensitized fibrotic lung fibroblasts to apoptosis, normal and IPF lung fibroblasts (n = 4 of each) were treated with Fas-Ab in the presence or absence of CCG-203971, and apoptosis was assessed after 8 hours (Figure 7E). Fas activation did not lead to a significant increase in apoptosis in either cohort of lung fibroblasts at this time point, but the addition of CCG-203971 significantly enhanced Fas-mediated apoptosis. These data suggest that the role of SRF/MRTF signaling in fibroblast resistance to apoptosis is not limited to cell lines and that regulation of SRF/MRTF signaling is sufficient to modulate apoptotic susceptibility in fibrotic lung fibroblasts.
TGF-β1 Does Not Confer Protection from Apoptosis Induced by Fas Activation in the Presence of CCG-203971
Among the multitude of profibrotic activities of TGF-β1, we have shown that TGF-β1 promotes fibroblast resistance to apoptosis through different mechanisms, including the induction of XIAP, survivin, and PAI-1 expression.36,40,41 To determine whether TGF-β1 could reverse the increased susceptibility to Fas-mediated apoptosis in normal lung fibroblasts exposed to CCG-203971, IMR-90 fibroblasts (Figure 8) and CCL-210 fibroblasts (Figure 9) were treated with Fas-activating antibodies in the presence/absence of 30 μmol/L CCG-203971 and/or 2 ng/mL TGF-β1. In each normal lung fibroblast cell line across multiple apoptosis assays, we found that TGF-β1 cotreatment had no significant impact on fibroblast susceptibility to Fas-mediated apoptosis in the presence of CCG-203971, indicating that the apoptosis-resistant fibroblast phenotype induced by TGF-β1 requires intact SRF/MRTF signaling.
Figure 8.

Transforming growth factor (TGF)-β1 fails to reduce Fas-mediated apoptosis in IMR-90 fibroblasts treated with CCG-203971. IMR-90 fibroblasts were cotreated with 250 ng/mL Fas-activating antibody (Fas-Ab) with/without 30 μmol/L CCG-203971 and/or 2 ng/mL TGF-β1 for 8 hours. Apoptosis was assessed by Western blot analysis for cleaved poly-(ADP-ribose) polymerase (PARP) (A) with densitometry (B) and by enzyme-linked immunosorbent assay for histone associated DNA fragments (C). Data are expressed as means ± SEM. n = 2 per condition (B and C). ∗P < 0.05 versus controls, TGF-β1 alone, Fas-Ab alone, and Fas-Ab + TGF-β1; †P < 0.05 versus Fas-Ab + CCG-203971, with or without TGF- β1; ‡P < 0.05 versus all groups that did not receive Fas-Ab + CCG-203971. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Figure 9.

Transforming growth factor (TGF)-β1 does not rescue CCL-210 fibroblasts from Fas-mediated apoptosis in the presence of CCG-203971 (CCG). CCL-210 fibroblasts were treated with 250 ng/mL Fas-activating antibody (Fas-Ab) with/without 30 μmol/L CCG-203971 and/or 2 ng/mL TGF-β1 for 8 hours. Apoptosis was assessed by Western blot analysis for cleaved poly-(ADP-ribose) polymerase (PARP) (A) with quantification by densitometry (B), enzyme-linked immunosorbent assay for histone associated DNA fragments (C), and cleavage of a fluorescent substrate by activated caspase-3/7 with image analysis at 2-hour intervals and quantification of the fluorescent signal using the IncuCyte software version 2011A, ×20 magnification (D). E: Representative images of each condition at baseline and after 10 hours. Data are expressed as means ± SEM. n = 2 per group (B); n = 3 per group (C); n = 2 with 16 independent images per replicate analyzed at each time point (D) (the detail of this analysis is given in Materials and Methods). ∗∗∗P < 0.001 versus control, TGF-β1 alone, Fas-Ab alone, and Fas-Ab + CCG-203971; †P < 0.05 versus Fas-Ab + CCG-203971 with or without TGF-β1; ‡P < 0.05 versus control, Fas-Ab alone, CCG-203971 alone, and Fas-Ab + TGF-β1; §P < 0.05, §§P < 0.01 versus control and CCG-203971 alone. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Having shown that TGF-β1 cotreatment did not protect fibroblasts from Fas-mediated apoptosis when SRF/MRTF signaling was blocked by CCG-203971, we next assessed the impact of CCG-203971 on the apoptosis susceptibility of fully differentiated myofibroblasts (Figure 10). CCL-210 fibroblasts were treated with/without TGF-β1 for 24 hours before treatment with Fas-Ab and/or CCG-203971 for 8 hours, and apoptosis was assessed by Western blot analysis for cleaved PARP (Figure 10A) and by cleavage of a fluorogenic substrate of caspase-3/7 (Figure 10B). Fas-Ab induced a small increase in cleaved PARP in fibroblasts, which was not seen in the TGF-β1 pretreated myofibroblasts. With image-based quantification, there was no statistically significant difference in apoptosis with Fas-treatment alone in either population of cells at the 8-hour time point. No apoptosis was seen in cells treated with CCG-203971 alone. In both fibroblasts and myofibroblasts, the combination of Fas-Ab and CCG-203971 was sufficient to induce robust apoptosis. Interestingly, differentiated myofibroblasts (pretreated with TGF-β1) demonstrated markedly increased apoptosis when treated with the combination; this finding was most evident using the image-based quantification of fluorogenic substrate, although a similar pattern of accentuated apoptosis in the myofibroblasts was also demonstrated by increased cleaved PARP along with decreased intact PARP (Figure 10A). These data indicate that CCG-203971 enhances Fas-mediated apoptosis in fibroblasts, regardless of state of differentiation.
Figure 10.

CCG-203971 sensitizes differentiated myofibroblasts to Fas-mediated apoptosis. CCL-210 fibroblasts were treated with/without 2 ng/mL transforming growth factor (TGF)-β1 for 24 hours and then treated with/without 250 ng/mL Fas-activating antibody (Fas-Ab) and/or 30 μmol/L CCG-203971. Apoptosis was assessed by Western blot analysis for poly-(ADP-ribose) polymerase (PARP) (A) and cleavage of a fluorescence substrate by activated caspase-3/7 with image analysis at 8 hours (B). Data are expressed as means ± SEM. n = 3 replicates of each condition with 16 separate images of each replicate analyzed. ∗∗P < 0.01, ∗∗∗P < 0.001 versus control; †††P < 0.001 versus Fas-Ab + CCG-203971. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
CCG-203971 Decreases TGF-β1 Induction of XIAP
To further examine the mechanisms by which the inhibition of SRF/MRTF signaling might enhance fibroblast susceptibility to apoptosis, we next assessed the effect of CCG-203971 on XIAP expression. We have shown that XIAP is induced by TGF-β1 in normal lung fibroblasts and serves to protect fibroblasts from Fas-mediated apoptosis.36 TGF-β1 treatment of CCL-210 fibroblasts that were exposed to CCG-203971 at doses that enhance fibroblast susceptibility to apoptosis did not result in an increase in XIAP (Figure 11, A and B), demonstrating that this inhibitor of apoptosis protein is regulated by the SRF/MRTF pathway.
Figure 11.
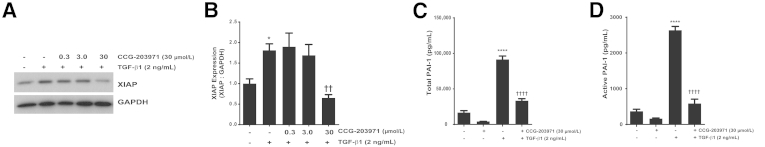
Inhibition of serum response factor/myocardin-related transcription factor with CCG-203971 prevents transforming growth factor (TGF)-β1 induction of X-linked inhibitor of apoptosis (XIAP) and secretion of plasminogen activator inhibitor (PAI)-1. CCL-210 fibroblasts were treated with 2 ng/mL TGF-β1 in the presence/absence of CCG-203971 (indicated doses) for 24 hours. Whole-cell lysates were assessed for XIAP expression by Western blot analysis (A) with densitometric analysis (B). Cell culture supernatants were assessed for total (C) and active (D) PAI-1. Data are expressed as means ± SEM. n = at least 3 replicates per condition (B); n = 3 for CCG-203971 alone and 12 for all other conditions (C and D). ∗P < 0.05, ∗∗∗∗P < 0.0001 versus control; ††P < 0.01, ††††P < 0.0001 versus TGF-β1. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Targeting the SRF/MRTF Pathway with CCG-203971 Decreases PAI-1 Expression by Fibroblasts
As another potential mechanism by which SRF/MRTF promotes fibrosis, we examined the participation of this signaling pathway in TGF-β1–induced fibroblast-mediated expression of the profibrotic molecule PAI-1. In this experiment, CCL-210 cells were treated with TGF-β1 in the presence or absence of 30 μmol/L CCG-203971. Consistent with findings from prior reports,40 TGF-β1 markedly increased the levels of total and active PAI-1 in the conditioned media of the fibroblast cultures at 24 hours (Figure 11, C and D). This increase in PAI-1 was significantly attenuated by the inhibition of the SRF/MRTF pathway.
Enhanced Fibroblast Apoptosis and Suppressed PAI-1 Expression in Vivo after the Administration of CCG-203971
Wild-type C57BL/6 mice were injured with intratracheal bleomycin for the examination of the mechanisms by which SRF/MRTF blockade reduced fibrosis in vivo. On day 11, CCG-203971 or DMSO treatment was initiated in subgroups of animals. On day 14, a time point at which we had observed an improvement in weight loss with drug treatment (Figure 4A), lungs were harvested for immunostaining of TUNEL as an indicator of apoptosis, and bronchoalveolar lavage fluid was collected for the quantification of PAI-1 levels. The inhibition of SRF/MRTF signaling with CCG-203971 resulted in a significant increase in the number of TUNEL-positive cells compared with that in the vehicle-treated control group (Figure 12, A–D). Having identified an increase in the number of TUNEL-positive cells in the lungs of mice treated with bleomycin and CCG-203971, we performed immunofluorescence colocalization with TUNEL and α-SMA as a qualitative assessment of which cells were undergoing apoptosis. The results show that almost all of the TUNEL-positive cells colocalized with α-SMA, suggesting myofibroblast apoptosis (Figure 12, E–G).
Figure 12.

Myofibroblast apoptosis is increased in vivo after the administration of CCG-203971 during the fibroproliferative phase of lung injury. Lung sections were analyzed for cell death by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining (red) 2 weeks after the instillation of intratracheal phosphate-buffered saline (A) or bleomycin (Bleo) (B and C) and 3 days after the initiation of dimethyl sulfoxide (B) or CCG-203971 (CCG971) (C). D: TUNEL-positive (pos) cells were quantified in 10 random 400× fields by a blinded observer (K.K.K.). E and F: Lungs from bleomycin-injured mice treated with CCG-203971 were co-stained for TUNEL (E) and α-smooth muscle actin (SMA; green; F). G: Merged images demonstrate colocalization of TUNEL and α-SMA. Data are expressed as means ± SEM. n = 3 per group (D). ∗P < 0.05 versus bleomycin alone.
To determine the in vivo contribution of the SRF/MRTF signaling pathway to PAI-1 production, we treated both targeted type II AEC– and bleomycin-injured mice with CCG-203971 and collected bronchoalveolar lavage fluid for PAI-1 quantification. In preliminary studies, we found no difference in active PAI-1 levels in the bronchoalveolar lavage fluid of uninjured mice treated with or without CCG-203971. Therefore, for our control group in these experiments, we used an uninjured/untreated group. In the bleomycin model, we observed a significant decrease in the mean active PAI-1 level after inhibition of the SRF/MRTF pathway (Figure 13A). Similarly, inhibition of SRF/MRTF after targeted type II AEC injury resulted in a significant decrease in the quantity of active PAI-1 (Figure 13B).
Figure 13.
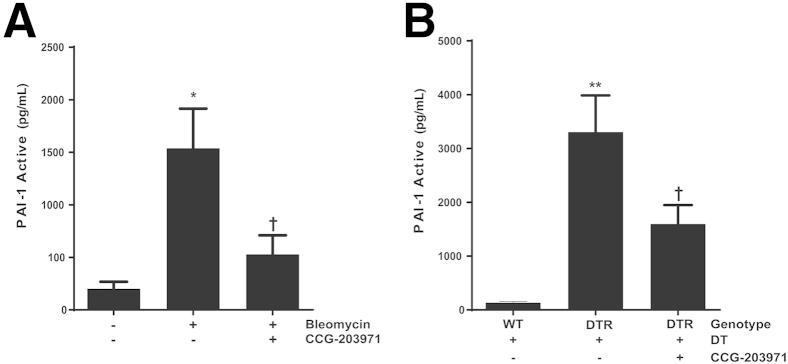
CCG-203971 treatment decreases alveolar plasminogen activator inhibitor (PAI)-1 in murine models of lung injury and fibrosis. Mice were injured with bleomycin (A) or targeted type II alveolar epithelial cell (AEC) injury (B), and CCG-203971 treatment was initiated on day 11. Bronchoalveolar lavage fluid was collected on day 21 after bleomycin injury and on day 15 in the AEC-injury model. Levels of active PAI-1 were quantified by using a carboxylated microsphere–based enzyme-linked immunosorbent assay. Data are expressed as means ± SEM. n = 5, control group (A); n = 9, bleomycin-treated groups (A); n = 5, all groups (B). ∗P < 0.05, ∗∗P < 0.01 versus controls; †P < 0.05 versus bleomycin or diphtheria toxin receptor (DTR) mice treated with diphtheria toxin (DT). WT, wild type.
Discussion
The accumulation and persistence of activated mesenchymal cells is integral to the development of fibrosis in the lung and other organs.4,44 Myofibroblasts have been morphologically and functionally characterized by their expression of organized stress fibers that incorporate α-SMA and their secretion of extracellular matrix constituents, including type 1 collagen and fibronectin. The elimination of myofibroblasts via apoptosis is crucial for the resolution of wound repair and the restoration of tissue homeostasis after injury.7,45 However, these cells characteristically display a phenotype of apoptosis resistance, especially in the setting of ongoing tissue fibrosis.4,45 The central roles that myofibroblast activation and survival play in the pathobiology of fibrosis highlight the attractiveness of these cells as targets for antifibrotic interventions.3,37 Based on the described role that SRF/MRTF signaling plays in myofibroblast activation, our initial goal was to determine the efficacy of targeting this pathway as a therapeutic intervention for in vivo lung fibrosis. Indeed, we found that CCG-203971, a novel nipecotic bis(amide) inhibitor of SRF/MRTF transcriptional activity, markedly attenuates lung fibrosis in two complementary murine models. After defining a crucial causal role for SRF/MRTF in fibrosis in vivo, our next goal was to investigate potential mechanisms by which this signaling pathway regulates fibrosis beyond myofibroblast differentiation. Importantly, we found that this pathway plays previously unrecognized roles in the regulation of PAI-1 production both in vitro and in vivo, in the regulation of XIAP expression by fibroblasts, and in fibroblast susceptibility to apoptosis.
Recent studies have shown that targeting SRF/MRTF signaling in fibroblasts derived from multiple organs, including the skin and colon, blocks myofibroblast differentiation and collagen production induced by TGF-β1.16,20 As an initial step in our investigation, we found that CCG-203971 blocked TGF-β1 induced expression of α-SMA and fibronectin in lung fibroblasts at concentrations consistent with those shown in skin and colon fibroblasts. Each of these proteins is crucial to myofibroblast function, as α-SMA expression confers cells with increased contractile activity,6 whereas cellular fibronectin is required for myofibroblast differentiation and serves as a link between organized stress fibers and the pericellular matrix.32,46 Although there was no clear effect on basal levels of α-SMA expression, blocking SRF/MRTF signaling with CCG-203971 did suppress basal levels of fibronectin, suggesting that this pathway has a previously unrecognized regulatory role in fibronectin homeostasis.
The inhibition of fibroblast function by the targeting of SRF/MRTF signaling was recently extended to an in vivo murine model of skin fibrosis induced by bleomycin, in which CCG-203971 led to a significant reduction in the development of dermal fibrosis.20 To investigate the efficacy of targeting this pathway with a therapeutic strategy in lung fibrosis, we used two distinct murine models (intratracheal bleomycin and targeted type II AEC injury).28,36 In the bleomycin model, we initiated the treatment during the fibrotic phase, and CCG-203971 administration was associated with a rapid reversal of the injury-induced weight loss and a significant reduction in the severity of fibrosis. Importantly, CCG-203971 had no detrimental effect on the weight of control mice. In the targeted type II AEC injury model of fibrosis, we similarly began treatment with CCG-203971 on day 11, such that there was limited overlap between DT-mediated injury of the alveolar epithelium and the drug administration. Although the effect of the drug on weight loss in this model was not as dramatic as in the bleomycin model, interference with the SRF/MRTF pathway resulted in a significant reduction in fibrosis. Of note, the fibrosis observed in this model is quite different from that seen in the bleomycin model. Instead of distinct areas of consolidation, the fibrosis is diffuse in nature and is observed as a more homogeneous thickening of alveolar interstitium, consistent with the pattern described in our original publication using this model of fibrosis.28 The demonstration of an antifibrotic effect of SRF/MRTF inhibition in two separate models highlights the importance of this pathway in fibrogenesis while demonstrating that these effects are not model dependent.36,37,47
Furthermore, our findings are consistent with those from recent studies evaluating the role of Rho/SRF/MRTF signaling in lung fibrosis. For example, one report showed evidence of increased Rho signaling in fibroblastic foci from IPF lung tissue and in fibrotic murine lungs after intratracheal bleomycin.13 In this same study, therapeutic administration of the Rho-kinase inhibitor fasudil led to a significant reduction in fibrosis. Moreover, mice deficient in MRTF-A had significantly decreased lung fibrosis 28 days after bleomycin administration. In a more recent study, noscapine, which targets microtubule polymerization and blocks TGF-β1–induced MRTF-A nuclear localization, was administered 7 days after bleomycin-induced injury and resulted in a significant reduction in lung collagen.48 Collectively, these studies support a crucial role for Rho/SRF/MRTF signaling in lung injury, repair, and fibrosis. These findings are consistent with those from prior studies showing that MRTF-A null mice have decreased fibrosis in the heart after myocardial infarction or activation of the renin–angiotensin system and decreased renal fibrosis in a model of diabetic nephropathy.49,50
To investigate the mechanisms underlying the decrease in lung fibrosis attributable to SRF/MRTF inhibition, we examined the impact of CCG-203971 administration on myofibroblast susceptibility to apoptosis. Clearance of myofibroblasts via apoptosis has been shown to be crucial for normal tissue repair,3,45 and studies have demonstrated that fibrotic lung fibroblasts, and normal lung fibroblasts exposed to TGF-β1, acquire an apoptosis-resistant phenotype in vitro.13,24,28,36,51,52 Furthermore, accumulating in vivo evidence shows that the regulation of prosurvival signaling can modulate lung fibrosis in several different murine models.35,53–58 Recent studies have shown that the inhibition of Rho/Rho-kinase signaling decreases fibroblast expression of antiapoptotic BCL-2 family proteins and promotes IPF lung fibroblast apoptosis while decreasing lung fibrosis in vivo.13,19 To investigate the effect of targeting the SRF/MRTF pathway on lung fibroblast susceptibility to Fas-mediated apoptosis, we used three distinct methods in two normal lung fibroblast cell lines and in normal and IPF patient–derived lung fibroblasts. In each cell line and with each method, we found that treatment with CCG-203971 led to a significantly enhanced apoptotic response to Fas activation. In contrast to the apoptosis-resistant phenotype of normal and IPF fibroblasts, murine fibroblasts lacking MRTF-A were sensitive to Fas-induced apoptosis in the absence of CCG-203971. Of note, TGF-β1 treatment failed to rescue fibroblasts from apoptosis induced by Fas-activating antibodies in the presence of CCG-203971, consistent with the role of SRF/MRTF as a downstream mediator of the antiapoptotic effects of TGF-β1.15 Importantly, CCG-203971 had a minimal effect on fibroblast apoptosis in the absence of the Fas-Ab.
The findings from our in vitro experiments indicate that the SRF/MRTF pathway regulates myofibroblast susceptibility to apoptosis, in part, through the expression of XIAP. Members of the inhibitor of apoptosis protein family have been shown to regulate resistance to apoptosis in normal and IPF lung fibroblasts.24,36,41,59,60 XIAP is the best studied of the inhibitor of apoptosis protein family and is the most likely to function as a direct caspase inhibitor in vivo.61 XIAP expression is increased in the fibroblast foci of IPF tissue and in explanted myofibroblasts from IPF lungs.36,60 Moreover, XIAP expression in normal lung fibroblasts is increased by TGF-β1 and the functional inhibition of XIAP sensitizes normal and IPF lung fibroblasts to Fas-mediated apoptosis.36 Here, we show that the inhibition of SRF/MRTF signaling with CCG-203971 completely suppressed TGF-β1–induced up-regulation of XIAP, suggesting one feasible mechanism whereby MRTF contributes to apoptosis resistance in fibroblasts. Supporting the hypothesis that sensitization of myofibroblasts to apoptosis might contribute to the reduction in fibrosis observed in our in vivo models, we found evidence of a statistically significant increase in myofibroblast apoptosis in the lungs of bleomycin-treated mice 3 days after the initiation of CCG-203971 treatment.
A robust literature demonstrates the strong profibrotic effect of PAI-1, and we have shown a crucial role for this multifunctional protein in the pathogenesis of both bleomycin and targeted type II AEC injury–induced fibrosis.26,30,62 Although the findings from a previous study suggested that TGF-β1–induced PAI-1 expression is independent of SRF/MRTF-mediated transcription, our data strongly implicate this pathway in the regulation of this profibrotic molecule.15 Specifically, CCG-203971 treatment blocked TGF-β1–mediated secretion of total and active PAI-1 into cell culture supernatants. Additionally, we found that inhibition of SRF/MRTF significantly reduces PAI-1 levels in the alveolar compartment of both bleomycin and targeted type II AEC–injured mice. Although the precise mechanisms by which PAI-1 promotes fibrosis are unclear, this molecule has been found to protect fibroblasts from apoptosis induced by plasmin while also inhibiting degradation of matrix fibronectin.40,63,64 Moreover, prior studies in vivo indicate that the vitronectin-binding function of PAI-1 is crucial to its profibrotic effect.30 Thus, the demonstration that CCG-203971 inhibits myofibroblast secretion of PAI-1 in vitro and decreases levels of active PAI-1 in vivo support another mechanism by which the SRF/MRTF signaling pathway might impact lung fibrosis.
Myofibroblast differentiation and activation in response to soluble and matrix-associated stimuli require Rho-dependent polymerization of the actin cytoskeleton, allowing for the nuclear localization of MRTF.14 In fibroblasts, nuclear localization of MRTF is induced by TGF-β1 and is required for myofibroblast differentiation and collagen synthesis.15,18,49 The profibrotic role of MRTF is not limited to TGF-β1–mediated signaling, however, as it is also required for myofibroblast differentiation and collagen synthesis induced by lysophosphatidic acid and by mechanotransduction pathways activated by stiff matrix substrates.13,25,65 This pathway is counter-regulated by known antifibrotic mediators. Specifically, the antifibrotic eicosanoid prostaglandin E2 signals through the prostaglandin E receptor 2 to prevent TGF-β1–mediated myofibroblast differentiation and collagen production, to increase myofibroblast susceptibility to Fas-mediated apoptosis, and to decrease fibroblast expression of the TGF-β1regulated inhibitor of apoptosis protein family members.36,41,59,60,66 Consistently, prostaglandin E2 has been shown to block nuclear localization of MRTF.31 The findings from another study showed that noscapine-mediated inhibition of myofibroblast differentiation in vitro and lung fibrosis in vivo are mediated by prostaglandin E receptor 2.48
Fibrosis of the lungs and other organs is estimated to contribute to up to 45% of deaths in developed countries, highlighting the pressing need for antifibrotic treatments.67 In diseases of the lung, fibrosis is typically advanced at the time of diagnosis. Promising results from recent clinical trials show that the rate of progression of fibrosis can be slowed, but the potential to reverse fibrotic changes in the adult lung remains poorly understood.37,68,69 The findings of our current study demonstrate that a novel compound targeting the SRF/MRTF signaling, when initiated during the fibroproliferative phase of two distinct animal models, can decrease the severity of lung fibrosis and increase myofibroblast apoptosis in vivo while blocking myofibroblast differentiation and enhancing myofibroblast susceptibility to apoptosis in vitro. Combined with studies showing that IPF lung fibroblasts have increased nuclear localization of MRTF,13 these studies demonstrate a central role for SRF/MRTF signaling in the pathogenesis of lung fibrosis, making this pathway an attractive therapeutic target.
Acknowledgments
Y.Z. provided Mkl1 knockout murine lung fibroblasts; S.L. provided N-(4-chlorophenyl)-1-[3-(2-furanyl)benzoyl]-3-piperidinecarboxamide (CCG-203971) synthesized by the Vahlteich Medicinal Chemistry Core (University of Michigan, Ann Arbor, MI).
Footnotes
Supported by NIH/NHLBI grants HL105489 (J.C.H.), HL108904 (K.K.K.), and HL078871 (T.H.S.), NIH grants 1S10RR27525 (V.G.K.) and R01AR066049 (S.D.L. and R.R.N.), and The Martin E. Galvin Fund for Pulmonary Fibrosis Research Pilot Grant (J.C.H., T.H.S., and K.K.K.).
Portions of this work were presented in abstract form at the 18th International Colloquium of Lung and Airway Fibrosis, September 20–24, 2014, Mont Tremblant, QC, Canada.
Disclosures: None declared.
A guest editor acted as editor-in-chief for this manuscript. No person at the University of Alabama, Birmingham was involved in the final disposition of this article.
See related Commentary on page 909
Contributor Information
Thomas H. Sisson, Email: tsisson@med.umich.edu.
Jeffrey C. Horowitz, Email: jchorow@umich.edu.
References
- 1.Raghu G., Chen S.Y., Yeh W.S., Maroni B., Li Q., Lee Y.C., Collard H.R. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med. 2014;2:566–572. doi: 10.1016/S2213-2600(14)70101-8. [erratum in: Lancet Respir Med 2014;2:e12] [DOI] [PubMed] [Google Scholar]
- 2.Raghu G., Collard H.R., Egan J.J., Martinez F.J., Behr J., Brown K.K., Colby T.V., Cordier J.F., Flaherty K.R., Lasky J.A., Lynch D.A., Ryu J.H., Swigris J.J., Wells A.U., Ancochea J., Bouros D., Carvalho C., Costabel U., Ebina M., Hansell D.M., Johkoh T., Kim D.S., King T.E., Jr., Kondoh Y., Myers J., Müller N.L., Nicholson A.G., Richeldi L., Selman M., Dudden R.F., Griss B.S., Protzko S.L., Schünemann H.J. ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis: An official Ats/Ers/Jrs/Alat statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thannickal V.J., Henke C.A., Horowitz J.C., Noble P.W., Roman J., Sime P.J., Zhou Y., Wells R.G., White E.S., Tschumperlin D.J. Matrix biology of idiopathic pulmonary fibrosis: a workshop report of the National Heart, Lung, and Blood Institute. Am J Pathol. 2014;184:1643–1651. doi: 10.1016/j.ajpath.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hinz B., Phan S.H., Thannickal V.J., Prunotto M., Desmoulière A., Varga J., De Wever O., Mareel M., Gabbiani G. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012;180:1340–1355. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Follonier L., Schaub S., Meister J.J., Hinz B. Myofibroblast communication is controlled by intercellular mechanical coupling. J Cell Sci. 2008;121:3305–3316. doi: 10.1242/jcs.024521. [DOI] [PubMed] [Google Scholar]
- 6.Hinz B., Celetta G., Tomasek J.J., Gabbiani G., Chaponnier C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol Biol Cell. 2001;12:2730–2741. doi: 10.1091/mbc.12.9.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tomasek J.J., Gabbiani G., Hinz B., Chaponnier C., Brown R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 8.Wipff P.J., Rifkin D.B., Meister J.J., Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol. 2007;179:1311–1323. doi: 10.1083/jcb.200704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsou P.S., Haak A.J., Khanna D., Neubig R.R. Cellular mechanisms of tissue fibrosis. 8. Current and future drug targets in fibrosis: focus on rho GTPase-regulated gene transcription. Am J Physiol Cell Physiol. 2014;307:C2–C13. doi: 10.1152/ajpcell.00060.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu F., Mih J.D., Shea B.S., Kho A.T., Sharif A.S., Tager A.M., Tschumperlin D.J. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol. 2010;190:693–706. doi: 10.1083/jcb.201004082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sandbo N., Ngam C., Torr E., Kregel S., Kach J., Dulin N. Control of myofibroblast differentiation by microtubule dynamics through a regulated localization of mDia2. J Biol Chem. 2013;288:15466–15473. doi: 10.1074/jbc.M113.464461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tomasek J.J., Vaughan M.B., Kropp B.P., Gabbiani G., Martin M.D., Haaksma C.J., Hinz B. Contraction of myofibroblasts in granulation tissue is dependent on rho/rho kinase/myosin light chain phosphatase activity. Wound Repair Regen. 2006;14:313–320. doi: 10.1111/j.1743-6109.2006.00126.x. [DOI] [PubMed] [Google Scholar]
- 13.Zhou Y., Huang X., Hecker L., Kurundkar D., Kurundkar A., Liu H., Jin T.H., Desai L., Bernard K., Thannickal V.J. Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J Clin Invest. 2013;123:1096–1108. doi: 10.1172/JCI66700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sandbo N., Dulin N. Actin cytoskeleton in myofibroblast differentiation: ultrastructure defining form and driving function. Transl Res. 2011;158:181–196. doi: 10.1016/j.trsl.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sandbo N., Lau A., Kach J., Ngam C., Yau D., Dulin N.O. Delayed stress fiber formation mediates pulmonary myofibroblast differentiation in response to TGF-beta. Am J Physiol Lung Cell Mol Physiol. 2011;301:L656–L666. doi: 10.1152/ajplung.00166.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson L.A., Rodansky E.S., Haak A.J., Larsen S.D., Neubig R.R., Higgins P.D. Novel Rho/MRTF/SRF inhibitors block matrix-stiffness and TGF-beta-induced fibrogenesis in human colonic myofibroblasts. Inflamm Bowel Dis. 2014;20:154–165. doi: 10.1097/01.MIB.0000437615.98881.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Small E.M. The actin-MRTF-SRF gene regulatory axis and myofibroblast differentiation. J Cardiovasc Transl Res. 2012;5:794–804. doi: 10.1007/s12265-012-9397-0. [DOI] [PubMed] [Google Scholar]
- 18.Scharenberg M.A., Pippenger B.E., Sack R., Zingg D., Ferralli J., Schenk S., Martin I., Chiquet-Ehrismann R. TGF-beta-induced differentiation into myofibroblasts involves specific regulation of two MKL1 isoforms. J Cell Sci. 2014;127:1079–1091. doi: 10.1242/jcs.142075. [DOI] [PubMed] [Google Scholar]
- 19.Huang X., Yang N., Fiore V.F., Barker T.H., Sun Y., Morris S.W., Ding Q., Thannickal V.J., Zhou Y. Matrix stiffness-induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am J Respir Cell Mol Biol. 2012;47:340–348. doi: 10.1165/rcmb.2012-0050OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haak A.J., Tsou P.S., Amin M.A., Ruth J.H., Campbell P., Fox D.A., Khanna D., Larsen S.D., Neubig R.R. Targeting the myofibroblast genetic switch: inhibitors of myocardin-related transcription factor/serum response factor-regulated gene transcription prevent fibrosis in a murine model of skin injury. J Pharmacol Exp Ther. 2014;349:480–486. doi: 10.1124/jpet.114.213520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bell J.L., Haak A.J., Wade S.M., Kirchhoff P.D., Neubig R.R., Larsen S.D. Optimization of novel nipecotic bis(amide) inhibitors of the Rho/MKL1/SRF transcriptional pathway as potential anti-metastasis agents. Bioorg Med Chem Lett. 2013;23:3826–3832. doi: 10.1016/j.bmcl.2013.04.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evelyn C.R., Wade S.M., Wang Q., Wu M., Iñiguez-Lluhí J.A., Merajver S.D., Neubig R.R. CCG-1423: a small-molecule inhibitor of RhoA transcriptional signaling. Mol Cancer Ther. 2007;6:2249–2260. doi: 10.1158/1535-7163.MCT-06-0782. [DOI] [PubMed] [Google Scholar]
- 23.Hayashi K., Watanabe B., Nakagawa Y., Minami S., Morita T. RPEL proteins are the molecular targets for CCG-1423, an inhibitor of Rho signaling. PLoS One. 2014;9:e89016. doi: 10.1371/journal.pone.0089016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horowitz J.C., Ajayi I.O., Kulasekaran P., Rogers D.S., White J.B., Townsend S.K., White E.S., Nho R.S., Higgins P.D., Huang S.K., Sisson T.H. Survivin expression induced by endothelin-1 promotes myofibroblast resistance to apoptosis. Int J Biochem Cell Biol. 2012;44:158–169. doi: 10.1016/j.biocel.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson L.A., Rodansky E.S., Sauder K.L., Horowitz J.C., Mih J.D., Tschumperlin D.J., Higgins P.D. Matrix stiffness corresponding to strictured bowel induces a fibrogenic response in human colonic fibroblasts. Inflamm Bowel Dis. 2013;19:891–903. doi: 10.1097/MIB.0b013e3182813297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osterholzer J.J., Christensen P.J., Lama V., Horowitz J.C., Hattori N., Subbotina N., Cunningham A., Lin Y., Murdock B.J., Morey R.E., Olszewski M.A., Lawrence D.A., Simon R.H., Sisson T.H. PAI-1 promotes the accumulation of exudate macrophages and worsens pulmonary fibrosis following type II alveolar epithelial cell injury. J Pathol. 2012;228:170–180. doi: 10.1002/path.3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang J., Velikoff M., Canalis E., Horowitz J.C., Kim K.K. Activated alveolar epithelial cells initiate fibrosis through autocrine and paracrine secretion of connective tissue growth factor. Am J Physiol Lung Cell Mol Physiol. 2014;306:L786–L796. doi: 10.1152/ajplung.00243.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sisson T.H., Mendez M., Choi K., Subbotina N., Courey A., Cunningham A., Dave A., Engelhardt J.F., Liu X., White E.S., Thannickal V.J., Moore B.B., Christensen P.J., Simon R.H. Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am J Respir Crit Care Med. 2010;181:254–263. doi: 10.1164/rccm.200810-1615OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reka A.K., Kurapati H., Narala V.R., Bommer G., Chen J., Standiford T.J., Keshamouni V.G. Peroxisome proliferator-activated receptor-gamma activation inhibits tumor metastasis by antagonizing Smad3-mediated epithelial-mesenchymal transition. Mol Cancer Ther. 2010;9:3221–3232. doi: 10.1158/1535-7163.MCT-10-0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Courey A.J., Horowitz J.C., Kim K.K., Koh T.J., Novak M.L., Subbotina N., Warnock M., Xue B., Cunningham A.K., Lin Y., Goldklang M.P., Simon R.H., Lawrence D.A., Sisson T.H. The vitronectin-binding function of PAI-1 exacerbates lung fibrosis in mice. Blood. 2011;118:2313–2321. doi: 10.1182/blood-2010-12-324574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Penke L.R., Huang S.K., White E.S., Peters-Golden M. Prostaglandin E2 inhibits alpha-smooth muscle actin transcription during myofibroblast differentiation via distinct mechanisms of modulation of serum response factor and myocardin-related transcription factor-A. J Biol Chem. 2014;289:17151–17162. doi: 10.1074/jbc.M114.558130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singer I.I., Kawka D.W., Kazazis D.M., Clark R.A. In vivo co-distribution of fibronectin and actin fibers in granulation tissue: immunofluorescence and electron microscope studies of the fibronexus at the myofibroblast surface. J Cell Biol. 1984;98:2091–2106. doi: 10.1083/jcb.98.6.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hecker L., Logsdon N.J., Kurundkar D., Kurundkar A., Bernard K., Hock T., Meldrum E., Sanders Y.Y., Thannickal V.J. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci Transl Med. 2014;6:231ra47. doi: 10.1126/scitranslmed.3008182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hecker L., Vittal R., Jones T., Jagirdar R., Luckhardt T.R., Horowitz J.C., Pennathur S., Martinez F.J., Thannickal V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med. 2009;15:1077–1081. doi: 10.1038/nm.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vittal R., Horowitz J.C., Moore B.B., Zhang H., Martinez F.J., Toews G.B., Standiford T.J., Thannickal V.J. Modulation of prosurvival signaling in fibroblasts by a protein kinase inhibitor protects against fibrotic tissue injury. Am J Pathol. 2005;166:367–375. doi: 10.1016/S0002-9440(10)62260-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ajayi I.O., Sisson T.H., Higgins P.D., Booth A.J., Sagana R.L., Huang S.K., White E.S., King J.E., Moore B.B., Horowitz J.C. X-linked inhibitor of apoptosis regulates lung fibroblast resistance to Fas-mediated apoptosis. Am J Respir Cell Mol Biol. 2013;49:86–95. doi: 10.1165/rcmb.2012-0224OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blackwell T.S., Tager A.M., Borok Z., Moore B.B., Schwartz D.A., Anstrom K.J. Future directions in idiopathic pulmonary fibrosis research. An NHLBI workshop report. Am J Respir Crit Care Med. 2014;189:214–222. doi: 10.1164/rccm.201306-1141WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Horowitz J.C., Lee D.Y., Waghray M., Keshamouni V.G., Thomas P.E., Zhang H., Cui Z., Thannickal V.J. Activation of the pro-survival phosphatidylinositol 3-kinase/AKT pathway by transforming growth factor-beta1 in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor. J Biol Chem. 2004;279:1359–1367. doi: 10.1074/jbc.M306248200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Horowitz J.C., Rogers D.S., Sharma V., Vittal R., White E.S., Cui Z., Thannickal V.J. Combinatorial activation of FAK and AKT by transforming growth factor-beta1 confers an anoikis-resistant phenotype to myofibroblasts. Cell Signal. 2007;19:761–771. doi: 10.1016/j.cellsig.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horowitz J.C., Rogers D.S., Simon R.H., Sisson T.H., Thannickal V.J. Plasminogen activation induced pericellular fibronectin proteolysis promotes fibroblast apoptosis. Am J Respir Cell Mol Biol. 2008;38:78–87. doi: 10.1165/rcmb.2007-0174OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sisson T.H., Maher T.M., Ajayi I.O., King J.E., Higgins P.D., Booth A.J., Sagana R.L., Huang S.K., White E.S., Moore B.B., Horowitz J.C. Increased survivin expression contributes to apoptosis-resistance in IPF fibroblasts. Adv Biosci Biotechnol. 2012;3:657–664. doi: 10.4236/abb.2012.326085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elzi D.J., Song M., Hakala K., Weintraub S.T., Shiio Y. Wnt antagonist SFRP1 functions as a secreted mediator of senescence. Mol Cell Biol. 2012;32:4388–4399. doi: 10.1128/MCB.06023-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li J., Lee J.M., Johnson J.A. Microarray analysis reveals an antioxidant responsive element-driven gene set involved in conferring protection from an oxidative stress-induced apoptosis in IMR-32 cells. J Biol Chem. 2002;277:388–394. doi: 10.1074/jbc.M109380200. [DOI] [PubMed] [Google Scholar]
- 44.Phan S.H. Biology of fibroblasts and myofibroblasts. Proc Am Thorac Soc. 2008;5:334–337. doi: 10.1513/pats.200708-146DR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thannickal V.J., Horowitz J.C. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc Am Thorac Soc. 2006;3:350–356. doi: 10.1513/pats.200601-001TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Serini G., Bochaton-Piallat M.L., Ropraz P., Geinoz A., Borsi L., Zardi L., Gabbiani G. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J Cell Biol. 1998;142:873–881. doi: 10.1083/jcb.142.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thannickal V.J., Roman J. Challenges in translating preclinical studies to effective drug therapies in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;181:532–533. doi: 10.1164/rccm.200911-1740ED. [DOI] [PubMed] [Google Scholar]
- 48.Kach J., Sandbo N., La J., Denner D., Reed E.B., Akimova O., Koltsova S., Orlov S.N., Dulin N.O. Antifibrotic effects of noscapine through activation of prostaglandin E2 receptors and protein kinase A. J Biol Chem. 2014;289:7505–7513. doi: 10.1074/jbc.M113.546812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Small E.M., Thatcher J.E., Sutherland L.B., Kinoshita H., Gerard R.D., Richardson J.A., Dimaio J.M., Sadek H., Kuwahara K., Olson E.N. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ Res. 2010;107:294–304. doi: 10.1161/CIRCRESAHA.110.223172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu H., Wu X., Qin H., Tian W., Chen J., Sun L., Fang M., Xu Y. Myocardin-related transcription factor A epigenetically regulates renal fibrosis in diabetic nephropathy. J Am Soc Nephrol. 2014 doi: 10.1681/ASN.2014070678. 2014, [Epub ahead of print] doi: 10.1681/ASN.2014070678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang S.K., Scruggs A.M., Donaghy J., Horowitz J.C., Zaslona Z., Przybranowski S., White E.S., Peters-Golden M. Histone modifications are responsible for decreased Fas expression and apoptosis resistance in fibrotic lung fibroblasts. Cell Death Dis. 2013;4:e621. doi: 10.1038/cddis.2013.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wynes M.W., Edelman B.L., Kostyk A.G., Edwards M.G., Coldren C., Groshong S.D., Cosgrove G.P., Redente E.F., Bamberg A., Brown K.K., Reisdorph N., Keith R.C., Frankel S.K., Riches D.W. Increased cell surface Fas expression is necessary and sufficient to sensitize lung fibroblasts to Fas ligation-induced apoptosis: implications for fibroblast accumulation in idiopathic pulmonary fibrosis. J Immunol. 2011;187:527–537. doi: 10.4049/jimmunol.1100447. [DOI] [PubMed] [Google Scholar]
- 53.Ding Q., Cai G.Q., Hu M., Yang Y., Zheng A., Tang Q., Gladson C.L., Hayasaka H., Wu H., You Z., Southern B.D., Grove L.M., Rahaman S.O., Fang H., Olman M.A. FAK-related nonkinase is a multifunctional negative regulator of pulmonary fibrosis. Am J Pathol. 2013;182:1572–1584. doi: 10.1016/j.ajpath.2013.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kang H.R., Lee C.G., Homer R.J., Elias J.A. Semaphorin 7A plays a critical role in TGF-beta1-induced pulmonary fibrosis. J Exp Med. 2007;204:1083–1093. doi: 10.1084/jem.20061273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kinoshita K., Aono Y., Azuma M., Kishi J., Takezaki A., Kishi M., Makino H., Okazaki H., Uehara H., Izumi K., Sone S., Nishioka Y. Antifibrotic effects of focal adhesion kinase inhibitor in bleomycin-induced pulmonary fibrosis in mice. Am J Respir Cell Mol Biol. 2013;49:536–543. doi: 10.1165/rcmb.2012-0277OC. [DOI] [PubMed] [Google Scholar]
- 56.Lagares D., Busnadiego O., García-Fernández R.A., Kapoor M., Liu S., Carter D.E., Abraham D., Shi-Wen X., Carreira P., Fontaine B.A., Shea B.S., Tager A.M., Leask A., Lamas S., Rodríguez-Pascual F. Inhibition of focal adhesion kinase prevents experimental lung fibrosis and myofibroblast formation. Arthritis Rheum. 2012;64:1653–1664. doi: 10.1002/art.33482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lagares D., Busnadiego O., García-Fernández R.A., Lamas S., Rodríguez-Pascual F. Adenoviral gene transfer of endothelin-1 in the lung induces pulmonary fibrosis through the activation of focal adhesion kinase. Am J Respir Cell Mol Biol. 2012;47:834–842. doi: 10.1165/rcmb.2011-0446OC. [DOI] [PubMed] [Google Scholar]
- 58.Le Cras T.D., Korfhagen T.R., Davidson C., Schmidt S., Fenchel M., Ikegami M., Whitsett J.A., Hardie W.D. Inhibition of PI3K by PX-866 prevents transforming growth factor-alpha-induced pulmonary fibrosis. Am J Pathol. 2010;176:679–686. doi: 10.2353/ajpath.2010.090123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang S.K., White E.S., Wettlaufer S.H., Grifka H., Hogaboam C.M., Thannickal V.J., Horowitz J.C., Peters-Golden M. Prostaglandin E(2) induces fibroblast apoptosis by modulating multiple survival pathways. FASEB J. 2009;23:4317–4326. doi: 10.1096/fj.08-128801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maher T.M., Evans I.C., Bottoms S.E., Mercer P.F., Thorley A.J., Nicholson A.G., Laurent G.J., Tetley T.D., Chambers R.C., McAnulty R.J. Diminished prostaglandin E2 contributes to the apoptosis paradox in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;182:73–82. doi: 10.1164/rccm.200905-0674OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gyrd-Hansen M., Meier P. IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat Rev Cancer. 2010;10:561–574. doi: 10.1038/nrc2889. [DOI] [PubMed] [Google Scholar]
- 62.Sisson T.H., Simon R.H. The plasminogen activation system in lung disease. Curr Drug Targets. 2007;8:1016–1029. doi: 10.2174/138945007781662319. [DOI] [PubMed] [Google Scholar]
- 63.Chang W., Wei K., Jacobs S.S., Upadhyay D., Weill D., Rosen G.D. SPARC suppresses apoptosis of idiopathic pulmonary fibrosis fibroblasts through constitutive activation of beta-catenin. J Biol Chem. 2010;285:8196–8206. doi: 10.1074/jbc.M109.025684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang Y.P., Wang W.L., Liu J., Li W.B., Bai L.L., Yuan Y.D., Song S.X. Plasminogen activator inhibitor-1 promotes the proliferation and inhibits the apoptosis of pulmonary fibroblasts by Ca(2+) signaling. Thromb Res. 2013;131:64–71. doi: 10.1016/j.thromres.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 65.Sakai N., Chun J., Duffield J.S., Wada T., Luster A.D., Tager A.M. LPA1-induced cytoskeleton reorganization drives fibrosis through CTGF-dependent fibroblast proliferation. FASEB J. 2013;27:1830–1846. doi: 10.1096/fj.12-219378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kolodsick J.E., Peters-Golden M., Larios J., Toews G.B., Thannickal V.J., Moore B.B. Prostaglandin E2 inhibits fibroblast to myofibroblast transition via E. prostanoid receptor 2 signaling and cyclic adenosine monophosphate elevation. Am J Respir Cell Mol Biol. 2003;29:537–544. doi: 10.1165/rcmb.2002-0243OC. [DOI] [PubMed] [Google Scholar]
- 67.Wynn T.A. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.King T.E., Jr., Bradford W.Z., Castro-Bernardini S., Fagan E.A., Glaspole I., Glassberg M.K., Gorina E., Hopkins P.M., Kardatzke D., Lancaster L., Lederer D.J., Nathan S.D., Pereira C.A., Sahn S.A., Sussman R., Swigris J.J., Noble P.W., ASCEND Study Group A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–2092. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 69.Richeldi L., du Bois R.M., Raghu G., Azuma A., Brown K.K., Costabel U., Cottin V., Flaherty K.R., Hansell D.M., Inoue Y., Kim D.S., Kolb M., Nicholson A.G., Noble P.W., Selman M., Taniguchi H., Brun M., Le Maulf F., Girard M., Stowasser S., Schlenker-Herceg R., Disse B., Collard H.R., INPULSIS Trial Investigators Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–2082. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]