Auranofin exerts broad-spectrum bactericidal activities by targeting thiol-redox homeostasis (original) (raw)
Significance
The identification of new antibiotics with novel mechanisms of action has become a pressing need considering the growing threat of drug-resistant infections. We have identified auranofin, an FDA-approved drug, as having potent bactericidal activity against Gram-positive pathogenic bacteria. Auranofin inhibits an enzyme, thioredoxin reductase, not targeted by other antibiotics, and thus retains efficacy against many clinically relevant drug-resistant strains, including in a mouse model of infection. Because auranofin is an approved drug, its route to the clinic may be expedited with reduced cost. Our work suggests that auranofin is a candidate for drug repurposing in antibacterial therapy.
Keywords: auranofin, tuberculosis, MRSA, Gram-positive
Abstract
Infections caused by antibiotic-resistant bacteria are a rising public health threat and make the identification of new antibiotics a priority. From a cell-based screen for bactericidal compounds against Mycobacterium tuberculosis under nutrient-deprivation conditions we identified auranofin, an orally bioavailable FDA-approved antirheumatic drug, as having potent bactericidal activities against both replicating and nonreplicating M. tuberculosis. We also found that auranofin is active against other Gram-positive bacteria, including Bacillus subtilis and Enterococcus faecalis, and drug-sensitive and drug-resistant strains of Enterococcus faecium and Staphylococcus aureus. Our biochemical studies showed that auranofin inhibits the bacterial thioredoxin reductase, a protein essential in many Gram-positive bacteria for maintaining the thiol-redox balance and protecting against reactive oxidative species. Auranofin decreases the reducing capacity of target bacteria, thereby sensitizing them to oxidative stress. Finally, auranofin was efficacious in a murine model of methicillin-resistant S. aureus infection. These results suggest that the thioredoxin-mediated redox cascade of Gram-positive pathogens is a valid target for the development of antibacterial drugs, and that the existing clinical agent auranofin may be repurposed to aid in the treatment of several important antibiotic-resistant pathogens.
The emergence and spread of antibiotic-resistant bacterial infections is an ongoing and growing public health concern, resulting in an increased urgency to identify new drugs with novel mechanisms of action (1). Drug resistance has major clinical consequences, including increased healthcare costs and higher rates of mortality. On a worldwide basis, outbreaks of methicillin-resistant Staphylococcus aureus (MRSA), previously confined to hospitals, are now occurring frequently in community settings (2, 3). For tuberculosis (TB), the costly and lengthy course of combination antibiotic treatment required to sterilize Mycobacterium tuberculosis is associated with low patient compliance in developing countries, which contributes significantly to the emergence of drug-resistant strains (4). Clinical cure rates for individuals with multidrug-resistant TB in outpatient settings are <50% (5).
How M. tuberculosis bacilli can persist in their host despite prolonged chemotherapy is not well understood, although it is hypothesized that a subpopulation of bacteria emerges that is nonreplicating and displays phenotypic (nonheritable) tolerance to most anti-TB drugs (6, 7). These “persisters” are likely responsible for relapse rates as high as 5% even after extended chemotherapy (5). Likewise, reactivation of nonreplicating persisters in immunocompromised individuals, such as those infected with HIV, leads to high rates of morbidity and mortality and greater risk of disease transmission. To shorten treatment times, new TB drugs should have activity not only against resistant isolates, but also against dormant populations.
Recent studies have suggested that the activities of some antibacterial drugs are mediated not simply by the initial drug–enzyme interaction, but also by downstream adverse effects on metabolic and homeostatic networks within the bacteria (8). Therefore, the bactericidal potential of a drug can be significantly influenced by its mode of action within cellular metabolic and signaling networks (8). One vulnerable pathway is thiol-based redox metabolism, which is essential for many cellular processes, including protection of the bacterial cell against endogenous and exogenous reactive oxygen species, proper protein folding, and DNA synthesis (9–12). In many organisms, glutathione (GSH) and GSH reductase (GR) function in parallel with thioredoxin (Trx) and Trx reductase (TrxR) to provide the cell with a source of reducing equivalents (10). In such organisms, the two systems can compensate for one another, as only double mutants of the pathways are nonviable (13). Many Gram-positive bacteria, including M. tuberculosis (and certain other Actinobacteria), S. aureus, and certain Bacillus spp., as well as the Gram-negative Helicobacter pylori, lack the conventional redox couple GSH-GR normally present in most Gram-negative species (14–16). Thus, the Trx-TrxR system is often essential in these GSH-lacking organisms, as has been experimentally verified in S. aureus and Bacillus subtilis (17, 18).
In this work, we report that both replicating and nonreplicating M. tuberculosis are susceptible to the FDA-approved organogold drug auranofin, in addition to a variety of additional pathogenic Gram-positive bacteria. We show that auranofin is a potent inhibitor of bacterial TrxR and disrupts the redox balance in these bacteria, resulting in bacterial cell death, which translates to in vivo efficacy in a mouse model of systemic MRSA infection. These results validate Trx-based redox control in Gram-positive organisms as a viable antibacterial drug target, and suggest that auranofin is a candidate for repurposing in antibacterial therapy.
Results
Auranofin Has Activity Against Replicating and Nonreplicating M. tuberculosis.
To identify inhibitors with activity against replicating and nonreplicating M. tuberculosis, we undertook a cell-based screen of bioactive compounds under nutrient-deprivation conditions. M. tuberculosis strain H37Ra was transformed with the integration vector pMV306hsp+Lux, in which the bacterial luciferase operon is driven by the M. tuberculosis hsp60 promoter sequence (19). This strain constitutively expresses luciferase, which therefore can serve as a readout of bacteria viability. Compound efficacy was assayed against bacteria after a 24-h period of starvation in PBS [also known as the Loebel model (20)], an interval that induces transcriptional changes to nutrient deprivation but still provides a robust luminescent readout (21). From a screen of 1280 pharmacologically active compounds (the LOPAC library), auranofin was identified as an inhibitor of reporter activity under both nonreplicating and replicating conditions (Fig. 1 A and B). Indeed, auranofin appeared to have greater activity against the reporter strain under nonreplicating conditions than in medium that supports mycobacterial growth (EC50 = 450 nM vs. 4.6 μM, respectively). Further examination of auranofin activity in growth-supporting minimal medium showed that this difference is related to the presence of BSA in the 7H9 medium used for assays under replicating conditions and leads to a 20-fold increase in the minimum inhibitory concentration (MIC) relative to the same growth-supporting medium lacking BSA (SI Appendix, Table S1). Auranofin was originally used for the treatment of rheumatoid arthritis and recently has been shown to have potent activity against a variety of pathogens (22). Because of the known pharmacology and established safety profile of this drug in humans, we focused our efforts on further characterization of its antibacterial activity.
Fig. 1.
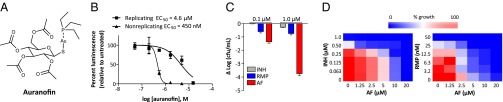
Auranofin displays potent activity against replicating and nonreplicating M. tuberculosis. (A) Structure of auranofin. (B) Auranofin activity against replicating and nonreplicating M. tuberculosis H37Ra constitutively expressing bacterial luciferase. Nonreplicating bacteria were starved in PBS for 24 h before treatment. Replicating cultures were grown in 7H9 medium. Cultures were treated for 24 h with auranofin and then assayed for luminescence. (C) Auranofin (AF) shows potent bactericidal activity against nonreplicating M. tuberculosis H37Ra relative to INH and RMP. Starvation-induced nonreplicating M. tuberculosis was treated with auranofin or the front-line anti-TB drugs INH and RMP for 5 d, followed by enumeration of viable colonies. (D) Combination treatment of M. tuberculosis H37Ra in growth assays in 7H9 medium with auranofin in combination with either INH or RMP. One compound was diluted along the ordinate of a 96-well plate and the other along the abscissa, resulting in a checkerboard of the two compounds.
We confirmed the activity of auranofin against nonreplicating M. tuberculosis (H37Ra) under carbon-starved culture conditions by CFU assays. Auranofin showed potent bactericidal activity against nonreplicating M. tuberculosis, resulting in 1.3-log and 3.7-log decreases in viable bacteria after 5 d of treatment with drug concentrations of 100 nM and 1.0 μM, respectively (Fig. 1_C_). Under the same assay conditions, the front-line anti-TB drugs rifampicin (RMP) and isoniazid (INH) showed only moderate activity, each reducing viable bacteria by <1 log at 1 μM concentration. These results suggest that auranofin’s antimicrobial efficacy is not limited to bacteria with highly active metabolic states, but extends to dormant bacterial populations with altered metabolism, an attractive feature for treatment of latent TB infection.
Combination therapy remains a cornerstone of modern TB treatment, and any novel drug must demonstrate compatibility with established anti-TB drugs (23). Thus, we assessed the activity of auranofin against M. tuberculosis in the presence of RMP and INH. Actively replicating M. tuberculosis in 7H9 (+ BSA) medium containing serial dilutions of auranofin were treated with serial dilutions of either RMP or INH (Fig. 1_D_). An inoculum of H37Ra was added to each well for a starting OD600 of 0.05, and after 5 d growth was assayed by measuring turbidity. Auranofin demonstrated additive effects on inhibiting M. tuberculosis growth in combination with RMP and INH, as assessed by determining the average fractional inhibitory concentration (FIC) index (average FIC index = 1.0 and 0.90, respectively) (Fig. 1_D_). These results suggest that auranofin is a suitable candidate for use with front-line anti-TB drugs.
Auranofin Has Different Potency Against Gram-Positive and Gram-Negative Bacteria.
In light of our results with M. tuberculosis, we profiled the activity of auranofin against a panel of Gram-positive and Gram-negative pathogenic bacterial species, including strains resistant to the contemporary drugs methicillin, vancomycin, and linezolid. Auranofin showed potent growth inhibition against each of the Gram-positive strains tested (Table 1). The activities against S. aureus are consistent with recent findings of auranofin’s antimicrobial activity against drug-sensitive and methicillin-resistant strains of S. aureus (24, 25). In addition, we found that auranofin inhibits growth of Enterococcus faecium and Enterococcus faecalis, with an MIC of 0.5 mg/L for both. In most instances, the drug showed equal efficacy against drug-resistant and drug-sensitive strains of the same species. In contrast to results observed against these diverse Gram-positive strains, auranofin lacked significant activity against Gram-negative bacteria, with an MIC ≥16 mg/L against multiple strains of Acinetobacter baumannii, Pseudomonas aeruginosa, and Klebsiella pneumoniae.
Table 1.
Auranofin displays potent activity against a panel of drug-sensitive and drug-resistant pathogenic Gram-positive bacteria
| Strain | Description | MIC, mg/L |
|---|---|---|
| H37Rv mc262320 | M. tuberculosis (Δ_panCD_, Δ_RD1_) | 0.5 |
| H37Ra | Reference M. tuberculosis | 0.5 |
| B. subtilis 168 | Reference strain | 0.05 |
| B. subtilis PY79 | Reference strain | 0.3 |
| MRSA Sanger 252 | Hospital-acquired MRSA | 0.5 |
| MRSA TCH1516 | USA300 MRSA | 0.5 |
| MRSA ST-59 | Hemolytic Taiwan clone | 0.5 |
| MRSA A7819 | Linezolid-resistant MRSA | 0.5 |
| VRSA-PA | vanA vancomycin-resistant | 0.5 |
| GISA D712 | Glycopeptide-intermediate S. aureus | 0.5 |
| GISA A5940 | Glycopeptide-intermediate S. aureus | 1 |
| X18311 MRSA | VISA isolate | 0.5 |
| PC-3 | VISA (NY) | 0.5 |
| HIP 5836 | VISA (NJ) | 0.5 |
| MSSA 29213 | Reference S. aureus | 0.5 |
| VRE8 WMC | Vancomycin-resistant E. faecium | 0.5 |
| VRE 12–15-19 UCLA | Vancomycin-resistant E. faecium | 1 |
| E. faecalis Belt | Non-VRE clinical isolate | 0.5 |
| K. pneumoniae 1100 | Respiratory isolate | >16 |
| A. baumannii 19606 | Urine isolate | >32 |
| A. baumannii 17978 | CNS isolate | 32 |
| A. baumannii 5075 | Multidrug-resistant strain | 32 |
| P. aeruginosa PA01 | Reference strain | >32 |
| P. aeruginosa PA103 | Reference strain | >32 |
Auranofin Inhibits M. tuberculosis and S. aureus TrxR in Vitro.
Previous work has shown that auranofin is highly thiol-reactive, with an affinity for redox-active cysteines (26, 27). For instance, it has been shown to inhibit TrxR from numerous organisms, such as Entamoeba histolytica, and the related enzyme Trx-GSH reductase of Schistosoma mansoni (28–30). Many Gram-positive bacteria, including M. tuberculosis and S. aureus, have the redox-active protein Trx but lack the low molecular weight counterpart GSH. Although M. tuberculosis and S. aureus do contain additional low molecular weight thiols, mycothiol (MSH) and ergothioneine in M. tuberculosis, and bacillithiol (BSH) in S. aureus and Bacillus spp., their functions remain relatively unknown (31). In addition, neither MSH nor BSH alone is essential for M. tuberculosis or S. aureus viability, respectively, and BSH-null B. subtilis still maintains proteins in a reduced state (32–34). Thus, these data suggest that Trx, along with its cognate oxidoreductase TrxR, are likely the key intracellular redox mediators in these species. This notion is supported by the fact that TrxR and Trx are essential for viability in S. aureus and B. subtilis, respectively (17, 18). Transposon mutagenesis studies and genetic knockout (below) also have suggested that the TrxR (TrxB2) of M. tuberculosis is essential (35, 36).
We further assessed the essentiality of trxB2 for M. tuberculosis in vitro growth by using a specialized transduction strategy for generating deletion mutants (37). Flanking sequences of the gene were cloned into a selectable/counterselectable cassette to generate an allelic exchange substrate and cloned into a phasmid for phage packaging and transduction into M. tuberculosis H37Rv and CDC1551. The transductants were checked for the deletion of the corresponding genes by PCR. None of the colonies obtained from the transductions of H37Rv or CDC1551 with the phage for trxB2 deletion were positive for the trxB2 deletion, supporting the notion that trxB2 has an essential function in the bacillus.
The TrxR enzymes of M. tuberculosis and S. aureus (TrxB) are part of the low molecular weight family of disulfide bacterial TrxRs and share <30% homology with human TrxR, which evolved from a separate lineage and is catalytically distinct from the bacterial enzyme (38). Because TrxB2 and TrxB appear to be essential for M. tuberculosis and S. aureus viability, respectively, we hypothesized that auranofin’s mechanism of action is mediated, at least in part, by inhibition of these proteins. To explore this possibility, we cloned both M. tuberculosis trxB2 and its substrate Trx (trxC) into expression vectors containing an N-terminal histidine tag and expressed them in Escherichia coli followed by purification using nickel-charged agarose resin (SI Appendix, Fig. S1). TrxR activity assays were performed in the presence of TrxC (39). Enzyme activity was assayed using a reaction that regenerates oxidized TrxC via the rapid intramolecular disulfide exchange reaction with 5–5′-dithiobis-(2-nitrobenzoic acid) (DTNB) (SI Appendix, Fig. S2) (40). In this assay, reaction progress was monitored by measuring the absorbance of the liberated TNB chromophore at 412 nm. Under these conditions, we obtained a K m value of 2.0 μM for TrxC with TrxB2, which matches previous findings (SI Appendix, Fig. S3) (39). Similarly, we cloned, expressed, and purified the S. aureus oxidoreductase TrxB and assayed its activity using TrxC, which gave a K m of 23 μM. When preincubated with TrxR and NADPH, auranofin potently inhibited both M. tuberculosis and S. aureus TrxR enzymes in a dose-dependent fashion, with IC50 values of 63 ± 3 nM and 90 ± 6 nM against TrxB2 and TrxB, respectively (Fig. 2 A and B), values well below the level needed to achieve its cellular effects.
Fig. 2.

Auranofin inhibits bacterial TrxR. (A) Purified recombinant M. tuberculosis TrxB2 was preincubated with NADPH and DTNB with or without auranofin for 15 min. Reactions were initiated by the addition of TrxC. Shown is a dose–response plot for the initial rate of TrxB2 activity in the presence or absence of auranofin. (Inset) Representative progress plot of TrxB2 activity in the presence or absence of auranofin used for generating the dose–response plot. The colors of the lines on the progress plot correspond to the same color points on the dose–response plot. The purple line on the progress plot corresponds to the untreated control, which is not graphed on the dose-response plot. Auranofin concentrations tested were 800, 400, 200, 100, 50, and 25 nM. (B) Dose–response plots for S. aureus TrxB activity with and without a 15-min preincubation with auranofin with corresponding progress plots showing that auranofin inhibits TrxB activity. Assays were carried out as with TrxB2. Dose–response plots represent at least three determinations ± SE.
Auranofin Treatment of Gram-Positive Bacteria Results in Thiol Depletion and Compromises Defense Against Oxidative Stress.
We hypothesized that inhibiting the function of TrxR would result in a reduced level of free thiols and decreased reducing potential in auranofin-treated cells. To test this notion, we assayed for free thiol content in both auranofin-treated M. tuberculosis and S. aureus. Indeed, M. tuberculosis and S. aureus treated with auranofin at the MIC showed a 23% and 38% decrease, respectively, in the amount of cellular free thiols (Fig. 3 A and B). In contrast, no decrease in free thiols was observed in S. aureus and M. tuberculosis treated by the antibiotics ampicillin and INH, respectively, at the MIC for each compound. These results suggest that auranofin-mediated inhibition of TrxR disrupts the bacteria’s thiol-redox homeostasis.
Fig. 3.

Auranofin depletes intracellular thiols and sensitizes S. aureus to oxidizing agents and oxidative stress. (A) S. aureus cultures treated with indicated concentrations of auranofin for 15 min show a decrease in free thiol concentration relative to untreated control. (B) M. tuberculosis mc26230 cultures treated with auranofin in minimal medium for 3 h at the indicated concentrations also show a similar dose-dependent depletion of thiols. (C) Combination treatment of S. aureus with auranofin and diamide has synergistic antimicrobial activities. S. aureus cultures were treated for 3 h with the indicated concentrations of diamide and 700 nM auranofin, alone or in combination. (D) Combined treatment of S. aureus with auranofin and paraquat affords synergistic antimicrobial activity. S. aureus cultures were treated for 3 h with the indicated concentrations of paraquat and 700 nM auranofin, alone or in combination. (E) Loss of GSH synthesis sensitizes E. coli to auranofin. WT or E. coli lacking GSH (gshA) or TrxR (trxB) were treated with the indicated concentrations of auranofin for 6 h, and growth was assayed by absorbance.
To further test this notion, we compared the antibacterial activity of diamide, a thiol-oxidizing agent, alone and in the presence of auranofin. Millimolar concentrations of diamide had only modest activity against S. aureus, resulting in a 1.1-log decrease in CFU at 10 mM (Fig. 3_C_). However, when combined with auranofin at 700 nM, the antimicrobial activity of the two compounds was synergistic and produced a 4.4-log decrease in CFU, more than 100-fold greater than what would be expected from dosewise additivity (Fig. 3_C_), consistent with the notion that auranofin impairs the bacteria’s ability to cope with thiol oxidation.
Loss of cellular reducing capacity also can compromise bacterial defense against reactive oxygen species, which involves multiple thiol-dependent enzymes (10, 41). We compared the antimicrobial activity of paraquat, which generates intracellular reactive oxygen species, in the presence or absence of auranofin. Paraquat alone showed little antimicrobial activity against S. aureus (0.8-log decrease in CFU at 5 mM), but when combined with 700 nM auranofin, the two compounds synergized (a ∼5-log decrease in CFU which is a greater than 3-log decrease beyond expected dosewise additivity) in their bacterial killing (Fig. 3_D_). These results demonstrate that auranofin severely impairs the bacterial defense against oxidative stress.
Defense Against Auranofin in Gram-Negative Bacteria Is Mediated by Glutathione.
Unlike M. tuberculosis and S. aureus, most Gram-negative species have both Trx and GSH systems, and the presence of GSH-GR in the Gram-negative E. coli model is redundant with the Trx system (10, 13). To determine whether the GSH system plays a role in E. coli resistance to auranofin, we evaluated the ability of strains of E. coli lacking in either GSH (ΔgshA) or TrxR (ΔtrxB) to grow in the presence of auranofin in M63 minimal medium. E. coli without TrxB showed slightly increased resistance to auranofin compared with WT E. coli (the parent K-12 strain of the knockouts), likely owing to an off-target effect, while E. coli lacking GSH showed a fourfold increase in sensitivity to killing by auranofin (Fig. 3_E_). These results suggest that the GSH system helps defend against a loss of TrxR-reducing capability in auranofin-treated E. coli, and further establishes TrxR as a relevant target against a number of bacterial species.
Auranofin Is Effective in a Murine Systemic Infection Model of S. aureus.
To determine auranofin’s in vivo efficacy, we tested its ability to protect mice from MRSA-induced mortality in a peritonitis model. Female CD1 mice were infected i.p. with ∼109 CFU of hospital-associated MRSA strain Sanger 252, a route and inoculum producing rapid bacteremia and subsequent lethality. At 1 h after infection, the mice (n = 8 per group) were treated with a single i.p. injection of two different dosages of auranofin (0.12 or 0.012 mg/kg, equivalent to human doses of 6 mg or 0.6 mg/day) or vehicle control. Treatment then continued once daily for the remainder of the study. These doses are significantly lower than the maximum tolerated dose in mice (70 mg/kg). Both doses of auranofin appeared to be well tolerated and were sufficient to provide significant protection against mortality (Fig. 4). Four of the eight mice receiving 0.12 mg/kg auranofin survived to 7 d, and three of the eight mice receiving 0.012 mg/kg survived to 7 d; none of the animals in the vehicle control group survived beyond 4 d. These results suggest that the potent in vitro activity of auranofin translates in vivo to a protective anti-MRSA activity at well-tolerated doses.
Fig. 4.

Auranofin shows efficacy in a murine peritonitis MRSA infection model. Survival of mice with an acute i.p. infection induced with an initial inoculum of 2 × 109 CFU/mouse MRSA Sanger 252 (n = 8 per group). Mice were treated with one dose at 1 h post infection, followed by once-daily treatment with i.p. auranofin. P < 0.01, Mantel–Cox test.
Discussion
The identification of safe, efficacious, and novel antibacterial drugs is a pressing need brought about by growing drug resistance and the limited pipeline for these infections. The repurposing of approved drugs for new uses may provide a more rapid and cost-effective route to the clinic for new therapeutic strategies. From a cell-based screen of bioactive compounds under nutrient-deprivation conditions we identified auranofin, an orally bioavailable FDA-approved antirheumatic drug, as having potent bactericidal activities against both M. tuberculosis and a number of other clinically important Gram-positive bacterial species. Because auranofin exerts its effects through a unique mechanism involving inhibition of TrxR, it retains activity against contemporary antibiotic-resistant strains. Intriguingly, auranofin showed significant activity against both replicating and nonreplicating M. tuberculosis and compatibility with other front-line TB drugs. The combination of these properties suggests that auranofin may meet the expectations for the target product profile for a new TB drug (5). As such, we are currently investigating whether these properties translate into longer-term in vivo treatment models of acute and latent M. tuberculosis infection.
Our data suggest that the bacterial flavoenzyme TrxR is the primary target for auranofin. In M. tuberculosis, Trx provides electrons for enzymes involved in protecting against oxidative and nitrosative stress, such as alkyl hydroperoxide reductase and thiol peroxidase. Thus, inhibition of TrxR would compromise the cell’s ability to cope with reactive species, especially in the oxidative environment of the macrophage phagosome (42). In addition, Trx is necessary for DNA synthesis and protein repair via the reduction of ribonucleotide reductase and methionine sulfoxide reductase, respectively (10). Therefore, the effect of inhibiting TrxR is likely multifaceted, impacting a variety of functions necessary for bacterial survival and proliferation. Such activity likely accounts for auranofin’s efficacy against both replicating and nonreplicating M. tuberculosis.
Auranofin consists of a gold(I) center coordinated to a thiosugar and triethylphosphine. The mechanism of TrxR inhibition by auranofin likely occurs through displacement of the two ligands from gold by the formation of a tight complex between the metal and the active site cysteines of TrxR. Crystallographic evidence for gold(I) binding to catalytic cysteine residues has previously been provided for the flavoenzymes Trx-GR and the trypanothione reductase from Shistosoma mansoni and Leishmania infantum, respectively (26, 43). Mercuric ion reductase, a related NADPH-dependent oxidoreductase, also is known to bind mercuric ion in a similar fashion (44). In addition, given the thiophilic nature of the compound, it is possible that auranofin may react with other enzymes bearing reactive cysteine residues, such as cysteine proteases and phosphatases (45, 46). Indeed, we found that auranofin can inhibit M. tuberculosis mycothione reductase in vitro, albeit with greatly reduced potency relative to its inhibition of TrxB2 (SI Appendix, Fig. S4). Mycothione reductase is an NADPH-dependent oxidoreductase with a catalytically important redox-active disulfide that reduces MSH (47). The inhibition of multiple enzymes by auranofin also may help explain our inability to isolate spontaneous auranofin-resistant mutants despite numerous attempts with M. tuberculosis and would likely reduce the risk of emergent resistance in the clinical setting. In addition, the possibility of formation of other forms of gold in vivo that are also biologically active cannot be excluded.
One potential concern regarding auranofin stems from its apparently low in vitro therapeutic index (HepG2 CC50 = 4.5 μM) (SI Appendix, Fig. S5). The mechanisms of auranofin’s cytotoxicity and anti-inflammatory actions are possibly mediated by its inhibition of several signaling pathways involved in inflammation and cell growth, in addition to inhibition of thiol-redox enzymes (48). However, auranofin has been used extensively, and its safety is well documented (49). Auranofin is approved for long-term daily dosage at 6 mg/day, and serious side effects are rare, the most common being gastrointestinal distress that is easily manageable (50). Patients on auranofin therapy have been monitored in clinical trials for longer than 5 years, a much greater interval than would be expected for a normal course of antibiotic therapy or even drug-resistant TB cases, and have shown no cumulative toxicity (51). At the current FDA-approved human dose (6 mg/day), a mean steady-state blood gold concentration of 3.5 μM is achieved in 12 wk. A Phase II clinical trial seeking to determine the safety and effectiveness of an increased dose of auranofin (12 mg/day) is currently underway (ClinicalTrials.gov identifier: NCT01419691).
Gold salts have been used for medicinal purposes for centuries, and specifically were among the first drugs used for TB therapy through the 1920s, although their use was discontinued owing to inadequate efficacy in poorly defined clinical trials (48). Auranofin, which was first approved in 1985 as an oral gold therapy, is currently one of only three gold complexes approved for use in the clinic, although it has fallen out of favor because of the improved efficacy of newer antirheumatic agents. Interest in its potential clinical applications has been sparked by recent studies showing significant antiparasitic and antineoplastic activity (48). The fact that auranofin is an approved and off-patent drug means that it could provide a more rapid and cost-effective route for the compound into clinical trials and, if successful, to patients. In addition to auranofin’s antibacterial efficacy against MRSA, its efficacy in acute and latent animal models of TB infection is currently under investigation. Given the differences in the pathologies and courses of infection between animal models of TB and humans with TB, information on auranofin’s effectiveness may be limited in these studies (52); therefore, small-scale clinical trials using doses already optimized as safe and efficacious may be warranted to evaluate the anti-TB efficacy of auranofin as a monotherapy or part of combination therapies.
Materials and Methods
General Methods.
M. tuberculosis strain H37Ra and the attenuated strain mc26230 (H37Rv Δ_panCD_, Δ_RD-1_) were grown in Middlebrook 7H9 broth (BD Diagnostics) supplemented with Middlebrook oleic, albumin, catalase, dextrose (OADC) Growth Supplement (10% vol/vol; BD Diagnostics), glycerol (0.2% vol/vol), and Tween 80 (0.05% vol/vol) or Roisin’s medium supplemented with glycerol (0.2%). For mc26230, pantothenic acid (100 mg/mL) was added. MICs were determined by broth microdilution methodology according to Clinical and Laboratory Standards Institute guidelines.
Nonreplicating M. tuberculosis Screen.
M. tuberculosis strain H37Ra was transfected with the integration vector pMV306hsp+Lux in which the bacterial luciferase operon is driven by the M. tuberculosis hsp60 promoter sequence (a gift from Brian Robertson, Medical Research Council Centre for Molecular Bacteriology and Infection, Department of Medicine, Imperial College London, London, and Siouxsie Wiles, Department of Molecular Medicine and Pathology, University of Auckland, Auckland, New Zealand; Addgene plasmid 26159). For screening, bacteria were washed three times in PBS with tyloxapol (0.05% vol/vol) and then incubated in PBS/tyloxapol for 24 h, followed by addition to white 384-well plates. Compound was added at a final concentration of 10 μM, and plates were read for luminescence 24 h later.
Deletion of TrxR (Rv3913, trxB2) in M. tuberculosis.
The Rv3913 gene was replaced by a γδ(sacB-hyg)γδ cassette as described by Jain et al. (37). Details are provided in SI Appendix.
Protein Expression, Purification, and Activity Assays.
M. tuberculosis trxb2 (Rv3913), trxC (Rv3914), and S. aureus trxB were cloned into the pET28a or pET28b expression vectors (EMD Millipore) and purified by Ni-NTA chromatography. TrxR activity assays were performed in a total volume of 50 μL in black clear-bottom 384-well plates at 37 °C. Standard reaction mixtures contained 25 nM TrxR, 100 μM DTNB, 100 μM NADPH, and 10 and 50 μM TrxC for TrxB2 and TrxB, respectively, unless noted otherwise, in 50 mM Hepes, pH 7.5, with 2 mM EDTA. Absorbance at 412 nm was read on a Spectramax M5 plate reader as an indication of enzyme activity.
Murine Peritonitis Infection Model.
Auranofin was tested in vivo in a murine model of MRSA peritonitis essentially as described previously (53). Details are provided in SI Appendix.
Supplementary Material
Supplementary File
Acknowledgments
We thank Drs. Tim Wright and Case McNamara for helpful discussions. This work was supported by funding from the Bill and Melinda Gates Foundation and National Institutes of Health Grant R01 AI097548.
Footnotes
The authors declare no conflict of interest.
References
- 1.Boucher HW, et al. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48(1):1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 2.Arias CA, Murray BE. Antibiotic-resistant bugs in the 21st century—a clinical super-challenge. N Engl J Med. 2009;360(5):439–443. doi: 10.1056/NEJMp0804651. [DOI] [PubMed] [Google Scholar]
- 3.Klevens RM, et al. Active Bacterial Core surveillance (ABCs) MRSA Investigators Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA. 2007;298(15):1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 4.Abubakar I, et al. Drug-resistant tuberculosis: Time for visionary political leadership. Lancet Infect Dis. 2013;13(6):529–539. doi: 10.1016/S1473-3099(13)70030-6. [DOI] [PubMed] [Google Scholar]
- 5.Koul A, Arnoult E, Lounis N, Guillemont J, Andries K. The challenge of new drug discovery for tuberculosis. Nature. 2011;469(7331):483–490. doi: 10.1038/nature09657. [DOI] [PubMed] [Google Scholar]
- 6.Stewart GR, Robertson BD, Young DB. Tuberculosis: A problem with persistence. Nat Rev Microbiol. 2003;1(2):97–105. doi: 10.1038/nrmicro749. [DOI] [PubMed] [Google Scholar]
- 7.Sacchettini JC, Rubin EJ, Freundlich JS. Drugs versus bugs: In pursuit of the persistent predator Mycobacterium tuberculosis. Nat Rev Microbiol. 2008;6(1):41–52. doi: 10.1038/nrmicro1816. [DOI] [PubMed] [Google Scholar]
- 8.Kohanski MA, Dwyer DJ, Collins JJ. How antibiotics kill bacteria: From targets to networks. Nat Rev Microbiol. 2010;8(6):423–435. doi: 10.1038/nrmicro2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ritz D, Beckwith J. Roles of thiol-redox pathways in bacteria. Annu Rev Microbiol. 2001;55:21–48. doi: 10.1146/annurev.micro.55.1.21. [DOI] [PubMed] [Google Scholar]
- 10.Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med. 2014;66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 11.Lundström J, Holmgren A. Protein disulfide-isomerase is a substrate for thioredoxin reductase and has thioredoxin-like activity. J Biol Chem. 1990;265(16):9114–9120. [PubMed] [Google Scholar]
- 12.Holmgren A, Sengupta R. The use of thiols by ribonucleotide reductase. Free Radic Biol Med. 2010;49(11):1617–1628. doi: 10.1016/j.freeradbiomed.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 13.Prinz WA, Aslund F, Holmgren A, Beckwith J. The role of the thioredoxin and glutaredoxin pathways in reducing protein disulfide bonds in the Escherichia coli cytoplasm. J Biol Chem. 1997;272(25):15661–15667. doi: 10.1074/jbc.272.25.15661. [DOI] [PubMed] [Google Scholar]
- 14.Newton GL, et al. Distribution of thiols in microorganisms: Mycothiol is a major thiol in most actinomycetes. J Bacteriol. 1996;178(7):1990–1995. doi: 10.1128/jb.178.7.1990-1995.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fahey RC, Brown WC, Adams WB, Worsham MB. Occurrence of glutathione in bacteria. J Bacteriol. 1978;133(3):1126–1129. doi: 10.1128/jb.133.3.1126-1129.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tomb JF, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388(6642):539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 17.Uziel O, Borovok I, Schreiber R, Cohen G, Aharonowitz Y. Transcriptional regulation of the Staphylococcus aureus thioredoxin and thioredoxin reductase genes in response to oxygen and disulfide stress. J Bacteriol. 2004;186(2):326–334. doi: 10.1128/JB.186.2.326-334.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scharf C, et al. Thioredoxin is an essential protein induced by multiple stresses in Bacillus subtilis. J Bacteriol. 1998;180(7):1869–1877. doi: 10.1128/jb.180.7.1869-1877.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andreu N, et al. Optimisation of bioluminescent reporters for use with mycobacteria. PLoS ONE. 2010;5(5):e10777. doi: 10.1371/journal.pone.0010777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loebel RO, Shorr E, Richardson HB. The influence of foodstuffs upon the respiratory metabolism and growth of human tubercle bacilli. J Bacteriol. 1933;26(2):139–166. doi: 10.1128/jb.26.2.139-166.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Betts JC, Lukey PT, Robb LC, McAdam RA, Duncan K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol Microbiol. 2002;43(3):717–731. doi: 10.1046/j.1365-2958.2002.02779.x. [DOI] [PubMed] [Google Scholar]
- 22.Finkelstein AE, et al. Auranofin: New oral gold compound for treatment of rheumatoid arthritis. Ann Rheum Dis. 1976;35(3):251–257. doi: 10.1136/ard.35.3.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zumla AI, et al. New antituberculosis drugs, regimens, and adjunct therapies: Needs, advances, and future prospects. Lancet Infect Dis. 2014;14(4):327–340. doi: 10.1016/S1473-3099(13)70328-1. [DOI] [PubMed] [Google Scholar]
- 24.Cassetta MI, Marzo T, Fallani S, Novelli A, Messori L. Drug repositioning: Auranofin as a prospective antimicrobial agent for the treatment of severe staphylococcal infections. Biometals. 2014;27(4):787–791. doi: 10.1007/s10534-014-9743-6. [DOI] [PubMed] [Google Scholar]
- 25.Hokai Y, et al. Auranofin and related heterometallic gold(I)-thiolates as potent inhibitors of methicillin-resistant Staphylococcus aureus bacterial strains. J Inorg Biochem. 2014;138:81–88. doi: 10.1016/j.jinorgbio.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Angelucci F, et al. Inhibition of Schistosoma mansoni thioredoxin-glutathione reductase by auranofin: Structural and kinetic aspects. J Biol Chem. 2009;284(42):28977–28985. doi: 10.1074/jbc.M109.020701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeon KI, Byun MS, Jue DM. Gold compound auranofin inhibits IkappaB kinase (IKK) by modifying Cys-179 of IKKbeta subunit. Exp Mol Med. 2003;35(2):61–66. doi: 10.1038/emm.2003.9. [DOI] [PubMed] [Google Scholar]
- 28.Gromer S, Arscott LD, Williams CH, Jr, Schirmer RH, Becker K. Human placenta thioredoxin reductase: Isolation of the selenoenzyme, steady-state kinetics, and inhibition by therapeutic gold compounds. J Biol Chem. 1998;273(32):20096–20101. doi: 10.1074/jbc.273.32.20096. [DOI] [PubMed] [Google Scholar]
- 29.Tejman-Yarden N, et al. A reprofiled drug, auranofin, is effective against metronidazole-resistant Giardia lamblia. Antimicrob Agents Chemother. 2013;57(5):2029–2035. doi: 10.1128/AAC.01675-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuntz AN, et al. Thioredoxin glutathione reductase from Schistosoma mansoni: An essential parasite enzyme and a key drug target. PLoS Med. 2007;4(6):e206. doi: 10.1371/journal.pmed.0040206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fahey RC. Glutathione analogs in prokaryotes. Biochim Biophys Acta. 2013;1830(5):3182–3198. doi: 10.1016/j.bbagen.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 32.Vilchèze C, et al. Mycothiol biosynthesis is essential for ethionamide susceptibility in Mycobacterium tuberculosis. Mol Microbiol. 2008;69(5):1316–1329. doi: 10.1111/j.1365-2958.2008.06365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Posada AC, et al. Importance of bacillithiol in the oxidative stress response of Staphylococcus aureus. Infect Immun. 2014;82(1):316–332. doi: 10.1128/IAI.01074-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaballa A, et al. Biosynthesis and functions of bacillithiol, a major low-molecular-weight thiol in Bacilli. Proc Natl Acad Sci USA. 2010;107(14):6482–6486. doi: 10.1073/pnas.1000928107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high-density mutagenesis. Mol Microbiol. 2003;48(1):77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 36.Griffin JE, et al. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011;7(9):e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jain P, et al. Specialized transduction designed for precise high-throughput unmarked deletions in Mycobacterium tuberculosis. MBio. 2014;5(3):e01245-14. doi: 10.1128/mBio.01245-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arscott LD, Gromer S, Schirmer RH, Becker K, Williams CH., Jr The mechanism of thioredoxin reductase from human placenta is similar to the mechanisms of lipoamide dehydrogenase and glutathione reductase and is distinct from the mechanism of thioredoxin reductase from Escherichia coli. Proc Natl Acad Sci USA. 1997;94(8):3621–3626. doi: 10.1073/pnas.94.8.3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Akif M, Khare G, Tyagi AK, Mande SC, Sardesai AA. Functional studies of multiple thioredoxins from Mycobacterium tuberculosis. J Bacteriol. 2008;190(21):7087–7095. doi: 10.1128/JB.00159-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moore EC, Reichard P, Thelander L. Enzymatic synthesis of deoxyribonucleotides, V: Purification and properties of thioredoxin reductase from Escherichia coli B. J Biol Chem. 1964;239:3445–3452. [PubMed] [Google Scholar]
- 41.Mishra S, Imlay J. Why do bacteria use so many enzymes to scavenge hydrogen peroxide? Arch Biochem Biophys. 2012;525(2):145–160. doi: 10.1016/j.abb.2012.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jaeger T, et al. Multiple thioredoxin-mediated routes to detoxify hydroperoxides in Mycobacterium tuberculosis. Arch Biochem Biophys. 2004;423(1):182–191. doi: 10.1016/j.abb.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 43.Ilari A, et al. A gold-containing drug against parasitic polyamine metabolism: The X-ray structure of trypanothione reductase from Leishmania infantum in complex with auranofin reveals a dual mechanism of enzyme inhibition. Amino Acids. 2012;42(2-3):803–811. doi: 10.1007/s00726-011-0997-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Engst S, Miller SM. Alternative routes for entry of HgX2 into the active site of mercuric ion reductase depend on the nature of the X ligands. Biochemistry. 1999;38(12):3519–3529. doi: 10.1021/bi982680c. [DOI] [PubMed] [Google Scholar]
- 45.Chircorian A, Barrios AM. Inhibition of lysosomal cysteine proteases by chrysotherapeutic compounds: A possible mechanism for the antiarthritic activity of Au(I) Bioorg Med Chem Lett. 2004;14(20):5113–5116. doi: 10.1016/j.bmcl.2004.07.073. [DOI] [PubMed] [Google Scholar]
- 46.Krishnamurthy D, et al. Gold(I)-mediated inhibition of protein tyrosine phosphatases: A detailed in vitro and cellular study. J Med Chem. 2008;51(15):4790–4795. doi: 10.1021/jm800101w. [DOI] [PubMed] [Google Scholar]
- 47.Patel MP, Blanchard JS. Expression, purification, and characterization of Mycobacterium tuberculosis mycothione reductase. Biochemistry. 1999;38(36):11827–11833. doi: 10.1021/bi991025h. [DOI] [PubMed] [Google Scholar]
- 48.Madeira JM, Gibson DL, Kean WF, Klegeris A. The biological activity of auranofin: Implications for novel treatment of diseases. Inflammopharmacology. 2012;20(6):297–306. doi: 10.1007/s10787-012-0149-1. [DOI] [PubMed] [Google Scholar]
- 49.Kean WF, Hart L, Buchanan WW. Auranofin. Br J Rheumatol. 1997;36(5):560–572. doi: 10.1093/rheumatology/36.5.560. [DOI] [PubMed] [Google Scholar]
- 50.Suarez-Almazor ME, Spooner CH, Belseck E, Shea B. Auranofin versus placebo in rheumatoid arthritis. Cochrane Database Syst Rev. 2000;(2):CD002048. doi: 10.1002/14651858.CD002048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blodgett RC, Jr, Pietrusko RG. Long-term efficacy and safety of auranofin: A review of clinical experience. Scand J Rheumatol Suppl. 1986;63:67–78. [PubMed] [Google Scholar]
- 52.Basaraba RJ. Experimental tuberculosis: The role of comparative pathology in the discovery of improved tuberculosis treatment strategies. Tuberculosis (Edinb) 2008;88(Suppl 1):S35–S47. doi: 10.1016/S1472-9792(08)70035-0. [DOI] [PubMed] [Google Scholar]
- 53.Hensler ME, et al. Anthracimycin activity against contemporary methicillin-resistant Staphylococcus aureus. J Antibiot (Tokyo) 2014;67(8):549–553. doi: 10.1038/ja.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary File