Redox Signal-mediated Enhancement of the Temperature Sensitivity of Transient Receptor Potential Melastatin 2 (TRPM2) Elevates Glucose-induced Insulin Secretion from Pancreatic Islets (original) (raw)
Background: Transient receptor potential melastatin 2 (TRPM2) is a temperature-sensitive Ca2+-permeable ion channel involved in glucose-induced insulin secretion.
Results: Hydrogen peroxide treatment caused a TRPM2-dependent increase in intracellular Ca2+ concentration. Glucose-induced insulin secretion from pancreatic islets was temperature-, antioxidant-, and TRPM2-dependent.
Conclusion: Redox signal-mediated TRPM2 sensitization elevates glucose-induced insulin secretion.
Significance: These results provide new insights into the involvement of TRPM2 sensitization in insulin secretion.
Keywords: calcium channel, glucose, insulin, ion channel, redox signaling
Abstract
Transient receptor potential melastatin 2 (TRPM2) is a thermosensitive Ca2+-permeable cation channel expressed by pancreatic β cells where channel function is constantly affected by body temperature. We focused on the physiological functions of redox signal-mediated TRPM2 activity at body temperature. H2O2, an important molecule in redox signaling, reduced the temperature threshold for TRPM2 activation in pancreatic β cells of WT mice but not in TRPM2KO cells. TRPM2-mediated [Ca2+]i increases were likely caused by Ca2+ influx through the plasma membrane because the responses were abolished in the absence of extracellular Ca2+. In addition, TRPM2 activation downstream from the redox signal plus glucose stimulation enhanced glucose-induced insulin secretion. H2O2 application at 37 °C induced [Ca2+]i increases not only in WT but also in TRPM2KO β cells. This was likely due to the effect of H2O2 on KATP channel activity. However, the _N_-acetylcysteine-sensitive fraction of insulin secretion by WT islets was increased by temperature elevation, and this temperature-dependent enhancement was diminished significantly in TRPM2KO islets. These data suggest that endogenous redox signals in pancreatic β cells elevate insulin secretion via TRPM2 sensitization and activity at body temperature. The results in this study could provide new therapeutic approaches for the regulation of diabetic conditions by focusing on the physiological function of TRPM2 and redox signals.
Introduction
The transient receptor potential (TRP)2 ion channel superfamily consists of 28 channels in six subfamilies in mammals. Some channels detect a wide range of environmental factors, including thermal, mechanical, and chemical stimuli, in various species (1). Among them, nine members (TRPA1, TRPV1, TRPV2, TRPV3, TRPV4, TRPM2, TRPM4, TRPM5, and TRPM8) have been reported to have temperature sensitivity and are called thermoTRPs (2). TRPM3 has recently been reported to be activated by elevated temperature (3). The ambient temperature around peripheral sensory nerve endings in the skin can change dynamically and is detected by several thermoTRPs, such as TRPA1, TRPV1, TRPV2, and TRPM8, covering a wide range of temperatures from noxious cold to noxious heat (4). In addition to sensory neurons, skin keratinocytes reportedly detect ambient temperature with TRPV3, one of the thermoTRPs, and transmit the temperature information to sensory neurons (5, 6). Furthermore, many deep organs that are not normally exposed to dynamic temperature changes also express thermoTRPs whose activities are continuously affected by body temperature (1). Among them, the activity of TRPM2 at body temperature is regulated by intracellular endogenous ligands, including ADP-ribose and cyclic ADP-ribose (7, 8), and by environmental reactive oxygen species (ROS), so-called redox signals (9). Therefore, TRPM2 activity could be modulated by these regulatory molecules at body temperature and could be involved in physiological functions of cells or tissues in which TRPM2 is expressed.
The TRPM2 channel is expressed in the brain, liver, spleen, and pancreas, where its functions are continuously affected by body temperature (10). We demonstrated previously that one of the molecular mechanisms for reducing the temperature threshold for TRPM2 activation utilized hydrogen peroxide (H2O2). H2O2 is a highly versatile and functional molecule in redox signaling systems (9), and it likely regulates a number of physiological functions as a second messenger in vivo. In the absence of H2O2, the temperature threshold for TRPM2 activation is ∼47 °C. H2O2 can lower the temperature threshold for TRPM2 activation toward physiological body temperature, depending on H2O2 concentration and treatment duration (9). We also explored its involvement in cytokine release and the phagocytic activity of mouse peritoneal macrophages, in which the binding of pathogen-associated molecular patterns to Toll-like receptors activates NADPH oxidase and elicits the production of ROS for microbicidal activity. These findings illustrate that TRPM2 activity at body temperature could be regulated by endogenous redox signals.
Pancreatic β cells play a crucial role in blood glucose regulation because they secrete hypoglycemic insulin when blood glucose levels are elevated. The primary pathway for glucose-induced insulin secretion is well known to be mediated by Ca2+ influx through voltage-gated Ca2+ channels upon membrane depolarization following ATP-sensitive K+ (KATP) channel closure. However, recent studies have revealed the significant contribution of many other ion channels to the increases in intracellular Ca2+ concentrations upon glucose stimulation (11). In particular, several thermoTRPs (TRPA1, TRPV2, TRPV4, TRPM2, TRPM3, TRPM4, and TRPM5) have been reported to be expressed in pancreatic β cells, although their functions are not yet fully elucidated (12). We have reported previously that TRPM2 has high Ca2+ permeability and is expressed in pancreatic β cells (7). It regulates insulin secretion evoked by glucose or glucose together with incretin hormones at body temperature (13, 14).
Pancreatic β cells produce ROS in response to many kinds of extracellular signals, including insulin, cytokines, hormones, and blood glucose elevation (15, 16). Interestingly, the expression levels of the ROS-eliminating enzymes catalase and glutathione reductase are extremely low in the pancreas compared with other tissues (17). Pharmacological and genetic inhibition of NADPH oxidase reportedly suppresses glucose-induced insulin secretion (18), and H2O2 application with low concentrations of glucose apparently enhances insulin secretion (16). Therefore, physiological concentrations of ROS could function as favorable signaling molecules in pancreatic β cells (15, 16). On the other hand, low levels of antioxidant enzymes in the pancreas might explain its high vulnerability to supranormal levels of ROS that lead to cellular malfunction or death of β cells and progression of type II diabetes (19). In this study, we focused on the physiological function of TRPM2 sensitization in glucose-induced insulin secretion from pancreatic islets and the effect of temperature on this process.
EXPERIMENTAL PROCEDURES
Animals
Male C57BL/6NCr mice (Japan SLC) and mutant TRPM2-deficient (TRPM2KO) mice were provided by Yasuo Mori (Kyoto University, Kyoto, Japan) (20) and were used at 10–16 weeks of age. WT and TRPM2KO mice were housed in a controlled environment (12-h light/12-h dark cycle; room temperature, 22 - 24 °C; relative humidity, 50–60%) with free access to food and water. All procedures involving the care and use of animals were approved by the National Institute for Physiological Science and carried out in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals.
Cell Culture
Islets of Langerhans were isolated from mouse pancreas by collagenase digestion as described previously (21) with minor modifications. Animals were anesthetized with sodium pentobarbital (80 mg/kg intraperitoneally, Dainippon Sumitomo Pharma Co., Ltd.) followed by injection of 1.14 mg/ml collagenase (Sigma-Aldrich, St. Louis, MO) into the common bile duct ligated upper and lower sites. The collagenase was dissolved in Ca2+-free HEPES-supplemented Krebs-Ringer bicarbonate buffer solution (HKRB(−) (129 mm NaCl, 5 mm NaHCO3, 4.7 mm KCl, 1.2 mm KH2PO4, 1.2 mm MgSO4, 10 mm HEPES, 3.3 mm glucose, and 0.1% BSA (pH 7.4)). The pancreas was removed by dissection and incubated in collagenase solution at 37 °C for 23 min. The islets were washed twice with HKRB(−) to remove collagenase and were then used for experiments. For [Ca2+]i imaging experiments, islets were incubated with 1 mm EGTA in HKRB(−) and dispersed into single cells. The dissociated single pancreatic β cells were suspended in RPMI 1640 medium (WAKO Pure Chemical Industries, Ltd.) containing 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 5.6 mm glucose unless indicated otherwise. Dispersed cells were seeded onto poly-l-lysine (100 μm)-coated glass coverslips and used for fluorescence measurements within 12–24 h of seeding. The concentration of glucose (5.6 mm) in culture medium matched the fasting blood glucose level (13).
Fluorescence Measurements
Fura-2 fluorescence of mouse pancreatic β cells was measured in 2 mm Ca2+-containing HKRB(+) (129 mm NaCl, 5 mm NaHCO3, 4.7 mm KCl, 1.2 mm KH2PO4, 1.2 mm MgSO4, 2.0 mm CaCl2, 10 mm HEPES, and 2.8 mm glucose (pH 7.4)). Ca2+-free HKRB(−) used in the Ca2+-free experiments was made by adding 5 mm EGTA instead of 2 mm CaCl2. Thermal stimulation was applied by increasing the bath temperature with preheated solution through an inline heater (SH-27B, Warner Instruments). The proximal temperature of the recording area was monitored with a thermocouple (TA-29, Warner Instruments). Fura-2 loaded in the cells was excited with 340- and 380-nm wavelengths, and emission was monitored at 510 nm with a (complementary metal-oxide-semiconductor) camera (Zyla 5.5, Andor Technology). Data were acquired using iQ2.8 software (Andor Technology) and analyzed by ImageJ (http://rsbweb.nih.gov/ij/). The cells that reacted to tolbutamide (300 μm) with a ratio increase over 0.3 from the basal ratio were identified as pancreatic β-cells. Ionomycin (5 μm) was applied to confirm cell viability, and ratio increases from basal level were normalized to those evoked by ionomycin for each experiment. In some experiments, [Ca2+]i was calculated according to an in vitro calibration using a Kd value of fura-2 (224 nm) at 37 °C.
Measurement of insulin release from mouse pancreatic islets of Langerhans
Islets were collected in RPMI of the same composition as that in cell culture and incubated for 2 h and then preincubated in Krebs-Ringer buffer, KRB(+) (129 mm NaCl, 5 mm NaHCO3, 5.2 mm KCl, 1.3 mm KH2PO4, 2.7 mm CaCl2, 1.3 mm MgSO4, 0.2% BSA, pH 7.4) containing 3.3 mm glucose for 30 min at 37 °C, and then 10 islets/10 μl were sorted into 1.5 ml tubes and used for the in vitro insulin secretion assay. All of the in vitro insulin secretion assays were conducted in triplicate and their average values were used. Insulin secretion was elicited by adding 400 μl of 16.7 mm glucose-containing KRB(+) and incubated for 60 min at temperatures of 33, 37 and 40 °C in the presence or absence of NAC (300 μm). KRB(+) with 3.3 mm glucose was used as the negative control. After 60 min incubation, the supernatants were collected and used for the measurement of insulin content by ELISA assay (Morinaga) following the manufacturer's instructions.
Statistical analysis
Data are presented as means ± S.E. or means ± S.D. Statistical analysis was performed using the Student t test, paired t test or two-way analysis of variance followed by the Bonferroni-type post-hoc multiple t tests. p values less than 0.05 were considered significant.
RESULTS
Temperature Sensitivity in Pancreatic β Cells Was Enhanced by H2O2 Treatment
We have reported previously that the temperature threshold for TRPM2 activation was reduced from a supraphysiological to a physiological temperature range by H2O2, a kind of ROS, termed “sensitization,” involved in macrophage functions (9). To examine whether TRPM2 sensitization was also observed in pancreatic β cells, we first compared heat-evoked changes in intracellular Ca2+ concentrations between WT and TRPM2KO β cells using a Ca2+ imaging method. β cells were identified by their reactivity to tolbutamide (300 μm), a KATP channel inhibitor. Heat-evoked responses in WT β cells were enhanced by H2O2 treatment in a dose-dependent manner (Fig. 1, A and C), similar to heterologously expressed TRPM2 channels and WT mouse peritoneal macrophages (9). On the other hand, TRPM2KO cells did not show any statistically significant enhancement of the heat-evoked responses even after treatment with high concentrations of H2O2 (Fig. 1, B and C), whereas the responses to a high concentration of K+ (40 mm) were comparable with those in WT cells (Fig. 1D). These data indicated that redox signal-mediated changes in the temperature threshold for TRPM2 activation could also occur in pancreatic β cells, suggesting that TRPM2 sensitization is a broad phenomenon in many kinds of tissues in which TRPM2 is expressed.
FIGURE 1.

H2O2-evoked elevation of temperature sensitivity in pancreatic β cells was dependent on TRPM2. A and B, H2O2 treatment enhanced the heat-evoked response in WT pancreatic β cells (A) but not in TRPM2KO cells (B) in the presence of 2.8 mm glucose. Tolbutamide (300 μm), a KATP channel inhibitor, was used to identify β cells, and 40 mm K+ was used as a positive control. Fura-2 ratio traces are shown as mean ± S.D. C, dose-response relation of H2O2-evoked enhancement of heat-evoked response in WT and TRPM2KO β cells. All ratio increases were normalized to that of ionomycin, a Ca2+ ionophore that was used to confirm cell viability. Data are mean ± S.E. (n = 5–7). Not only a statistical significance between WT and TRPM2KO (p < 0.001) but also a significant correlation between genotype and temperature (_p_ < 0.001) were observed (two-way analysis of variance). NS, _p_ > 0.05 and ***, p < 0.001 _versus_ the 0 μm group in each genotype; $, p < 0.001 between the WT and TRPM2KO (Bonferroni-type post hoc multiple _t_ test). _D_, high K+ (40 mm)-evoked responses were not different between the two genotypes. Data are mean ± S.E. (_n_ = 31 in WT and 15 in TRPM2KO). NS, _p_ > 0.05 (Student's t test) between the WT and TRPM2KO.
The Heat-evoked Response Enhanced by H2O2 Was Dependent on Extracellular Ca2+
The H2O2 effects on heat-evoked responses were not observed under Ca2+-free extracellular conditions in WT cells (Fig. 2A), indicating that Ca2+ influx from the extracellular space mediated the heat-evoked [Ca2+]i increases. In the experiment shown in Fig. 2A, we used a different concentration of glucose from the previous analyses. Therefore, we investigated the responses in the presence of 10 mm glucose both in the culture medium and extracellular bath solution, similar to a previous report (22). However, we again failed to observe [Ca2+]i increases under conditions in which the extracellular medium was Ca2+-free (Fig. 2B). As shown in Fig. 2B, the responses to tolbutamide were small in the presence of 10 mm glucose because [Ca2+]i levels remained high, probably because of the closure of the KATP channel in the presence of the high concentration of glucose. One group reported that TRPM2 acted as a lysosomal Ca2+ release channel in pancreatic β cells (22). Therefore, we compared the lysosomal Ca2+ levels between WT and TRPM2KO β cells under extracellular Ca2+-free conditions in the presence of glycyl-l-phenylalanine-2-nephthylamide (GPN), a reagent destructive for lysosomes. 10 mm glucose evoked Ca2+ oscillations in β cells of both genotypes, and removal of extracellular Ca2+ suppressed the oscillatory [Ca2+]i responses (Fig. 2, C and D). After the removal of extracellular Ca2+, 200 μm GPN was applied to elicit Ca2+ release from lysosomes. [Ca2+]i increases upon GPN application were similar in WT and TRPM2KO β cells (Fig. 2, C–E), arguing against the involvement of TRPM2 in controlling lysosomal Ca2+ levels.
FIGURE 2.

Ca2+ release from lysosomes is unlikely to be involved in the H2O2-enhanced heat responses in pancreatic β cells. A and B, heat-evoked changes in [Ca2+]i in the absence of extracellular Ca2+ under different glucose conditions in culture medium and extracellular bath solution; 5.6 mm glucose in culture (5.6G culture) and 2.8 mm glucose in bath (2.8G bath) (A) and 10 mm glucose in culture (10G culture) and in bath (10G bath) (B). After addition of extracellular Ca2+, sensitization of heat-evoked responses was observed under both conditions. C and D, [Ca2+]i increases evoked by a lysosome-destroying reagent, GPN (200 μm), in the absence of extracellular Ca2+ in the presence of 10 mm glucose (10G) in WT (C) and TRPM2KO pancreatic β cells (D) at 37 °C. E, comparison of GPN-evoked [Ca2+]i increases between WT and TRPM2KO pancreatic β cells. Data are mean ± S.E. (n = 6). NS, p > 0.05 (Student's t test) between the WT and TRPM2KO.
H2O2 Evoked [Ca2+]i Increases at a Constant Temperature of 37 °C in WT and TRPM2KO β Cells
To examine the H2O2-evoked [Ca2+]i responses under more physiological conditions, we compared low concentration H2O2-evoked [Ca2+]i increases in WT and TRPM2KO β cells at a constant temperature (37 °C). H2O2 ranging from 3–10 μm caused similar [Ca2+]i increases in both genotypes (Fig. 3, A–C), suggesting that H2O2 also affected proteins other than TRPM2, such as the KATP channel, to regulate [Ca2+]i in pancreatic β cells.
FIGURE 3.
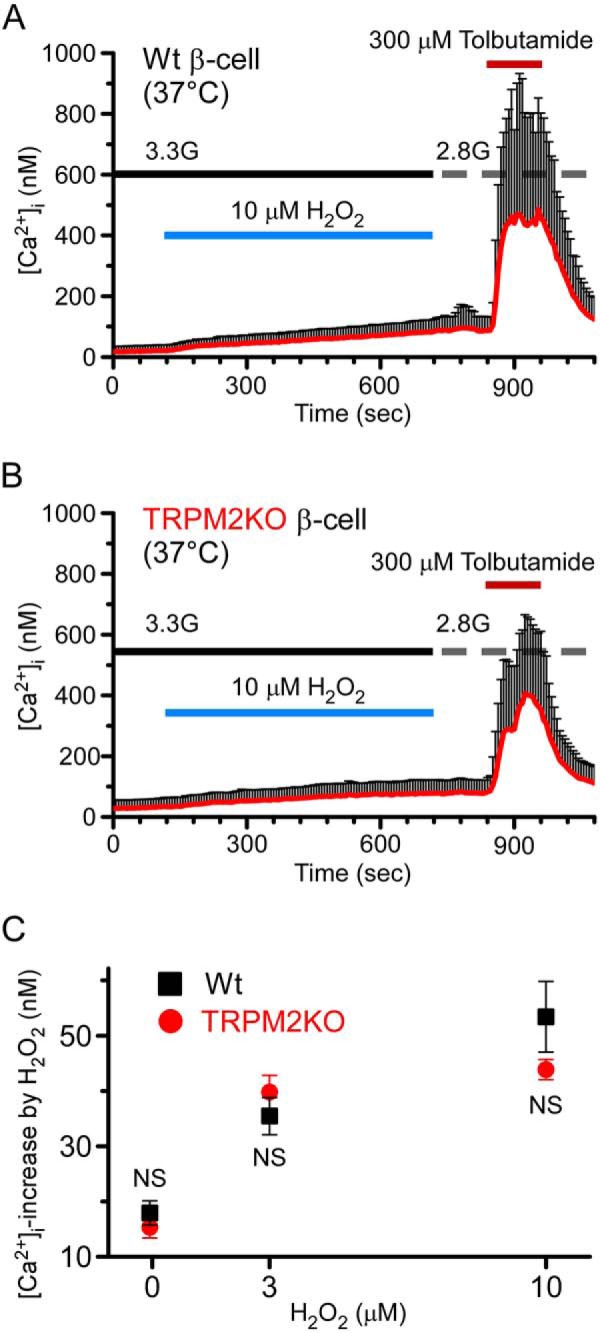
H2O2 increased [Ca2+]i in both WT and TRPM2KO pancreatic β cells at a constant temperature of 37 °C. A and B, [Ca2+]i increases evoked by 10 μm H2O2 in WT (A) and TRPM2KO pancreatic β-cells (B) at 37 °C in the presence of a low concentration of glucose (3.3 mm, 3.3G). C, comparison of dose dependence in H2O2-evoked [Ca2+]i increases between WT and TRPM2KO pancreatic β cells. Data are mean ± S.E. (n = 4). NS, p > 0.05 (Student's t test) between the WT and TRPM2KO at each concentration of H2O2.
Glucose-induced Insulin Secretion from pancreatic Islets Was Temperature-, Antioxidant-, and TRPM2-dependent
We have reported previously that glucose-induced insulin secretion was enhanced by the functional expression of TRPM2 in pancreatic β cells (13), although the detailed mechanisms have not been fully elucidated. In this study, we focused on the involvement of TRPM2 sensitization in glucose-induced insulin secretion because pancreatic β cells produce ROS in response to blood glucose elevation (16). We compared insulin secretion between WT and TRPM2KO pancreatic islets in the presence or absence of an antioxidant, NAC, to investigate the contribution of ROS and TRPM2 sensitization to insulin secretion. In addition, we conducted insulin secretion assays at three different temperatures (33, 37, and 40 °C) to clarify the involvement of TRPM2 sensitization. Consistent with our previous work (13), insulin secretion by TRPM2KO islets exposed to 16.7 mm glucose (16.7G) was significantly lower than that from WT islets at 37 and 40 °C but not at 33 °C, and the statistical significance of the decrease became larger in a temperature-dependent manner (Fig. 4, C, F, and I). In addition, NAC treatment significantly attenuated the 16.7 mm glucose-induced insulin secretion by WT islets but not by TRPM2KO islets (Fig. 4, A–I). Interestingly, the difference in insulin secretion between WT and TRPM2KO islets completely disappeared with NAC treatment, and no statistical significance among the WT (16.7G + NAC), TRPM2KO (16.7G), and TRPM2KO (16.7G + NAC) groups was observed at any of the temperatures. Specifically, we observed 17.4 ± 2.4 ng/10 islets, 19.8 ± 1.3 ng/10 islets, and 19.3 ± 1.8 ng/10 islets at 33 °C; 44.9 ± 5.3 ng/10 islets, 41.2 ± 3.9 ng/10 islets, and 39.8 ± 3.6 ng/10 islets at 37 °C; and 50.9 ± 3.5 ng/10 islets, 55.4 ± 2.9 ng/10 islets, and 51.3 ± 2.5 ng/10 islets at 40 °C in these three groups, respectively (two-way analysis of variance followed by the Bonferroni-type post hoc multiple t tests).
FIGURE 4.

Glucose-induced insulin secretion was attenuated by an antioxidant, _N_-acetyl cysteine, in a temperature-dependent manner in WT pancreatic islets but not in TRPM2KO islets. Effects of an antioxidant, NAC, were studied in WT (A, D, and G) and TRPM2KO (B, E, and H) islets at three different temperatures, 33 °C (A and B), 37 °C (C and D), or 40 °C (E and F). Glucose (3.3 mm, 3.3G) was used as a negative control, and 16.7 mm glucose (16.7G) was used to induce insulin secretion from pancreatic islets (n = 7–9). Each data pair of 16.7G and 16.7G + NAC was from a single animal and the side-by-side experimental batch (n = 3). NS, p > 0.05; *, p < 0.05; ***, _p_ < 0.001 (paired Student's _t_ test). Glucose-induced insulin secretion was attenuated in TRPM2KO islets compared with the WT, and the statistical significance of the difference between genotypes increased with temperature elevation (_C_, _F_, and _I_). _J_, NAC-sensitive fractions of glucose-induced insulin secretion at each temperature and genotype. Not only statistical significance between the WT and TRPM2KO (_p_ < 0.001) but also a significant correlation between genotype and temperature (_p_ < 0.05) was observed (two-way analysis of variance). NS, _p_ > 0.05; **, p < 0.01 between the WT and TRPM2KO under each temperature condition (Student's t test).
To more clearly evaluate the effects of TRPM2 sensitization and temperature on insulin secretion, we extracted the NAC-sensitive fraction from each experimental batch (Fig. 4J). WT islets showed clear temperature-dependent increases in NAC-sensitive insulin secretion, whereas such temperature-dependent NAC-sensitive insulin secretion was almost gone in TRPM2KO islets. In addition, a statistically significant correlation between temperature and genotype (p = 0.022, two-way analysis of variance) was also observed, suggesting that temperature has an important role in the physiological function of TRPM2 under conditions where a redox signal is present. These results are consistent with our previous study using mouse peritoneal macrophages (9).
DISCUSSION
We found that the heat-evoked responses in pancreatic β cells could be regulated by redox signaling that could occur in β cells in response to in vivo physiological stimuli like cytokines, insulin, and blood glucose elevation. The heat-evoked responses in β cells disappeared when extracellular Ca2+ was lacking or when TRPM2KO cells were examined (Figs. 1B and 2, A and B). Therefore, the heat-evoked responses observed in WT β cells were likely mediated by Ca2+ influx through TRPM2 channels.
We did not observe heat-mediated increases in [Ca2+]i upon H2O2 treatment under extracellular Ca2+-free conditions (Fig. 2, A and B) regardless of glucose concentrations (5.6 mm or 10 mm) in culture medium or extracellular bath solution. These results suggest little involvement of lysosomes in the H2O2-induced [Ca2+]i increases. Another group reported that TRPM2 functioned as a Ca2+ release channel in the lysosomes of pancreatic β cells (22), results that apparently contradict our results. However, we note that the experimental conditions were different, especially regarding glucose concentrations. However, similar GPN-evoked [Ca2+]i increases between WT and TRPM2KO β cells suggest that lysosomal Ca2+ levels are not different between the two genotypes. In addition, the fact that lysosomal lumina are maintained at an acidic pH (< 5.0) for enzyme reactions (23) and that TRPM2 activity is almost completely inhibited under acidic conditions (24) would make the involvement of lysosome TRPM2 in [Ca2+]i mobilization in β cells unlikely. Other possible factors might contribute to the difference between our results and the work mentioned above. In addition to TRPM2, other thermoTRPs (TRPV2, TRPV4, TRPM3, TRPM4, and TRPM5) are reportedly expressed in pancreatic β cells (12), and, among them, TRPV4 and TRPM3 are sensitive to physiological temperature and have Ca2+ permeability. However, these channels are not likely to contribute to the heat-evoked responses we observed in this study because the responses were completely lost in TRPM2KO β cells. In addition, activation of TRPM4 and TRPM5, monovalent-selective channels, could cause [Ca2+]i increases upon heat stimulation because their activation leads to membrane depolarization followed by the activation of voltage-gated Ca2+ channels. However, the fact that the phenomenon was lost in TRPM2KO β cells argues against that possibility as well. Accordingly, participation of TRPM2 channels is the most likely explanation for the H2O2-evoked events.
We asked whether H2O2 actually acted as a signaling molecule and regulated intracellular Ca2+ and insulin secretion from pancreatic β cells because H2O2 evoked [Ca2+]i increases both in WT and TRPM2KO β cells (Fig. 3) and because β cells highly express KATP channels whose activity is largely affected by ROS. ATP depletion following oxidative stress enhances the opening of KATP channels (25), whereas oxidation of sulfhydryl groups on KATP channels suppresses channel activity (26). Therefore, [Ca2+]i increases observed in TRPM2KO cells could be due to KATP channel closure in response to ROS and/or ROS actions on other proteins. Considering the extremely low expression level of ROS-quenching enzymes in pancreatic β cells (17), ROS concentrations could be dynamically changed in response to physiological stimuli, including blood glucose elevation initiated by food consumption. Chronic oxidative stress is well known to be involved in diabetic pathogenesis. However, ROS also have a role as signaling molecules regulating glucose-induced insulin secretion from pancreatic islets (27). Therefore, we investigated the involvement of TRPM2 sensitization in insulin secretion from pancreatic islets and found that the sensitization of TRPM2 mediated by redox signals that accompanied glucose stimulation enhanced glucose-induced insulin secretion. In addition, NAC-sensitive (redox signal-dependent) insulin secretion was elevated in a temperature-dependent manner in WT but not in TRPM2KO pancreatic islets (Fig. 4J). ROS modulate diverse protein functions as signaling molecules (28). Regarding the proteins involved in [Ca2+]i regulation in pancreatic islets, redox signals have dual effects on KATP channels (25, 26). Increases in insulin secretion from TRPM2KO islets exhibited a slight temperature dependence (Fig. 4, B, E, and H) that could be explained by the general temperature dependence of physicochemical reactions because of the change in molecular movement (29) as well as activation of other thermosensitive TRP channels, such as TRPM5 (30). However, we found that the NAC-sensitive fraction of insulin secretion was drastically diminished in TRPM2KO islets and that no statistically meaningful difference was observed between WT (16.7 mm glucose + NAC), TRPM2 (16.7 mm glucose), and TRPM2KO (16.7 mm glucose + NAC). Therefore, TRPM2 can be viewed as one of the main targets of redox signaling upon glucose stimulation, causing changes in the temperature sensitivity of insulin secretion from β cells, although the effects of TRPM2-mediated depolarization and [Ca2+]i increases on the function of other molecules cannot be excluded.
Along with the ROS-mediated regulation of TRPM2 function, the activity of TRPM2 is also regulated by incretin hormone through Gs-coupled receptor activation, which changes insulin secretion from pancreatic islets (13, 14). Therefore, TRPM2 function is thought to be regulated by various mechanisms working in concert to correctly modulate insulin secretion from pancreatic islets. Elucidating the detailed mechanisms controlling the role of TRPM2 in islet function could lead to a better understanding of the pathogenesis of diabetes and also provide new therapeutic approaches to regulate diabetic conditions.
Acknowledgments
We thank Dr. Kunitoshi Uchida (National Institute for Physiological Sciences) for advice.
*
This work was supported by Japanese Ministry of Education, Culture, Sports, Science and Technology Grants 24111561 (to M. T.) 25860172 (to M. K.) and by The Salt Science Research Foundation.
2
The abbreviations used are:
TRP
transient receptor potential
ROS
reactive oxygen species
GPN
glycyl-l-phenylalanine-2-nephthylamide
NAC
_N_-acetylcysteine.
REFERENCES
- 1.Venkatachalam K., Montell C. (2007) TRP channels. Annu. Rev. Biochem. 76, 387–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Voets T. (2014) TRP channels and thermosensation. Handb. Exp. Pharmacol. 223, 729–741 [DOI] [PubMed] [Google Scholar]
- 3.Vriens J., Owsianik G., Hofmann T., Philipp S. E., Stab J., Chen X., Benoit M., Xue F., Janssens A., Kerselaers S., Oberwinkler J., Vennekens R., Gudermann T., Nilius B., Voets T. (2011) TRPM3 is a nociceptor channel involved in the detection of noxious heat. Neuron 70, 482–494 [DOI] [PubMed] [Google Scholar]
- 4.Vay L., Gu C., McNaughton P. A. (2012) The thermo-TRP ion channel family: properties and therapeutic implications. Br. J. Pharmacol 165, 787–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mandadi S., Sokabe T., Shibasaki K., Katanosaka K., Mizuno A., Moqrich A., Patapoutian A., Fukumi-Tominaga T., Mizumura K., Tominaga M. (2009) TRPV3 in keratinocytes transmits temperature information to sensory neurons via ATP. Pflugers Arch. 458, 1093–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang S. M., Lee H., Chung M. K., Park U., Yu Y. Y., Bradshaw H. B., Coulombe P. A., Walker J. M., Caterina M. J. (2008) Overexpressed transient receptor potential vanilloid 3 ion channels in skin keratinocytes modulate pain sensitivity via prostaglandin E2. J. Neurosci. 28, 13727–13737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Togashi K., Hara Y., Tominaga T., Higashi T., Konishi Y., Mori Y., Tominaga M. (2006) TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J. 25, 1804–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sano Y., Inamura K., Miyake A., Mochizuki S., Yokoi H., Matsushime H., Furuichi K. (2001) Immunocyte Ca2+ influx system mediated by LTRPC2. Science 293, 1327–1330 [DOI] [PubMed] [Google Scholar]
- 9.Kashio M., Sokabe T., Shintaku K., Uematsu T., Fukuta N., Kobayashi N., Mori Y., Tominaga M. (2012) Redox signal-mediated sensitization of transient receptor potential melastatin 2 (TRPM2) to temperature affects macrophage functions. Proc. Natl. Acad. Sci. U.S.A. 109, 6745–6750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faouzi M., Penner R. (2014) Trpm2. Handb. Exp. Pharmacol. 222, 403–426 [DOI] [PubMed] [Google Scholar]
- 11.Islam M. S. (2010) Calcium signaling in the islets. Adv. Exp. Med. Biol. 654, 235–259 [DOI] [PubMed] [Google Scholar]
- 12.Uchida K., Tominaga M. (2014) The role of TRPM2 in pancreatic β-cells and the development of diabetes. Cell Calcium 56, 332–339 [DOI] [PubMed] [Google Scholar]
- 13.Uchida K., Dezaki K., Damdindorj B., Inada H., Shiuchi T., Mori Y., Yada T., Minokoshi Y., Tominaga M. (2011) Lack of TRPM2 impaired insulin secretion and glucose metabolisms in mice. Diabetes 60, 119–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yosida M., Dezaki K., Uchida K., Kodera S., Lam N. V., Ito K., Rita R. S., Yamada H., Shimomura K., Ishikawa S. E., Sugawara H., Kawakami M., Tominaga M., Yada T., Kakei M. (2014) Involvement of cAMP-EPAC-TRPM2 activation in glucose- and incretin-induced insulin secretion. Diabetes 63, 3394–3403 [DOI] [PubMed] [Google Scholar]
- 15.Paulsen C. E., Carroll K. S. (2010) Orchestrating redox signaling networks through regulatory cysteine switches. ACS Chem. Biol. 5, 47–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pi J., Bai Y., Zhang Q., Wong V., Floering L. M., Daniel K., Reece J. M., Deeney J. T., Andersen M. E., Corkey B. E., Collins S. (2007) Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 56, 1783–1791 [DOI] [PubMed] [Google Scholar]
- 17.Lenzen S., Drinkgern J., Tiedge M. (1996) Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic. Biol. Med. 20, 463–466 [DOI] [PubMed] [Google Scholar]
- 18.Morgan D., Rebelato E., Abdulkader F., Graciano M. F., Oliveira-Emilio H. R., Hirata A. E., Rocha M. S., Bordin S., Curi R., Carpinelli A. R. (2009) Association of NAD(P)H oxidase with glucose-induced insulin secretion by pancreatic β-cells. Endocrinology 150, 2197–2201 [DOI] [PubMed] [Google Scholar]
- 19.Newsholme P., Haber E. P., Hirabara S. M., Rebelato E. L., Procopio J., Morgan D., Oliveira-Emilio H. C., Carpinelli A. R., Curi R. (2007) Diabetes associated cell stress and dysfunction: role of mitochondrial and non-mitochondrial ROS production and activity. J. Physiol. 583, 9–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamamoto S., Shimizu S., Kiyonaka S., Takahashi N., Wajima T., Hara Y., Negoro T., Hiroi T., Kiuchi Y., Okada T., Kaneko S., Lange I., Fleig A., Penner R., Nishi M., Takeshima H., Mori Y. (2008) TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat. Med. 14, 738–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sutton R., Peters M., McShane P., Gray D. W., Morris P. J. (1986) Isolation of rat pancreatic islets by ductal injection of collagenase. Transplantation 42, 689–691 [DOI] [PubMed] [Google Scholar]
- 22.Lange I., Yamamoto S., Partida-Sanchez S., Mori Y., Fleig A., Penner R. (2009) TRPM2 functions as a lysosomal Ca2+-release channel in β cells. Sci. Signal 2, ra23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohkuma S., Poole B. (1978) Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc. Natl. Acad. Sci. U.S.A. 75, 3327–3331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Starkus J. G., Fleig A., Penner R. (2010) The calcium-permeable non-selective cation channel TRPM2 is modulated by cellular acidification. J. Physiol. 588, 1227–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gier B., Krippeit-Drews P., Sheiko T., Aguilar-Bryan L., Bryan J., Düfer M., Drews G. (2009) Suppression of KATP channel activity protects murine pancreatic β cells against oxidative stress. J. Clin. Invest. 119, 3246–3256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Islam M. S., Berggren P. O., Larsson O. (1993) Sulfhydryl oxidation induces rapid and reversible closure of the ATP-regulated K+ channel in the pancreatic β-cell. FEBS Lett. 319, 128–132 [DOI] [PubMed] [Google Scholar]
- 27.Maechler P., Li N., Casimir M., Vetterli L., Frigerio F., Brun T. (2010) Role of mitochondria in β-cell function and dysfunction. Adv. Exp. Med. Biol. 654, 193–216 [DOI] [PubMed] [Google Scholar]
- 28.Spickett C. M., Pitt A. R., Morrice N., Kolch W. (2006) Proteomic analysis of phosphorylation, oxidation and nitrosylation in signal transduction. Biochim. Biophys. Acta 1764, 1823–1841 [DOI] [PubMed] [Google Scholar]
- 29.Escolar J. C., Hoo-Paris R., Castex C., Sutter B. C. (1990) Effect of low temperatures on glucose-induced insulin secretion and glucose metabolism in isolated pancreatic islets of the rat. J. Endocrinol. 125, 45–51 [DOI] [PubMed] [Google Scholar]
- 30.Colsoul B., Schraenen A., Lemaire K., Quintens R., Van Lommel L., Segal A., Owsianik G., Talavera K., Voets T., Margolskee R. F., Kokrashvili Z., Gilon P., Nilius B., Schuit F. C., Vennekens R. (2010) Loss of high-frequency glucose-induced Ca2+ oscillations in pancreatic islets correlates with impaired glucose tolerance in Trpm5−/− mice. Proc. Natl. Acad. Sci. U.S.A. 107, 5208–5213 [DOI] [PMC free article] [PubMed] [Google Scholar]