Use of the CRISPR/Cas9 System to Produce Genetically Engineered Pigs from In Vitro-Derived Oocytes and Embryos (original) (raw)
ABSTRACT
Targeted modification of the pig genome can be challenging. Recent applications of the CRISPR/Cas9 system hold promise for improving the efficacy of genome editing. When a designed CRISPR/Cas9 system targeting CD163 or CD1D was introduced into somatic cells, it was highly efficient in inducing mutations. When these mutated cells were used with somatic cell nuclear transfer, offspring with these modifications were created. When the CRISPR/Cas9 system was delivered into in vitro produced presumptive porcine zygotes, the system was effective in creating mutations in e_GFP, CD163,_ and CD1D (100% targeting efficiency in blastocyst stage embryos); however, it also presented some embryo toxicity. We could also induce deletions in CD163 or CD1D by introducing two types of CRISPRs with Cas9. The system could also disrupt two genes, CD163 and eGFP, simultaneously when two CRISPRs targeting two genes with Cas9 were delivered into zygotes. Direct injection of CRISPR/Cas9 targeting CD163 or CD1D into zygotes resulted in piglets that have mutations on both alleles with only one CD1D pig having a mosaic genotype. We show here that the CRISPR/Cas9 system can be used by two methods. The system can be used to modify somatic cells followed by somatic cell nuclear transfer. System components can also be used in in vitro produced zygotes to generate pigs with specific genetic modifications.
Keywords: blastocyst, CRISPR/Cas9, embryo, genetic engineering, porcine/pig, somatic cell nuclear transfer
INTRODUCTION
Pigs are both useful models for many human conditions [1, 2] as well as an important source of protein for a healthy diet in humans [3]. Historically, genetic engineering (GE) has been an inefficient process in pigs. Conventionally GE pigs were produced first by pronuclear injection [4]. Subsequent technologies involved GE somatic cells for use as nuclear donors in somatic cell nuclear transfer (SCNT). The first pigs with a targeted modification were generated through this approach [5, 6]. However, the efficiency of producing the mutation in these pigs was low.
Recent reports describing meganucleases, such as zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and components in the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated (Cas9) system suggest that GE in pigs might now be more efficient. Targeted meganucleases can induce double-strand breaks (DSBs) at specific locations in the genome and cause either random mutations through nonhomologous end joining (NHEJ) or stimulation of homologous recombination (HR) if donor DNA is provided [7–10]. Targeted modification of the genome through HR can be achieved with meganucleases if donor DNA is provided along with the targeted nuclease. After introducing specific modifications in somatic cells, these cells were used to produce GE pigs for various purposes via SCNT. Thus, meganucleases are a useful tool in generating GE pigs. Among the different meganucleases, the CRISPR/Cas9 system, adapted from prokaryotes where it is used as a defense mechanism [11], appears to be an effective approach. In nature, the Cas9 system requires three components, an RNA (∼20 bases) that contains a region that is complementary to the target sequence (cis-repressed RNA [crRNA]), an RNA that contains a region that is complementary to the crRNA (trans-activating crRNA [tracrRNA]), and Cas9, the enzymatic protein component in this complex. A single guide RNA (gRNA) can be constructed to serve the roles of the base-paired crRNA and tracrRNA. The gRNA/protein complex can scan the genome and catalyze a DSB at regions that are complementary to the crRNA/gRNA [12]. Unlike other designed nucleases, only a short oligomer needs to be designed to construct the reagents required to target a gene of interest whereas a series of cloning steps are required to assemble ZFNs and TALENs. The CRISPR/Cas9 system has been successfully used to generate GE animals in various vertebrates including zebrafish [13], monkeys [14], mice [15], rats [16], and pigs [17].
Unlike current standard methods for gene disruption, the use of designed nucleases offers the opportunity to use zygotes as starting material for GE. Standard methods for gene disruption in livestock involve HR in cultured cells and subsequent reconstruction of embryos by SCNT. Because cloned animals produced through SCNT sometimes show signs of developmental defects [18, 19], progeny of the SCNT/GE founders are typically used for research to avoid confounding SCNT anomalies and phenotype that could occur if founder animals are used for experiments. Considering the longer gestation period and higher housing costs of pigs compared to rodents, there are time and cost benefits to the reduced need for breeding. A recent report demonstrated that direct injection of ZFNs and TALENs into porcine zygotes could disrupt an endogenous gene and produce piglets with the desired mutations [20]. However, only about 10% of piglets showed biallelic modification of the target gene, and some presented mosaic genotypes. A recent article demonstrated that CRISPR/Cas9 system could induce mutations in developing embryos and produce GE pigs at a higher efficiency than ZFNs or TALENs [17]. However, GE pigs produced from the CRISPR/Cas9 system also possessed mosaic genotypes. In addition, all the above-mentioned studies used in vivo derived zygotes for the experiments, which require intensive labor and numerous sows to obtain a sufficient number of zygotes.
Here we describe an efficient approach to use the CRISPR/Cas9 system in generating GE pigs via both injection of in vitro derived zygotes and modification of somatic cells followed by SCNT. Two endogenous genes (CD163 and CD1D) and one transgene (eGFP) were targeted, and only in vitro derived oocytes or zygotes were used for SCNT or RNA injections, respectively. CD163 appears to be required for productive infection by porcine reproductive and respiratory syndrome virus [21, 22], a virus known to cause a significant economic loss to swine industry [23]. CD1D is considered a nonclassical major histocompatibility complex protein and is involved in presentation of lipid antigens to invariant natural killer T cells [24]. Pigs deficient in these genes were designed to be models for agriculture and biomedicine. The eGFP transgene was used as a target for preliminary proof-of-concept experiments and optimizations of methods.
MATERIALS AND METHODS
Chemical and Reagents
Unless otherwise stated, all of the chemicals used in this study were purchased from Sigma.
Animal and Recombinant DNA Usage
The use of animals was approved by University of Missouri Animal Care and Use Committee. The use of recombinant DNA was approved by the Institutional Biosafety Committee.
Design of gRNAs to Build Specific CRISPRs
Guide RNAs were designed to regions within exon 7 of CD163 that were unique to the wild-type (WT) CD163 and not present in the domain swap (DS)-targeting vector (described below), so that the CRISPR would result in DSBs within WT CD163 but not in the DS-targeting vector. There were only four locations in which the targeting vector would introduce a single nucleotide polymorphism that would alter an _Streptococcus pyogenes (Spy) protospacer adjacent motif (PAM) [25]. All four targets were selected, including GGAAACCCAGGCTGGTTGGAgGG (CRISPR 10), GGAACTACAGTGCGGCACTGtGG (CRISPR 134), CAGTAGCACCCCGCCCTGACgGG (CRISPR 256), and TGTAGCCACAGCAGGGACGTcGG (CRISPR 282). The PAM can be identified by the bold font in each gRNA.
For CD1D mutations, the search for CRISPR targets was arbitrarily limited to the coding strand within the first 1000 bp of the primary transcript. However, RepeatMasker [26] (pig repeat library) identified a repetitive element beginning at base 943 of the primary transcript. The search for CRISPR targets was then limited to the first 942 bp of the primary transcript. The search was further limited to the first 873 bp of the primary transcript because the last Spy PAM is located at base 873. The first target (CRISPR 4800) was selected because it overlapped with the start codon located at base 42 in the primary transcript (CCAGCCTCGCCCAGCGACATgGG). Two additional targets (CRISPR 5620, 5626) were selected because they were the most distal to the first selection within our arbitrarily selected region (CTTTCATTTATCTGAACTCAgGG and TTATCTGAACTCAGGGTCCCcGG). These targets overlap. In relation to the start codon, the most proximal Spy PAMs were located in simple sequence that contained an extensively homopolymeric sequence as determined by visual appraisal. The fourth target (CRISPR 5350) was selected because, in relation to the first target selection, it was the most proximal target that did not contain extensive homopolymeric regions (CAGCTGCAGCATATATTTAAgGG). Specificity of the designed crRNAs was confirmed by searching for similar porcine sequences in GenBank. The oligonucleotides (Supplemental Table S1; all the supplemental data are available online at www.biolreprod.org) were annealed and cloned into the p330X vector that contains two expression cassettes, a human codon-optimized S. pyogenes (h_Spy) Cas9 and the chimeric gRNA. P330X was digested with BbsI (New England Biolabs [NEB]) following the Zhang laboratory protocol (http://www.addgene.org/crispr/zhang/).
To target eGFP, two specific gRNAs targeting the eGFP coding sequence was designed within the first 60 bp of the eGFP start codon. Both _eGFP_1 and _eGFP_2 gRNA were on the antisense strand, and eGFP1 directly targeted the start codon. The _eGFP_1 gRNA sequence was CTCCTCGCCCTTGCTCACCAtGG, and the _eGFP_2 gRNA sequence was GACCAGGATGGGCACCACCCcGG.
Synthesis of Donor DNA for CD163 and CD1D Genes
Both porcine CD163 and CD1D were amplified by PCR from DNA isolated from the fetal fibroblasts that would be used for later transfections to ensure an isogenic match between the targeting vector and the transfected cell line. Briefly, LA taq (Clontech) using the forward primer CTCTCCCTCACTCTAACCTACTT, and the reverse primer TATTTCTCTCACATGGCCAGTC were used to amplify a 9538 bp fragment of CD163. The fragment was DNA sequence validated and used to build the domain-swap targeting vector (Supplemental Fig. S1). This vector included 33 point mutations within exon 7 so that it would encode the same amino acid sequence as human CD163L from exon 11. The replacement exon was 315 bp. In addition, the subsequent intron was replaced with a modified myostatin intron B that housed a selectable marker gene that could be removed with Cre-recombinase (Cre) and had previously demonstrated normal splicing when harboring the retained loxP site (Wells, unpublished results). The long arm of the construct was 3469 bp and included the DS exon. The short arm was 1578 bp and included exons 7 and 8 (Supplemental Fig. S1B). This plasmid was used to attempt to replace the coding region of exon 7 in the first transfection experiments and allowed for selection of targeting events via the selectable marker (G418). If targeting were to occur, the marker could be deleted by Cre-recombinase. The CD163 DS-targeting vector was then modified for use with cell lines that already contained a SIGLEC1 gene disrupted with Neo that could not be Cre deleted [22]. In this targeting vector, the Neo cassette, loxP and myostatin intron B, were removed, and only the DS exon remained with the WT long and short arm (Supplemental Fig. S1C).
The genomic sequence for porcine CD1D was amplified with LA taq using the forward primer CTCTCCCTCACTCTAACCTACTT and reverse primer GACTGGCCATGTGAGAGAAATA, resulting in an 8729 bp fragment. The fragment was DNA sequenced and used to build the targeting vector shown in Supplemental Figure S2. The Neo cassette is under the control of a phosphoglycerol kinase (PGK) promoter and flanked with loxP sequences, which were introduced for selection. The long arm of the construct was 4832 bp and the short arm was 3563 bp, and included exons 6 and 7. If successful HR occurred, exons 3, 4, and 5 would be removed and replaced with the Neo cassette. If NHEJ repair occurred incorrectly, then exon 3 would be disrupted.
Fetal Fibroblast Collection
Porcine fetal tissue was collected on Day 35 of gestation to create cell lines. Two WT male and female fetal fibroblast cell lines were established from a large white domestic cross. Male and female fetal fibroblasts that had previously been modified to contain a Neo cassette (SIGLEC1−/− genetics) were also used in these studies [22]. Fetal fibroblasts were collected as described with minor modifications [27]; minced tissue from each fetus was digested in 20 ml of digestion media (Dulbecco-modified Eagle medium [DMEM] containing L-glutamine and 1 g/L D-glucose [Cellgro] supplemented with 200 units/ml collagenase and 25 Kunitz units/ml DNaseI) for 5 h at 38.5°C. After digestion, fetal fibroblast cells were washed and cultured with DMEM, 15% fetal bovine serum (FBS), and 40 μg/ml gentamicin. After overnight culture, the cells were typsinized and frozen at −80°C in aliquots in FBS with 10% dimethyl sulfoxide and stored in liquid nitrogen.
Cell Transfection and Genotyping
Transfection conditions were essentially as previously reported [28]. The donor DNA was always used at a constant amount of 1 μg with varying amounts of CRISPR/Cas9 plasmid (listed below). Donor DNA was linearized with MLUI (CD163) (NEB) or AFLII (CD1D) (NEB) prior to transfection. The gender of the established cell lines was determined by PCR as described previously prior to transfection [29]. Both male and female cell lines were transfected, and genome modification data was analyzed together between the transfections. Fetal fibroblast cell lines of similar passage number (2–4) were cultured for 2 days and grown to 75%–85% confluency in DMEM containing L-glutamine and 1 g/L D-glucose (Cellgro) supplemented with 15% FBS, 2.5 ng/ml basic fibroblast growth factor, and 10 mg/ml gentamicin. Fibroblast cells were washed with phosphate-buffered saline (PBS) (Life Technologies) and trypsinized. As soon as cells detached, the cells were rinsed with an electroporation medium (75% cytosalts [120 mM KCl, 0.15 mM CaCl2, 10 mM K2HPO4, pH 7.6, 5 Mm MgCl2]) [30] and 25% Opti-MEM (Life Technologies). Cell concentration was quantified by using a hemocytometer. Cells were pelleted at 600 × g for 5 min and resuspended at a concentration of 1 × 106 in electroporation medium. Each electroporation used 200 μl of cells in 2 mm gap cuvettes with three (1 msec) square-wave pulses administered through a BTX ECM 2001 at 250 V. After the electroporation, cells were resuspended in DMEM described above. For selection, 600 μg/ml G418 (Life Technologies) was added 24 h after transfection, and the medium was changed on Day 7. Colonies were picked on Day 14 after transfection. Fetal fibroblasts were plated at 10 000 cells/plate if G418 selection was used and at 50 cells/plate if no G418 selection was used [31]. Fetal fibroblast colonies were collected by applying 10 mm autoclaved cloning cylinders sealed around each colony by autoclaved vacuum grease. Colonies were rinsed with PBS and harvested via trypsin; then resuspended in DMEM culture medium. A part (1/3) of the resuspended colony was transferred to a 96-well PCR plate, and the remaining (2/3) cells were cultured in a well of a 24-well plate. The cell pellets were resuspended in 6 μl of lysis buffer (40 mM Tris, pH 8.9, 0.9% Triton X-100, 0.4 mg/ml proteinase K [NEB]), incubated at 65°C for 30 min for cell lysis, followed by 85°C for 10 min to inactivate the proteinase K.
PCR Screening for DS and Large and Small Deletions
Detection of HR-directed repair.
Long-range PCRs were used to identify mutations on either CD163 or CD1D. Three different PCR assays were used to identify HR events: PCR amplification of regions spanning from the CD163 or CD1D sequences in the donor DNA to the endogenous CD163 or CD1D sequences on either the right or left side and a long-range PCR that amplified large regions of CD163 or CD1D encompassing the designed donor DNAs. An increase in the size of a PCR product, either 1.8 kb (CD1D) or 3.5 kb (CD163), arising from the addition of exogenous Neo sequences, was considered evidence for HR-directed repair of the genes. All the PCR conditions included an initial denaturation of 95°C for 2 min followed by 33 cycles of 30 sec at 94°C, 30 sec at 50°C, and 7–10 min at 68°C. LA taq was used for all the assays following the manufacturers' recommendations. Primers are shown in Supplemental Table S2.
Small deletions assay (NHEJ).
Small deletions were determined by PCR amplification of CD163 or CD1D flanking a projected cutting site introduced by the CRISPR/Cas9 system. The size of the amplicons was 435 bp and 1244 bp for CD163 and CD1D, respectively. Lysates from both embryos and fetal fibroblasts were PCR amplified with LA taq. PCR conditions of the assays were an initial denaturation of 95°C for 2 min followed by 33 cycles of 30 sec at 94°C, 30 sec at 56°C, and 1 min at 72°C. For genotyping of the transfected cells, insertions and deletions (INDELs) were identified by separating PCR amplicons by agarose gel electrophoresis. For embryo genotyping, the resulting PCR products were subsequently DNA sequenced to identify small deletions using forward primers used in the PCR. Primer information is shown in the Supplemental Table S3.
SCNT
To produce SCNT embryos, either sow-derived oocytes (ART, Inc.) or gilt-derived oocytes from a local slaughter house were used. The sow-derived oocytes were shipped overnight in maturation medium (TCM-199 with 2.9 mM Hepes, 5 μg/ml insulin, 10 ng/ml epidermal growth factor [EGF], 0.5 μg/ml porcine follicle-stimulating hormone [p-FSH], 0.91 mM pyruvate, 0.5 mM cysteine, 10% porcine follicular fluid, and 25 ng/ml gentamicin) and transferred into fresh medium after 24 h. After 40–42 h of maturation, cumulus cells were removed from the oocytes by vortexing in the presence of 0.1% hyaluronidase. The gilt-derived oocytes were matured as described below for in vitro fertilization (IVF). During manipulation, oocytes were placed in the manipulation medium (TCM-199 [Life Technologies] with 0.6 mM NaHCO3, 2.9 mM Hepes, 30 mM NaCl, 10 ng/ml gentamicin, and 3 mg/ml BSA, with osmolarity of 305 mOsm) supplemented with 7.0 μg/ml cytochalasin B. The polar body along with a portion of the adjacent cytoplasm, presumably containing the metaphase II plate, was removed, and a donor cell was placed in the perivitelline space by using a thin glass capillary [32]. The reconstructed embryos were then fused in a fusion medium (0.3 M mannitol, 0.1 mM CaCl2, 0.1 mM MgCl2, and 0.5 mM Hepes) with two DC pulses (1-sec interval) at 1.2 kV/cm for 30 μsec using a BTX Electro Cell Manipulator (Harvard Apparatus). After fusion, fused embryos were fully activated with 200 μM thimerosal for 10 min in the dark and 8 mM dithiothreitol for 30 min [33]. Embryos were then incubated in modified porcine zygote medium PZM3-MU1 [34, 35] with 0.5 μM Scriptaid (S7817; Sigma-Aldrich), a histone deacetylase inhibitor, for 14–16 h, as described previously [36–38].
IVF
For IVF, ovaries from prepubertal gilts were obtained from an abattoir (Farmland Foods Inc.). Immature oocytes were aspirated from medium size (3–6 mm) follicles using an 18-gauge hypodermic needle attached to a 10 ml syringe. Oocytes with evenly dark cytoplasm and intact surrounding cumulus cells were then selected for maturation. Around 50 cumulus oocyte complexes were place in a well containing 500 μl of maturation medium, TCM-199 (Invitrogen) with 3.05 mM glucose, 0.91 mM sodium pyruvate, 0.57 mM cysteine, 10 ng/ml EGF, 0.5 μg/ml luteinizing hormone (LH), 0.5 μg/ml FSH, 10 ng/ml gentamicin (APP Pharm), and 0.1% polyvinyl alcohol for 42–44 h at 38.5°C, 5% CO2, in humidified air. At the end of the maturation, the surrounding cumulus cells were removed from the oocytes by vortexing for 3 min in the presence of 0.1% hyaluronidase. Then, in vitro matured oocytes were placed in 50 μl droplets of IVF medium (modified Tris-buffered medium containing 113.1 mM NaCl, 3 mM KCl, 7.5 mM CaCl2, 11 mM glucose, 20 mM Tris, 2 mM caffeine, 5 mM sodium pyruvate, and 2 mg/ml bovine serum albumin [BSA]) in groups of 25–30 oocytes. One 100 μl frozen semen pellet was thawed in 3 ml of Dulbecco PBS supplemented with 0.1% BSA. Either frozen WT or fresh eGFP semen was washed in 60% Percoll for 20 min at 650 × g and in modified Tris-buffered medium for 10 min by centrifugation. In some cases, freshly collected semen heterozygous for a previously described eGFP transgene [39] was washed three times in PBS. The semen pellet was then resuspended with IVF medium to 0.5 × 106 cells/ml. Fifty microliter of the semen suspension was introduced into the droplets with oocytes. The gametes were coincubated for 5 h at 38.5°C in an atmosphere of 5% CO2 in air. After fertilization, the embryos were incubated in PZM3-MU1 [34, 35] at 38.5°C and 5% CO2 in air.
Embryo Transfer
Embryos generated to produce GE CD163 or CD1D pigs were transferred into surrogates either on Day 1 (SCNT) or 6 (zygote injected) after first standing estrus. For Day 6 transfer, zygotes were cultured for five additional days in PZM3-MU1 [35] in the presence of 10 ng/ml ps48 (Stemgent, Inc.). The embryos were surgically transferred into the ampullary-isthmic junction of the oviduct of the surrogate [40].
In Vitro Synthesis of RNA for CRISPR/Cas9 System
Template DNA for in vitro transcription was amplified using PCR (Supplemental Table S4). CRISPR/Cas9 plasmid used for cell transfection experiments served as the template for the PCR. In order to express the Cas9 in the zygotes, the mMESSAGE mMACHINE Ultra Kit (Ambion) was used to produce mRNA of Cas9. Then a poly A signal was added to the Cas9 mRNA using a Poly (A) tailing kit (Ambion). CRISPR guide RNAs were produced by MEGAshortscript (Ambion). The quality of the synthesized RNAs were visualized on a 1.5% agarose gel and then diluted to a final concentration of 10 ng/μl (both gRNA and Cas9) and distributed into 3 μl aliquots.
Microinjection of Designed CRISPR/Cas9 System in Zygotes
Messenger RNA coding for Cas9 and gRNA was injected into the cytoplasm of fertilized oocytes at 14 h postfertilization (presumptive zygotes) using a FemtoJet microinjector (Eppendorf). Microinjection was performed in manipulation medium on the heated stage of a Nikon inverted microscope (Nikon Corporation; Tokyo, Japan). Injected zygotes were then transferred into the PZM3-MU1 with 10 ng/ml ps48 until further use.
Statistical Analysis
The number of colonies with a modified genome was classified as 1, and the colonies without a modification of the genome were classified as 0. Differences were determined by using PROC GLM (SAS) with a _P_-value of 0.05 being considered as significant. Means were calculated as least-square means. Data are presented as numerical means ± SEM.
RESULTS
CRISPR/Cas9-Mediated Knockout of CD163 and CD1D in Somatic Cells
Efficiency of four different CRISPRs plasmids (guides 10, 131, 256, and 282) targeting CD163 was tested at an amount of 2 μg/μl of donor DNA (Table 1). CRISPR 282 resulted in significantly more average colony formation than CRISPR 10 and 256 treatments (P < 0.05). From the long-range PCR assay described above, large deletions were found ranging from 503 bp to as much as 1506 bp instead of a DS through HR as was originally intended (Fig. 1A). This was not expected because previous reports with other DNA-editing systems showed much smaller deletions of 6–333 bp using ZFN in pigs [10]. CRISPR 10 and a mix of all four CRISPRs resulted in a higher number of colonies with a modified genome than CRISPR 256 and 282 (Table 1, P < 0.002). Transfection with CRISPR 10 and a plasmid containing Neo but no homology to CD163 resulted in no colonies presenting the large deletion. Interestingly, one monoallelic deletion was also detected when the donor DNA was introduced without any CRISPR. This assay likely represents an underestimation of the mutation rate because we did not screen for any potential small deletions by sequencing which could not be detected on an agarose gel in the transfected somatic cells.
TABLE 1.
Efficiency of four different CRISPRs plasmids (guides 10, 131, 256, and 282) targeting CD163. Four different CRISPRS were tested at an amount of 2 μg to 1 μg donor DNA (shown in Supplemental Fig. S1).

FIG. 1.

Generation of CD163 and CD1D knockout pigs by CRISPR/Cas9 and SCNT. A) Targeted deletion of CD163 in somatic cells after transfection with CRISPR/Cas9 and donor DNA. A wild-type (WT) genotype results in a 6545 bp band. Lanes 1–6 represent six different colonies from a single transfection with CRISPR 10 with Cas9 and donor DNA containing Neo. Lanes 1, 4, and 5 show a large homozygous deletion of 1500–2000 bp. Lane 2 represents a smaller homozygous deletion. Lanes 3 and 6 represent either a WT allele and a small deletion or a biallelic modification of both alleles. The exact modifications of each colony were only determined by sequencing for colonies used for SCNT. The faint WT band in some of the lanes may represent cross-contamination of fetal fibroblasts from a neighboring WT colony. NTC = no template control. B) Targeted deletion of CD1D in somatic cells after transfection with CRISPR/Cas9 and donor DNA. A WT genotype results in an 8729 bp band. Lanes 1–4 represent colonies with a 500–2000 bp deletion of CD1D. Lane 4 appears to be a WT colony. NTC = no template control. C) Image of CD163 knockout pig produced by SCNT during the study. This male piglet contains a homozygous 1506 bp deletion of CD163. D) Image of CD1D pigs produced during the study. This piglet contains a 1653 bp deletion of CD1D. E) Genotype of two SCNT litters containing the 1506 bp deletion of CD163. Lanes 1–4 represent the genotype for each piglet from each litter. Sow indicates the recipient female of the SCNT embryos, and WT represents a WT control. NTC = no template control. F) Genotype of two SCNT litters containing the 1653 bp deletion of CD1D. Lanes 1–7 (litter 158) and lanes 1–4 (litter 159) represent the genotype for each piglet.
Our initial goal was to obtain a DS-targeting event by HR for CD163, but CRISPRs did not increase our efficiency of targeting CD163. It should be noted that various combinations of this targeting vector had been used to modify CD163 by HR by traditional transfections and resulted in 0 targeting events after screening 3399 colonies (Whitworth and Prather, unpublished results). We did obtain two pigs with a partial DS resulting from HR that contained 16 of the 33 mutations that we were attempting to introduce by transfection with CRISPR 10 and the DS-targeting vector as donor DNA.
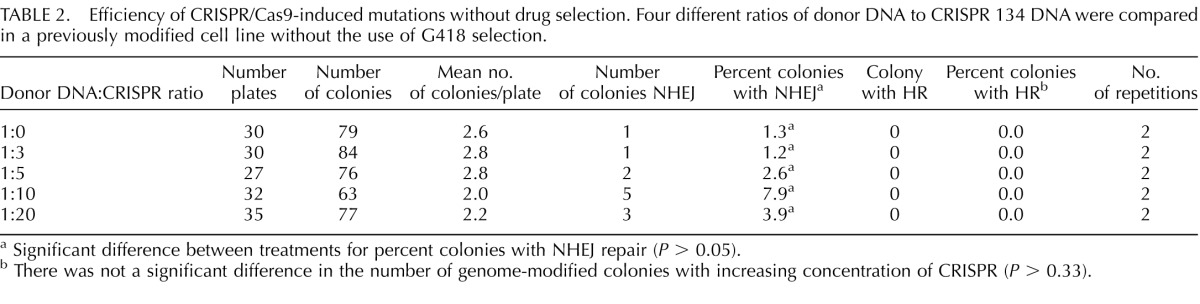
Next, the efficiency of CRISPR/Cas9-induced mutations without drug selection was tested; the fetal fibroblast cell line used in this study already had an integration of the Neo resistant cassette and a knockout of SIGLEC1 [22]. We also tested whether the ratio of CRISPR/Cas9 and donor DNA would increase genome modification or result in a toxic effect at a high concentration. CRISPR 134 was selected for this trial because in the previous experiment, it resulted in a high number of total colonies and an increased percentage of colonies possessing a modified genome. Increasing amounts of CRISPR 134 DNA from 3:1 to 20:1 did not have a significant effect on fetal fibroblast survivability. The percent of colonies with a genome modified by NHEJ was not significantly different between the various CRISPR concentrations but had the highest number of NHEJ at a 10:1 ratio (Table 2, P = 0.33). Even at the highest ratio of CRISPR DNA to donor DNA (20:1), HR was not observed.
TABLE 2.
Efficiency of CRISPR/Cas9-induced mutations without drug selection. Four different ratios of donor DNA to CRISPR 134 DNA were compared in a previously modified cell line without the use of G418 selection.
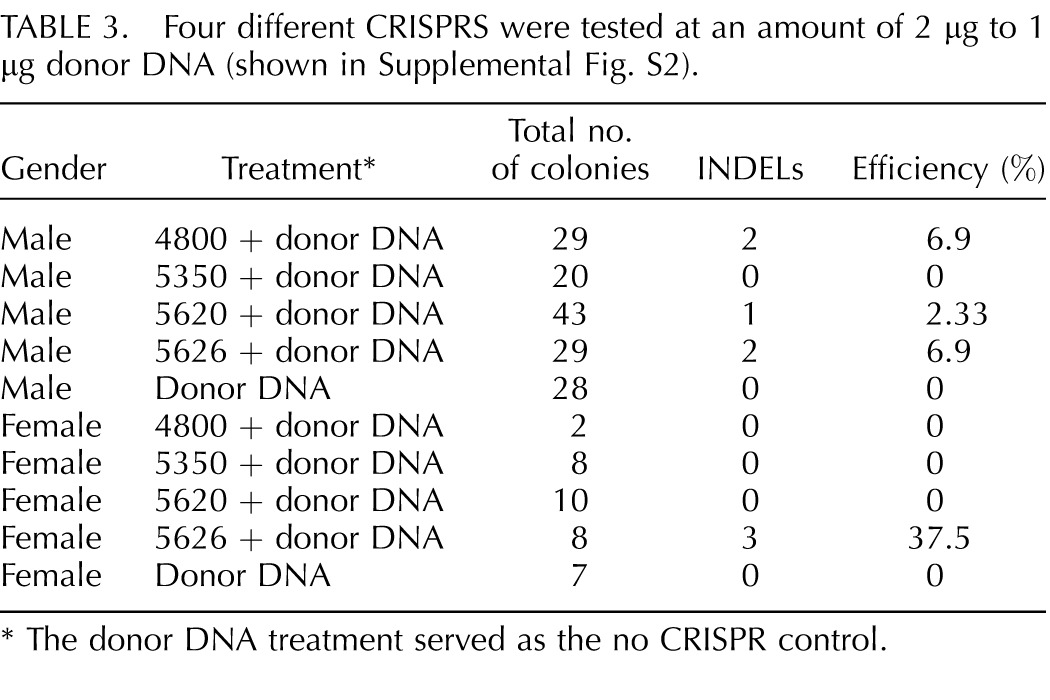
Based on this experience, targeted disruption of CD1D in somatic cell was attempted. Four different CRISPRs were designed and tested in both male and female cells. We could detect modifications of CD1D from three of the applied CRISPRs, but use of CRISPR 5350 did not result in modification of CD1D with a deletion large enough to detect by agarose gel electrophoresis (Table 3). Interestingly, we did not obtain any genetic modification through HR although donor DNA was provided. However, we did observed large deletions similar to the CD163 knockout experiments (Fig. 1B). No targeted modification of CD1D with a large deletion was detected when CRISPR/Cas9 was not used with the donor DNA. Modification of CD1D from CRISPR/Cas9-guided targeting was 4/121 and 3/28 in male and female colonies of cells, respectively. Only INDELs detectable by agarose gel electrophoresis were included in the transfection data.
TABLE 3.
Four different CRISPRS were tested at an amount of 2 μg to 1 μg donor DNA (shown in Supplemental Fig. S2).
Production of CD163 and CD1D Pigs Through SCNT Using the GE Cells
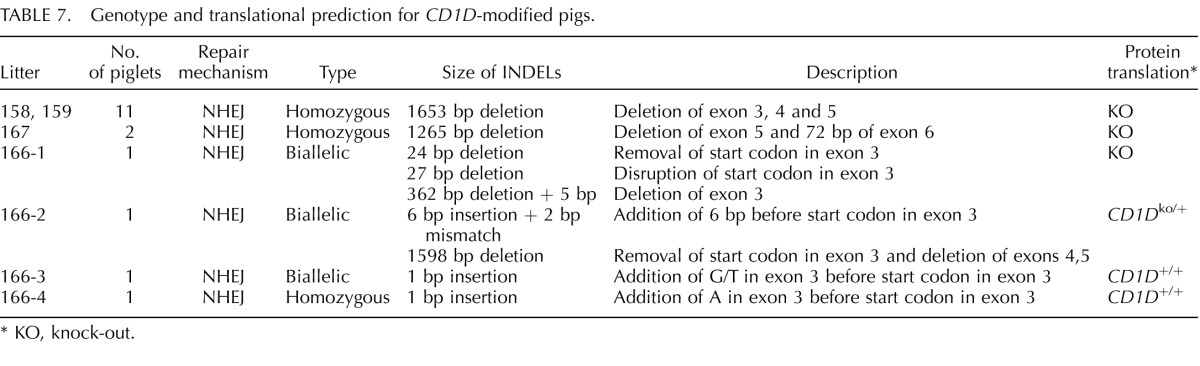
The cells presenting modification of CD163 or CD1D were used for SCNT to produce CD163 and CD1D knockout pigs (Fig. 1). Seven embryo transfers (CD163 Table 4), six embryo transfers (_CD163-_No Neo), and five embryo transfers (CD1D) into recipient gilts were performed with SCNT embryos from male and female fetal fibroblasts transfected with CRISPR/Cas9 systems. Six (CD163), two (_CD163_-No Neo), and four (CD1D) (Table 5) of the recipient gilts remained pregnant to term resulting in pregnancy rates of 85.7%. 33.3%, and 80%, respectively. Of the CD163 recipients, five delivered healthy piglets by cesarean section. One (O044) farrowed naturally. Litter size ranged from one to eight. Four pigs were euthanized because of failure to thrive after birth. One piglet was euthanized due to a severe cleft palate. All the remaining piglets appear healthy (Fig. 1C). Two litters of male piglets resulting from fetal fibroblasts transfected with CRISPR 10 and donor DNA described in Figure 1B had a 30 bp deletion in exon 7 adjacent to CRISPR 10 and an additional 1476 bp of the preceding intron, thus removing the intron 6/exon 7 junction of CD163 (Fig. 1E). The genotypes and predicted translations are summarized in Table 6. One male piglet and one female litter (4 piglets) were obtained from the _CD163_-No Neo transfection of previously modified SIGLEC1 cells. All five piglets were double knockouts for SIGLEC1 and CD163. The male piglet had a biallelic modification of CD163 with a 29 bp deletion in exon 7 and a 1382 bp deletion that included a partial deletion of exon 7 and complete deletion of exon 8 and the proceeding intron, thus removing the intron exon junction. The female piglets had a biallelic mutation of CD163, including a 1382 bp deletion with a 11 bp insertion on one allele and a 1720 bp deletion of CD163 on the other allele. A summary of the CD163 modifications and the predicted translations can be found in Table 6. A summary of the CD1D modifications and predicted translations by CRISPR modification can be found in Table 7. Briefly, one female and two male litters were born, resulting in 13 piglets. One piglet died immediately after birth. Twelve of the 13 piglets contained either a biallelic or homozygous deletion of CD1D (Fig. 1F). One piglet was WT.
TABLE 4.
Embryo Transfer data for CD163.
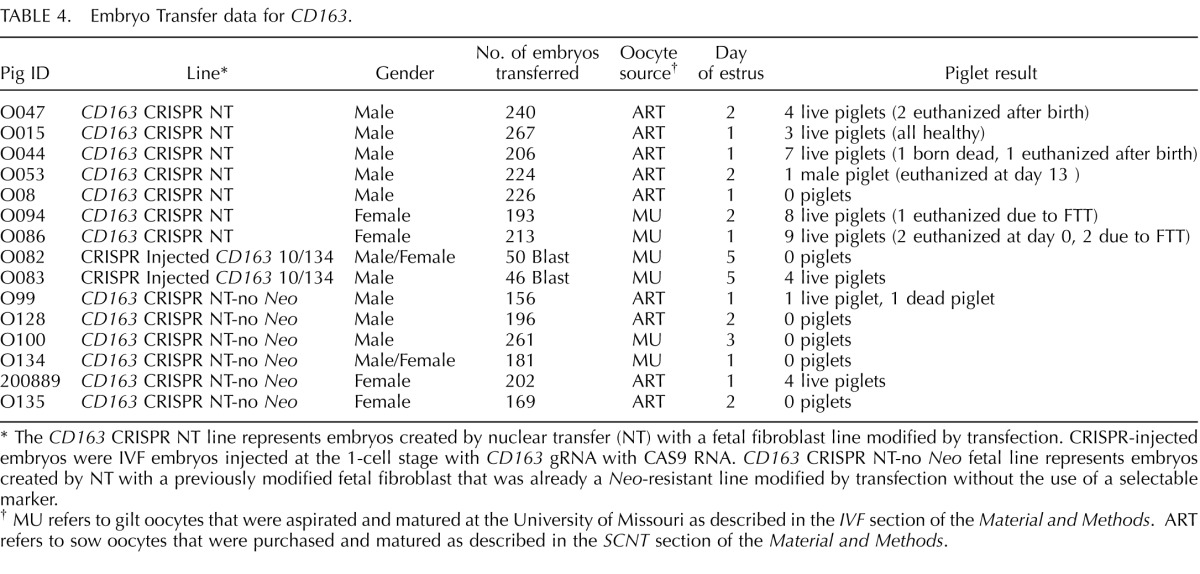
TABLE 5.
Embryo transfer data for CD1D.
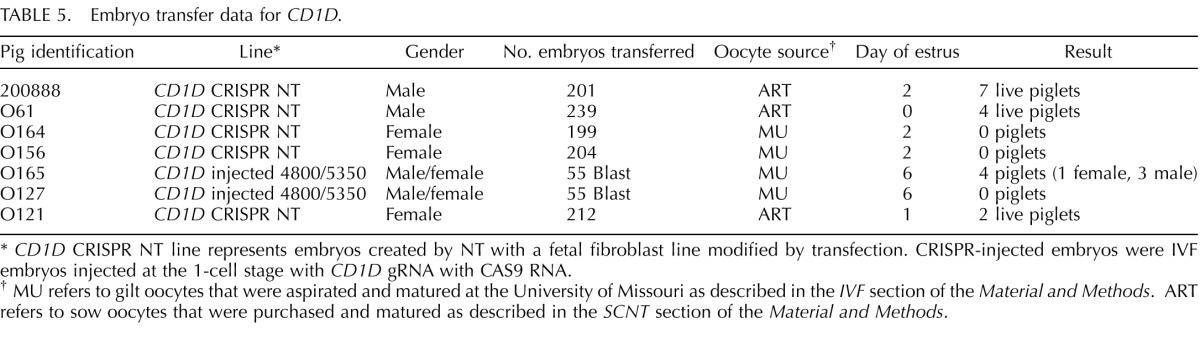
TABLE 6.
Genotype and translational prediction for _CD163_-modified pigs.
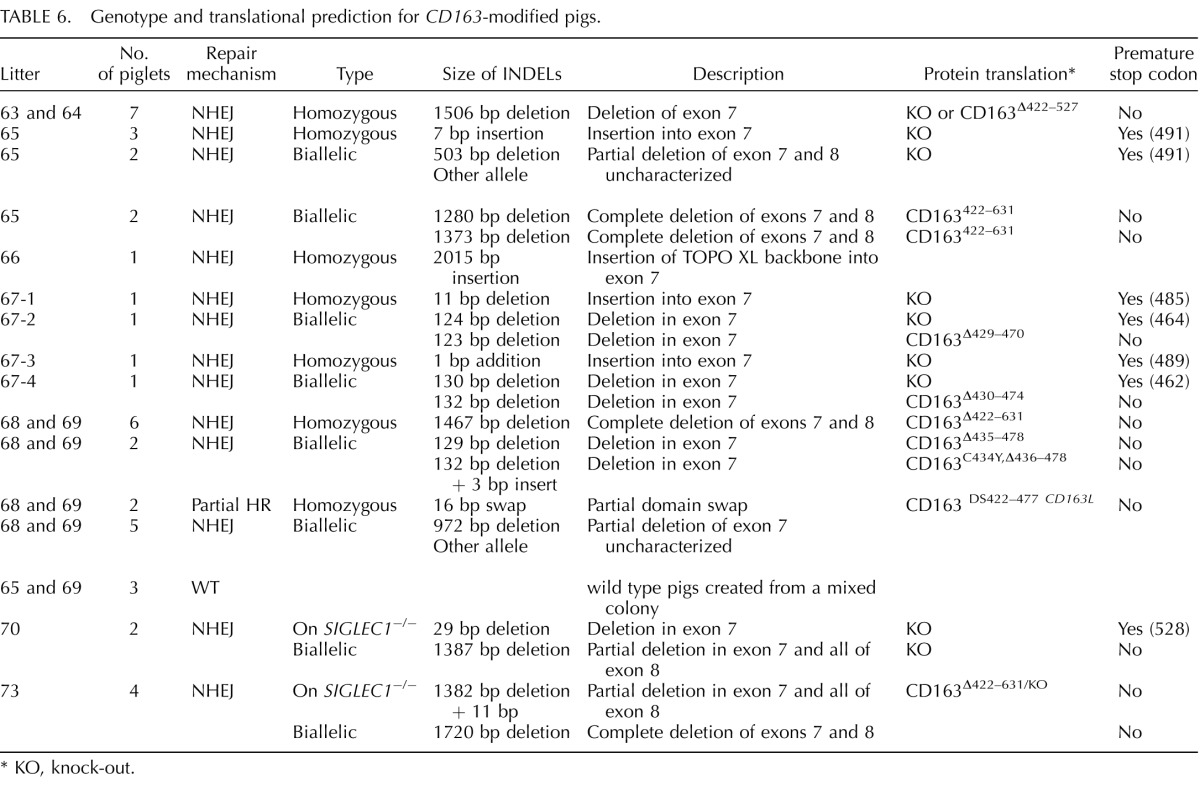
TABLE 7.
Genotype and translational prediction for _CD1D_-modified pigs.
Efficiency of CRISPR/Cas9 System in Porcine Zygotes
Based on targeted disruption of CD163 and CD1D in somatic cells using the CRISPR/Cas9 system, this approach was applied to porcine embryogenesis. First, we tested the effectiveness of the CRISPR/Cas9 system in developing embryos. CRISPR/Cas9 system targeting eGFP was introduced into zygotes fertilized with semen from a boar heterozygous for the eGFP transgene [39]. After the injection, subsequent embryos expressing eGFP were monitored. We tested various concentrations of the CRISPR/Cas9 system and observed cytotoxicity of the delivered CRISPR/Cas9 system (Fig. 2A); embryo development after CRISPR/Cas9 injection was lower compared to control. However, all the concentrations of CRISPR/Cas9 that were examined were effective in generating modification of eGFP because no embryos with eGFP expression were found in the CRISPR/Cas9-injected group (Fig. 2B); of the noninjected control embryos 67.7% were green. When individual blastocysts were genotyped, we could identify small mutations near the CRISPR binding sites (Fig. 2C). Based on the toxicity and effectiveness, 10 ng/μl of gRNA and Cas9 mRNA were used for the following experiments.
FIG. 2.

Effect of CRISPR/Cas9 system in porcine embryos. A) Frequency of blastocyst formation after injection of different concentrations of CRISPR/Cas9 system into zygotes. Toxicity of the CRISPR/Cas9 system was lowest at 10 ng/μl. B) CRISPR/Cas9 system can successfully disrupt expression of eGFP in blastocysts when introduced into zygotes. Original magnification ×4. C) Types of mutations on eGFP generated by the CRISPR/Cas9 system.
When CRISPR/Cas9 components designed to target CD163 were introduced into presumptive zygotes, we observed targeted modification of the genes in the subsequent blastocysts. When individual blastocysts were genotyped for mutation of CD163, specific mutations were found in all the embryos (100% GE efficiency). More importantly, while we could find embryos with homozygous or biallelic modifications (8/18 and 3/18, respectively) (Fig. 3), we also detected mosaic (monoallelic modifications) genotypes (4/18 embryos). Some embryos (8/10) from the pool were injected with 2 ng/μl Cas9 and 10 ng/μl CRISPR and no difference was found in the efficiency of mutagenesis. Next, based on the in vitro results, we introduced two CRISPRs representing different gRNA to disrupt CD163 or CD1D during embryogenesis to induce a specific deletion of the target genes. As a result, we could successfully induce a designed deletion of CD163 and CD1D by introducing two guides. A designed deletion is defined as a deletion that removes the genomic sequence between the two guides introduced. Among the embryos that received two CRISPRs targeting CD163, all but one embryo resulted in a targeted modification of CD163. In addition, we found 5/13 embryos having a designed deletion on CD163 (Fig. 4A) and 10/13 embryos appear to have modification of CD163 in either homozygous or biallelic fashion. Targeting CD1D with two CRISPRs was also effective because all the embryos (23/23) showed a modification of CD1D. However, we could only find the designed deletion of CD1D in two embryos (2/23) (Fig. 4B). We also found 5/23 embryos possessing mosaic genotypes, but the rest of embryos had either homozygous or biallelic modification of CD1D. Finally, we tested whether multiple genes can be targeted by the CRISPR/Cas9 system within the same embryo. For this purpose, targeting both CD163 and eGFP was performed in the zygotes that were fertilized with heterozygous eGFP semen. When blastocysts from the injected embryos were genotyped for CD163 and eGFP, we found that CD163 and eGFP were successfully targeted during embryogenesis. Sequencing results demonstrated that multiple genes can be targeted by introducing multiple CRISPRs with Cas9 (Fig. 4C).
FIG. 3.

Effect of CRISPR/Cas9 system in targeting CD163 in porcine embryos. A) Examples of mutations generated on CD163 by the CRISPR/Cas9 system. All the embryos examined by DNA sequencing showed mutation on the CD163 (18/18). CRISPR 134 is highlighted in bold. B) Sequencing read of a homozygous deletion caused by the CRISPR/Cas9 system. The image represents no. 1–4 carrying 2 bp deletion of CD163.
FIG. 4.

Effect of CRISPR/Cas9 system when introduced with two types of CRISPRs. A) PCR amplification of CD163 in blastocysts injected with CRISPR/Cas9 as zygotes. Lanes 1, 3, 6, and 12 show the designed deletion between two different CRISPRs. B) PCR amplification of CD1D in blastocysts injected with CRISPR/Cas9 as zygotes. CD1D had a lower frequency of deletion as determined by gel electrophoresis when compared to CD163 (3/23); lanes 1, 8, and 15 show obvious deletions in CD1D. C) CRISPR/Cas9 system could successfully target two genes when the system is provided with two CRISPRs targeting CD163 and eGFP. The modifications of CD163 and eGFP are shown.
Production of CD163 and CD1D Mutants from CRISPR/Cas9-Injected Zygotes
Based on the success from our previous in vitro study, we produced some CRISPR/Cas9-injected zygotes and transferred 46–55 blastocysts per recipient (because this number has been shown to be effective in producing pigs from our in vitro derived embryos) [40]. We performed four embryo transfers, two each for CD163 and CD1D, and obtained a pregnancy for each modification. We produced four healthy piglets carrying modifications on CD163 (Table 4). All the piglets, litter 67 from recipient sow ID O083 showed either homozygous or biallelic modification of CD163 (Fig. 5). Two piglets showed the designed deletion of CD163 by the two CRISPRs delivered. All the piglets were healthy. For CD1D, one pregnancy also produced four piglets (litter 166 from recipient sow identification no. O165): one female and three males (Table 5). One piglet (166-1) did carry a mosaic mutation of CD1D, including a 362 bp deletion that completely removed exon 3 that contains the start codon (Fig. 6). One piglet contained a 6 bp insertion with a 2 bp mismatch on one allele with a large deletion on the other allele. Two additional piglets had a biallelic single bp insertion. There were no mosaic mutations detected for CD163.
FIG. 5.

CD163 knockout pigs generated by CRISPR/Cas9 system injected into zygotes. A) PCR amplification of CD163 from the knockout pigs; a clear sign of deletion was detected in 67-2 and 67-4. B) Image of CD163 knockout pigs with a surrogate. All the animals are healthy and show no signs of abnormalities. C) Genotype of CD163 knockout pigs. Two animals (67-1 and 67-3) are carrying a homozygous deletion or insertion in CD163. The other two animals (67-2 and 67-4) are carrying a biallelic modification of CD163. The deletion was cause by introducing two different CRISPRs with Cas9 system. No animals from the zygote injection for CD163 showed a mosaic genotype.
FIG. 6.

CD1D knockout pigs generated by CRISPR/Cas9 system injected into zygotes. A) PCR amplification of CD1D from knockout pigs; 166-1 shows a mosaic genotype for CD1D. 166-2, 166-3, and 166-4 do not show a change in size for the amplicon, but sequencing of the amplicon revealed modifications. B) PCR amplification of the long-range assay showed a clear deletion of one allele in piglets 166-1 and 166-2. C) Image of CD1D knockout pigs with surrogate. D) Sequence data of CD1D knock out pigs. The atg start codon in exon 3 is in bold and also lower case.
DISCUSSION
An increase in efficiency of GE pig production can have a wide impact by providing more GE pigs for agriculture and biomedicine. Our study shows that by using the CRISPR/Cas9 system, GE pigs with specific mutations can be produced at a high efficiency. We successfully applied the CRISPR/Cas9 system to modify genes in both somatic cells and in preimplantation embryos.
When the CRISPR/Cas9 system was introduced into somatic cells, it successfully induced targeted disruption of our target genes by NHEJ but did not increase our ability to target by HR. Targeting efficiency of individual CRISPR/Cas9 in somatic cells was variable, which indicated that the design of the guide can affect the targeting efficiency. Specifically, we could not find any targeted modification of CD1D when CRISPR 5350 and Cas9 were introduced into somatic cells. This suggests that it could be beneficial to design multiple gRNAs and validate their efficiencies prior to producing pigs. A reason for the lack of HR-directed repair with the presence of donor DNA is still unclear. After screening 886 colonies (both CD163 and CD1D) transfected with CRISPR and donor DNA, only one colony had evidence for a partial HR event. Our results demonstrated that the CRISPR/Cas9 system worked with introduced donor DNA to cause unexpected large deletions on the target genes but did not increase HR efficiency for these two particular targeting vectors. However, a specific mechanism for the large deletion observation is not known. Previous reports from our group suggested that a donor DNA can be effectively used with a ZFN to induce HR-directed repair [41, 42]. Similarly, we did see an increase in the targeting efficiency when donor DNA was used with CRISPR/Cas9 system, but complete HR directed repair was not observed. In our previous study using ZFN, we observed that targeted modification can occur through a combination of HR and NHEJ because we found a partial recombination of the introduced donor DNA after induced DSBs by the ZFN. One explanation might be that HR and NHEJ pathways are not independent but can act together to complete the repair process after DSBs induced by meganucleases.
Higher concentrations of CRISPRs might improve targeting efficiency in somatic cells although no statistical difference was found in these experimental results. This may suggest that CRISPR is a limiting factor in CRISPR/Cas9 system, but further validation is needed. Targeted cells were successfully used to produce GE pigs through SCNT, indicating the application of CRISPR/Cas9 does not affect the ability of the cells to be cloned. A few piglets were euthanized because of health issues; however, this is not uncommon in SCNT-derived piglets.
When the CRISPR/Cas9 system was introduced into developing embryos by zygote injection, nearly 100% of embryos and pigs contained an INDEL in the targeted gene, demonstrating that the technology is very effective during embryogenesis. The efficiency we observed during our study surpasses frequencies reported in other studies utilizing meganucleases during embryogenesis [17, 20]. A decrease in the number of embryos reaching the blastocyst stage suggested that the concentration of CRISPR/Cas9 we introduced in this study may be toxic to embryos. Further optimization of the delivery system may increase survivability of embryos and thus improve the overall efficiency of the process. The nearly 100% mutagenesis rate observed here was different from a previous report in CRISPR/Cas9-mediated knockout in pigs [17]; however, the difference in efficiency between the studies could be a combination of the guide and target that was selected. In our study, lower concentrations of CRISPR/Cas9 (10 ng/μl each) was effective in generating mutations in developing embryos and producing GE pigs. The concentration is lower than previously reported in pig zygotes (125 ng/μl of Cas9 and 12.5 ng/μl of CRISPR) [17]. The lower concentration of CRISPR/Cas9 components could be beneficial to developing embryos because introducing excess amounts of nucleic acid into developing embryos can be toxic [43]. We did see some mosaic genotypes in CRISPR/Cas9-injected embryos from our in vitro assays; however, only one piglet produced through the approach had a mosaic genotype. Potentially, an injection with CRISPR/Cas9 components may be more effective than introduction of other meganucleases [17, 20] because the mosaic genotype was considered to be a main hurdle of using the CRISPR/Cas9 system in zygotes. Another benefit of using the CRISPR/Cas9 system demonstrated by our results is that we did not lose any CD163 knockout pigs produced from IVF-derived zygotes injected with CRISPR/Cas9 system whereas a few piglets resulting from SCNT were euthanized after a few days. This suggests that the technology could not only bypass the need of SCNT in generating knockout pigs but could also overcome the common health issues associated with SCNT. Now that injection of CRISPR/Cas9 mRNA into zygotes has been optimized, future experiments will include coinjection of donor DNA as well.
We have demonstrated that introducing two CRISPRs with Cas9 in zygotes can induce chromosomal deletions in developing embryos and produce pigs with an intended deletion, that is, specific deletion between the two CRISPR guides. This designed deletion can be beneficial because we can specify the size of the deletion rather than relying on random events caused by NHEJ. Specifically, if there is insertion/deletion of nucleotides in a multiple of three caused by a meganuclease, the mutation may rather result in a hypomorphic mutation because no frame shift would occur. However, by introducing two CRISPRs, we can cause larger deletions that will have a higher chance of generating nonfunctional protein. Interestingly, CD1D CRISPRs were designed across a greater area in the genome than CD163; there was a 124 bp distance between CD163 CRISPR 10 and 134 while there was a distance of 550 bp between CRISPR 4800 and 5350 for CD1D. The longer distance between CRISPRs was not very effective in generating a deletion as shown in the study. However, because we have only limited number of observations and need to consider the efficacy of individual CRISPRs, which we have not address here, further study is need to verify the relationship between the distance between CRISPRs and probability of causing intended deletions.
The CRISPR/Cas9 system was also effective in targeting two genes simultaneously within the same embryo with the only extra step being the introduction of one additional CRISPR with crRNA. This illustrates the ease of disrupting multiples genes compared to other meganucleases. These results suggest that this technology may be used to target gene clusters or gene families that may have a compensatory effect, thus proving difficult to determine the role of individual genes unless all the genes are disrupted. Our results demonstrate that CRISPR/Cas9 technology can be applied in generating GE pigs by increasing the efficiency of gene targeting in somatic cells and by direct zygote injection.
ACKNOWLEDGMENT
The authors would like to thank those who assisted with piglet care related to this project, including: Keith Giroux, Tricia Meyer, Jason Dowell, Lonnie Dowell, Mykel Anderson, Madison Hennessey, Katie White, Dahrea Hays, Rachel Bardot, Mykel Anderson, Brittany Elliot, and Sabrina Hammond. We would also like to thank Mariah Thomas for laboratory assistance for this project.
Footnotes
4
Current Address: Department of Animal and Poultry Sciences, Virginia Tech, Blacksburg, Virginia.
1
Supported by the National Swine Resource and Research Center (NSRRC) via funding from the National Institutes of Health for targeted disruption of CD1D (OD011140 to R.S.P., K.D.W., and E.M.W.), Genus, plc, (to R.S.P. and K.D.W.) and the Christopher Columbus Fellowship Foundation (R.S.P.) for targeted disruption of CD163, and Food for the 21st Century at the University of Missouri (K.D.W. and R.S.P.).
3
These authors contributed equally to the work.
REFERENCES
- Whyte JJ, Prather RS. Genetic modifications of pigs for medicine and agriculture. Mol Reprod Dev. 2011;78:879–891. doi: 10.1002/mrd.21333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters EM, Prather RS. Advancing swine models for human health and diseases. Mo Med. 2013;110:212–215. [PMC free article] [PubMed] [Google Scholar]
- U.S. Department of Agriculture and U.S. Department of Health and Human Services Dietary Guidelines for Americans, 2010 7th Edition, Washington, DC: U.S. Government Printing Office; December 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer RE, Pursel VG, Rexroad CE, Jr, , Wall RJ, Bolt DJ, Ebert KM, Palmiter RD, Brinster RL. Production of transgenic rabbits, sheep and pigs by microinjection. Nature. 1985;315:680–683. doi: 10.1038/315680a0. [DOI] [PubMed] [Google Scholar]
- Lai L, Kolber-Simonds D, Park KW, Cheong HT, Greenstein JL, Im GS, Samuel M, Bonk A, Rieke A, Day BN, Murphy CN, Carter DB, et al. Production of alpha-1,3-galactosyltransferase knockout pigs by nuclear transfer cloning. Science. 2002;295:1089–1092. doi: 10.1126/science.1068228. [DOI] [PubMed] [Google Scholar]
- Dai Y, Vaught TD, Boone J, Chen SH, Phelps CJ, Ball S, Monahan JA, Jobst PM, McCreath KJ, Lamborn AE, Cowell-Lucero JL, Wells KD, et al. Targeted disruption of the alpha1,3-galactosyltransferase gene in cloned pigs. Nat Biotechnol. 2002;20:251–255. doi: 10.1038/nbt0302-251. [DOI] [PubMed] [Google Scholar]
- Gaj T, Gersbach CA, Barbas CF., III. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauschild J, Petersen B, Santiago Y, Queisser AL, Carnwath JW, Lucas-Hahn A, Zhang L, Meng X, Gregory PD, Schwinzer R, Cost GJ, Niemann H. Efficient generation of a biallelic knockout in pigs using zinc-finger nucleases. Proc Natl Acad Sci U S A. 2011;108:12013–12017. doi: 10.1073/pnas.1106422108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Yang H, Li W, Zhao B, Ouyang Z, Liu Z, Zhao Y, Fan N, Song J, Tian J, Li F, Zhang J et al. Generation of PPARgamma mono-allelic knockout pigs via zinc-finger nucleases and nuclear transfer cloning. Cell Res. 2011;21:979–982. doi: 10.1038/cr.2011.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte JJ, Zhao J, Wells KD, Samuel MS, Whitworth KM, Walters EM, Laughlin MH, Prather RS. Gene targeting with zinc finger nucleases to produce cloned eGFP knockout pigs. Mol Reprod Dev. 2011;78:2. doi: 10.1002/mrd.21271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331–338. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- Terns MP, Terns RM. CRISPR-based adaptive immune systems. Curr Opin Microbiol. 2011;14:321–327. doi: 10.1016/j.mib.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh JR, Joung JK. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol. 2013;31:227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu Y, Shen B, Cui Y, Chen Y, Wang J, Wang L, Kang Y, Zhao X, Si W, Li W, Xiang AP, Zhou J, et al. Generation of gene-modified Cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell. 2014;156:836–843. doi: 10.1016/j.cell.2014.01.027. [DOI] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Qiu Z, Shao Y, Chen Y, Guan Y, Liu M, Li Y, Gao N, Wang L, Lu X, Zhao Y, Liu M. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat Biotechnol. 2013;31:681–683. doi: 10.1038/nbt.2661. [DOI] [PubMed] [Google Scholar]
- Hai T, Teng F, Guo R, Li W, Zhou Q. One-step generation of knockout pigs by zygote injection of CRISPR/Cas system. Cell Res. 2014;24:372–375. doi: 10.1038/cr.2014.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter DB, Lai L, Park KW, Samuel M, Lattimer JC, Jordan KR, Estes DM, Besch-Williford C, Prather RS. Phenotyping of transgenic cloned piglets. Cloning Stem Cells. 2002;4:131–145. doi: 10.1089/153623002320253319. [DOI] [PubMed] [Google Scholar]
- Shimozawa N, Ono Y, Kimoto S, Hioki K, Araki Y, Shinkai Y, Kono T, Ito M. Abnormalities in cloned mice are not transmitted to the progeny. Genesis. 2002;34:203–207. doi: 10.1002/gene.10143. [DOI] [PubMed] [Google Scholar]
- Lillico SG, Proudfoot C, Carlson DF, Stverakova D, Neil C, Blain C, King TJ, Ritchie WA, Tan W, Mileham AJ, McLaren DG, Fahrenkrug SC et al. Live pigs produced from genome edited zygotes. Sci Rep. 2013;3:2847. doi: 10.1038/srep02847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Breedam W, Delputte PL, Van Gorp H, Misinzo G, Vanderheijden N, Duan X, Nauwynck HJ. Porcine reproductive and respiratory syndrome virus entry into the porcine macrophage. J Gen Virol. 2010;91:1659–1667. doi: 10.1099/vir.0.020503-0. [DOI] [PubMed] [Google Scholar]
- Prather RS, Rowland RR, Ewen C, Trible B, Kerrigan M, Bawa B, Teson JM, Mao J, Lee K, Samuel MS, Whitworth KM, Murphy CN et al. An intact sialoadhesin (Sn/SIGLEC1/CD169) is not required for attachment/internalization of the porcine reproductive and respiratory syndrome virus. J Virol. 2013;87:9538–9546. doi: 10.1128/JVI.00177-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann EJ, Kliebenstein JB, Johnson CD, Mabry JW, Bush EJ, Seitzinger AH, Green AL, Zimmerman JJ. Assessment of the economic impact of porcine reproductive and respiratory syndrome on swine production in the United States. J Am Vet Med Assoc. 2005;227:385–392. doi: 10.2460/javma.2005.227.385. [DOI] [PubMed] [Google Scholar]
- Borg NA, Wun KS, Kjer-Nielsen L, Wilce MC, Pellicci DG, Koh R, Besra GS, Bharadwaj M, Godfrey DI, McCluskey J, Rossjohn J. CD1d-lipid-antigen recognition by the semi-invariant NKT T-cell receptor. Nature. 2007;448:44–49. doi: 10.1038/nature05907. [DOI] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. RepeatMasker Open-3.0. 1996–2010 Current Version: open-4.0.5 (RMLib: 20140131 and Dfam: 1.2).http://www.repeatmasker.org>. CD163: accessed July 25, 2014; CD1D: accessed August 27, 2013. [Google Scholar]
- Lai L, Prather RS. Creating genetically modified pigs by using nuclear transfer. Reprod Biol Endocrinol. 2003;1:82. doi: 10.1186/1477-7827-1-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross JW, Whyte JJ, Zhao J, Samuel M, Wells KD, Prather RS. Optimization of square-wave electroporation for transfection of porcine fetal fibroblasts. Transgenic Res. 2010;19:611–620. doi: 10.1007/s11248-009-9345-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitworth KM, Spate LD, Li R, Rieke A, Sutovsky P, Green JA, Prather RS. Activation method does not alter abnormal placental gene expression and development in cloned pigs. Mol Reprod Dev. 2010;77:1016–1030. doi: 10.1002/mrd.21235. [DOI] [PubMed] [Google Scholar]
- van den Hoff MJ, Moorman AF, Lamers WH. Electroporation in ‘intracellular' buffer increases cell survival. Nucleic Acids Res. 1992;20:2902. doi: 10.1093/nar/20.11.2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaton BP, Wells Kevin D. Compound transgenics: recombinase-mediated gene stacking In: Pinkert CA. (ed.). Transgenic Animal Technology: A Laboratory Handbook, 3rd ed. Waltham, MA: Elsevier; 2014. 565 578 [Google Scholar]
- Lai L, Prather RS. Production of cloned pigs by using somatic cells as donors. Cloning Stem Cells. 2003;5:233–241. doi: 10.1089/153623003772032754. [DOI] [PubMed] [Google Scholar]
- Machaty Z, Wang WH, Day BN, Prather RS. Complete activation of porcine oocytes induced by the sulfhydryl reagent, thimerosal. Biol Reprod. 1997;57:1123–1127. doi: 10.1095/biolreprod57.5.1123. [DOI] [PubMed] [Google Scholar]
- Yoshioka K, Suzuki C, Tanaka A, Anas IM, Iwamura S. Birth of piglets derived from porcine zygotes cultured in a chemically defined medium. Biol Reprod. 2002;66:112–119. doi: 10.1095/biolreprod66.1.112. [DOI] [PubMed] [Google Scholar]
- Bauer BK, Spate LD, Murphy CN, Prather RS. Arginine supplementation in vitro increases porcine embryo development and affects mRNA transcript expression. Reprod Fertil Dev. 2011;23:107. [Google Scholar]
- Zhao J, Hao Y, Ross JW, Spate LD, Walters EM, Samuel MS, Rieke A, Murphy CN, Prather RS. Histone deacetylase inhibitors improve in vitro and in vivo developmental competence of somatic cell nuclear transfer porcine embryos. Cell Reprogram. 2010;12:75–83. doi: 10.1089/cell.2009.0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Ross JW, Hao Y, Spate LD, Walters EM, Samuel MS, Rieke A, Murphy CN, Prather RS. Significant improvement in cloning efficiency of an inbred miniature pig by histone deacetylase inhibitor treatment after somatic cell nuclear transfer. Biol Reprod. 2009;81:525–530. doi: 10.1095/biolreprod.109.077016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitworth KM, Zhao J, Spate LD, Li R, Prather RS. Scriptaid corrects gene expression of a few aberrantly reprogrammed transcripts in nuclear transfer pig blastocyst stage embryos. Cell Reprogram. 2011;13:191–204. doi: 10.1089/cell.2010.0087. [DOI] [PubMed] [Google Scholar]
- Whitworth KM, Li R, Spate LD, Wax DM, Rieke A, Whyte JJ, Manandhar G, Sutovsky M, Green JA, Sutovsky P, Prather RS. Method of oocyte activation affects cloning efficiency in pigs. Mol Reprod Dev. 2009;76:490–500. doi: 10.1002/mrd.20987. [DOI] [PubMed] [Google Scholar]
- Lee K, Redel BK, Spate L, Teson J, Brown AN, Park KW, Walters E, Samuel M, Murphy CN, Prather RS. Piglets produced from cloned blastocysts cultured in vitro with GM-CSF. Mol Reprod Dev. 2013;80:145–154. doi: 10.1002/mrd.22143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon DN, Lee K, Kang MJ, Choi YJ, Park C, Whyte JJ, Brown AN, Kim JH, Samuel M, Mao J, Park KW, Murphy CN et al. Production of biallelic CMP-Neu5Ac hydroxylase knock-out pigs. Sci Rep. 2013;3:1981. doi: 10.1038/srep01981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Kwon DN, Ezashi T, Choi YJ, Park C, Ericsson AC, Brown AN, Samuel MS, Park KW, Walters EM, Kim DY, Kim JH et al. Engraftment of human iPS cells and allogeneic porcine cells into pigs with inactivated RAG2 and accompanying severe combined immunodeficiency. Proc Natl Acad Sci U S A. 2014;111:7260–7265. doi: 10.1073/pnas.1406376111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinster RL, Chen HY, Trumbauer ME, Yagle MK, Palmiter RD. Factors affecting the efficiency of introducing foreign DNA into mice by microinjecting eggs. Proc Natl Acad Sci U S A. 1985;82:4438–4442. doi: 10.1073/pnas.82.13.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]