Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation (original) (raw)
. Author manuscript; available in PMC: 2015 Jun 8.
Published in final edited form as: Nature. 2000 Nov 2;408(6808):106–111. doi: 10.1038/35040593
Abstract
Members of the myocyte enhancer factor-2 (MEF2) family of transcription factors associate with myogenic basic helix–loop– helix transcription factors such as MyoD to activate skeletal myogenesis1. MEF2 proteins also interact with the class II histone deacetylases HDAC4 and HDAC5, resulting in repression of MEF2-dependent genes2–4. Execution of the muscle differentiation program requires release of MEF2 from repression by HDACs, which are expressed constitutively in myoblasts and myotubes5. Here we show that HDAC5 shuttles from the nucleus to the cytoplasm when myoblasts are triggered to differentiate. Calcium/ calmodulin-dependent protein kinase (CaMK) signalling, which stimulates myogenesis5 and prevents formation of MEF2–HDAC complexes4, also induces nuclear export ofHDAC4 and HDAC5 by phosphorylation of these transcriptional repressors. An HDAC5 mutant lacking two CaMK phosphorylation sites is resistant to CaMK-mediated nuclear export and acts as a dominant inhibitor of skeletal myogenesis, whereas a cytoplasmic HDAC5 mutant is unable to block efficiently the muscle differentiation program. Our results highlight a mechanism for transcriptional regulation through signal- and differentiation-dependent nuclear export of a chromatin-remodelling enzyme, and suggest that nucleo cytoplasmic trafficking of HDACs is involved in the control of cellular differentiation.
Histone acetylation/deacetylation has emerged as a fundamental mechanism for the control of gene expression6. Histone acetyltransferases (HATs) stimulate transcription through acetylation of histones, resulting in relaxation of nucleosomes; and HDACs antagonize this activity and repress transcription6. HDAC4 and HDAC5 block myogenesis by associating with and inhibiting the activity of the MEF2 transcription factor, and CaMK signalling overcomes this inhibition by dissociating MEF2–HDAC complexes, with consequent stimulation of myogenesis4,5.
To investigate further the mechanism for suppression of myogenesis by HDAC4 and HDAC5, and the blockade to this suppression by CaMK signalling, we raised antibodies against HDAC5 and examined the subcellular distribution of the endogenous protein during differentiation of the C2 skeletal muscle cell line. In proliferating undifferentiated myoblasts, HDAC5 was predominantly nuclear (Fig. 1a), whereas after initiation of differentiation by removal of serum from the medium, HDAC5 became progressively localized in the cytoplasm, although residual nuclear staining was evident (Fig. 1b–d). In contrast, MEF2 was exclusively nuclear in myotubes (Fig. 1e), consistent with the essential role for this transcription factor in skeletal muscle gene expression. In residual unfused myoblasts that failed to differentiate, HDAC5 remained in the nucleus, suggesting that cytoplasmic accumulation of HDAC5 is coupled to activation of the muscle differentiation program.
Figure 1.
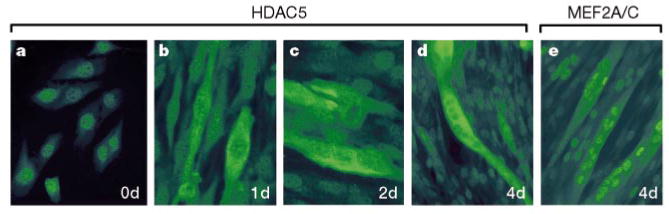
Shuttling of HDAC5 from the nucleus to the cytoplasm during myogenic differentiation. Undifferentiated C2 myoblasts maintained in growth medium (a), and cells exposed to differentiation medium for one (b), two (c) or four (d) days were stained with antibodies directed against the N terminus of HDAC5 and a fluorescein-conjugated secondary antibody. We note that HDAC5 staining remains mostly nuclear in myoblasts that fail to differentiate into myotubes, whereas it is mostly cytoplasmic in adjacent myotubes. Myotubes were also stained with antiserum that recognizes both MEF2A and MEF2C (e). Images in b and c were magnified about 1.5 times relative to those in a, d and e to enable better visualization of elongated cells in the early stages of differentiation.
As activated CaMK can overcome HDAC-mediated repression of myogenesis5, we investigated whether CaMK signalling also alters the subcellular distribution of HDAC5. In the absence of activated CaMK, epitope-tagged derivatives of MEF2C and HDAC5 were coexpressed exclusively in the nuclei of Cos cells (Fig. 2a). In contrast, HDAC5 was cytoplasmic in cells expressing a constitutively active form of CaMKI. MEF2C remained nuclear in CaMKI-expressing cells, consistent with the finding that CaMK signalling disrupts MEF2–HDAC complexes and stimulates MEF2-dependent transcription4.
Figure 2.
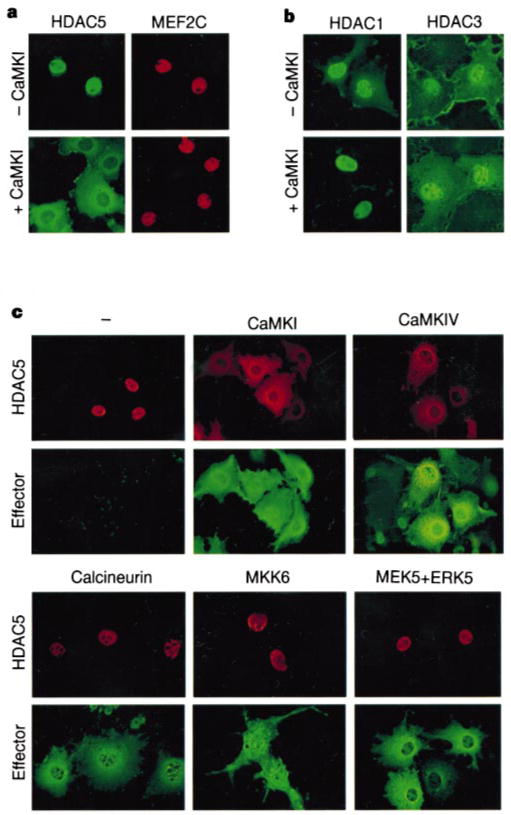
HDAC5 is excluded from the nucleus in cells expressing activated forms of CaMK. a, b, Cos cells were co-transfected with expression vectors encoding Flag-tagged HDAC5 and Myc-tagged MEF2C (a) or Flag-tagged HDAC1 or HDAC3 (b) in the absence or presence of a plasmid for activated CaMKI. HDACs and MEF2C were detected by indirect immunofluorescence using antibodies against the epitope tags and fluorescein-conjugated (HDACs) and rhodamine-conjugated (MEF2C) secondary antibodies. c, Cos cells were co-transfected with expression vectors for Myc-tagged HDAC5 and epitope-tagged derivatives of the indicated MEF2 activators. Cells were analysed by indirect immunofluorescence using antibodies against the epitope tags and rhodamine-conjugated (HDAC5) and fluorescein-conjugated (Effector) secondary antibodies. ERK5 staining is shown for cells co-expressing MEK5 and ERK5.
Association of HDAC5 with MEF2 is mediated by an 18-amino-acid motif near the amino terminus2–4 (Fig. 3e). A similar region is contained in HDAC4, but not in the class I HDAC1 or HDAC3. Nuclear exclusion in response to CaMK signalling was also observed with HDAC4 (data not shown), but not with HDAC1 and HDAC3 (Fig. 2b). Identical results were obtained in Cos, HeLa, 293 and 10T1/2 fibroblasts, as well as in C2 and L6 myoblasts (data not shown); results with Cos cells are shown because they express the exogenous proteins at highest levels.
Figure 3.
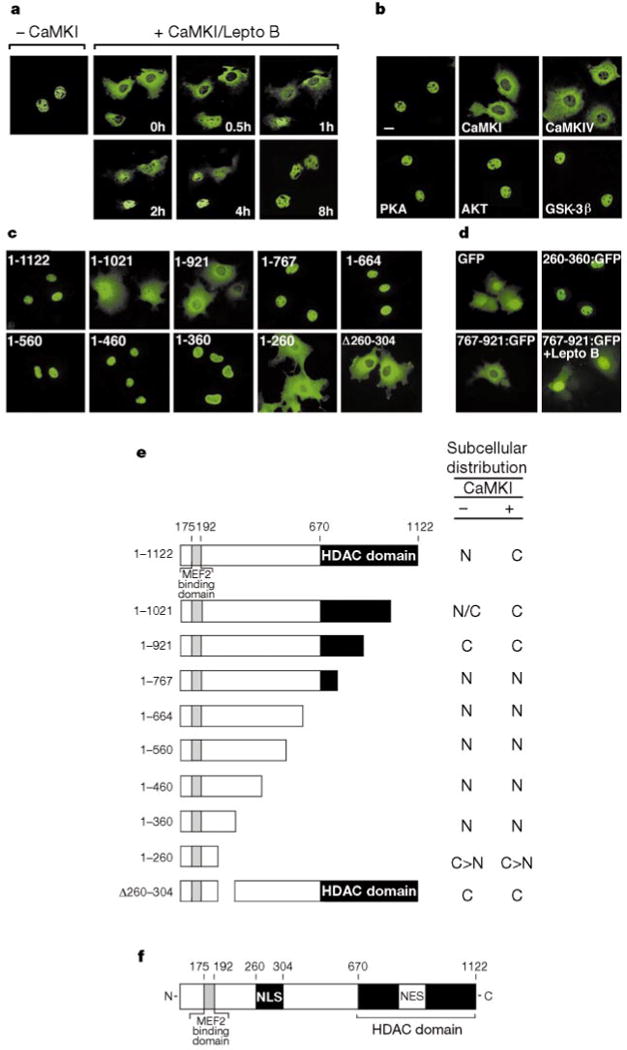
CaMK signalling stimulates nuclear export of HDAC5. a, Cos cells were transfected with an expression vector encoding HDAC5 fused to GFP in the absence or presence of a vector for activated CaMKI. GFP-positive cells were photographed before and after addition of leptomycin B (20 ng ml−1) to the medium for the indicated times. b, c, Cos cells were transfected with expression vectors for Flag-tagged HDAC5 and activated AKT, wild-type PKA, or wild-type GSK-3β (b) or Flag-tagged HDAC5 deletion mutants (c). HDAC5 was detected by indirect immunofluorescence using anti-Flag antibodies and a fluorescein-conjugated secondary antibody. d, Cos cells were transfected with expression vectors for the indicated HDAC5–GFP fusion proteins. GFP-positive cells were photographed 24 h after transfection. Leptomycin B (20 ng ml−1) was added to HDAC5(767–921)–GFP expressing cells 8 h before analysis. e, Subcellular distribution of HDAC5 mutants in the absence and presence of activated CaMK. C, cytoplasmic; N, nuclear; N/C, whole-cell; C ⟩ N, cytoplasmic greater than nuclear. f, Diagram of HDAC5.
In addition to CaMK, mitogen-activated protein kinases such as MKK6 and MEK5, which stimulate phosphorylation of the MEF2 transactivation domain through p38 (ref. 7) and ERK5 (ref. 8), respectively, activate MEF2. The calcium/calmodulin-dependent phosphatase calcineurin also stimulates MEF2 activity9,10. We therefore investigated whether these MEF2 activators also promote cytoplasmic accumulation of HDAC5. CaMKIV, like CaMKI, caused nuclear exclusion of HDAC5, whereas activated forms of MKK6, ERK5 and calcineurin had no effect on HDAC5 nuclear localization (Fig. 2c), further establishing the specificity of this signalling process.
Nuclear exclusion of HDAC5 could result from inhibition of nuclear import or stimulation of nuclear export. To distinguish between these possibilities, the subcellular distribution of an HDAC5–green fluorescent protein (GFP) chimaeric molecule was monitored in cells exposed to leptomycin B, a fungal toxin that blocks nuclear export by the exportin protein Crm1 (refs 11, 12). HDAC5–GFP normally resides in the nucleus, but is mostly cytoplasmic in cells expressing activated CaMKI (Fig. 3a). Treatment of CaMKI-expressing cells with leptomycin B resulted in translocation of HDAC5 from the cytoplasm to the nucleus over a time course of 4 h (Fig. 3a). Similar results were obtained in cells translationally arrested with cycloheximide (data not shown), indicating that newly synthesized, as well as pre-existing, cytoplasmic HDAC5 enters the nucleus after leptomycin B treatment. These results indicate that CaMK signalling stimulates nuclear export of HDAC5 through a Crm1-dependent mechanism.
Protein kinase A (PKA) and glycogen synthase kinase-3 (GSK-3) promote nuclear export of the Msn2 (ref. 13) transcription factor and two members of the NFAT family14 of transcription factors, respectively, and the AKT kinase stimulates nuclear export of the forkhead transcription factor15. Of note, the consensus phosphorylation sites for PKA and AKT (R/K-x-x-S/T) overlap precisely with that of CaMK16. Despite their overlapping substrate specificity, however, neither PKA, AKT nor GSK-3 was capable of driving HDAC5 from the nucleus to the cytoplasm (Fig. 3b).
To identify sequences in HDAC5 that control nuclear localization and responsiveness to CaMK signalling, we generated serial carboxy-terminal truncation mutants of HDAC5 and examined them by indirect immunofluorescence. Full-length HDAC5 (amino acids 1–1122) was localized exclusively in the nuclei of transfected Cos cells (Fig. 3c). However, removal of about 100 C-terminal amino acids from HDAC5 (mutant 1–1021) led to a diffuse, whole-cell pattern of HDAC5 localization, and a mutant lacking an additional 100 amino acids (mutant 1–921) was completely excluded from the nucleus. Further deletion of HDAC5 C-terminal sequences up to residue 360 resulted in a nuclear pattern of HDAC5 localization, whereas a mutant comprising the first 260 amino acids of HDAC5 (mutant 1–260) was primarily cytoplasmic. These results suggest that sequences in the N and C termini of HDAC5 regulate nuclear import and export, respectively (see Fig. 3f). Consistent with these predictions, an HDAC5 mutant lacking amino acids 260–304 was cytoplasmic (Fig. 3c; D260–304), and all of the HDAC5 mutants lacking sequences C-terminal to amino acid 767 remained predominantly nuclear in the presence of activated CaMKI (Fig. 3e; data not shown).
To assess whether the above domains in HDAC5 act as true nuclear import and export signals, we fused amino acids 260–360 and 761–921 of HDAC5 to the N terminus of GFP and determined the subcellular distribution of the resultant fusion proteins. Unmodified GFP was localized in both the nucleus and cytoplasm of transfected cells (Fig. 3d). When fused to amino acids 260–360 of HDAC5, GFP was driven into the nucleus. In contrast, a GFP fusion protein containing amino acids 761–921 of HDAC5 was exclusively cytoplasmic, and partially relocated to the nucleus in the presence of leptomycin B. These results show that specific portions of HDAC5 can act as nuclear import or export signals when fused to a heterologous protein.
HDAC5 contains six consensus CaMK phosphorylation sites that are conserved in HDAC4 (Fig. 4a). To determine whether HDACs can act as direct substrates for CaMK, we immunoprecipitated ectopic HDACs from transfected Cos cells and incorporated them into reaction mixtures containing [γ-32P]ATP in the absence or presence of activated CaMKIV. HDAC4, -5 and -3 were phosphorylated in the absence of exogenous CaMK (Fig. 4b), indicating that these HDACs may associate stably with one or more endogenous kinases present in Cos cells. In the presence of activated CaMKIV, the level of phosphorylation of HDAC4 and -5 was significantly increased, whereas the phosphorylation status of HDAC1 and -3 was unchanged. Analysis of deletion mutants localized the CaMK phosphorylation sites to a segment of the N-terminal extension of HDAC5 (amino acids 248–615), which contains three consensus CaMK phosphorylation sites: Ser 259, Ser 279 and Ser 498 (see Fig. 4a). Similar sites influence constitutive cytoplasmic localization of HDAC4 in U20S cells17.
Figure 4.
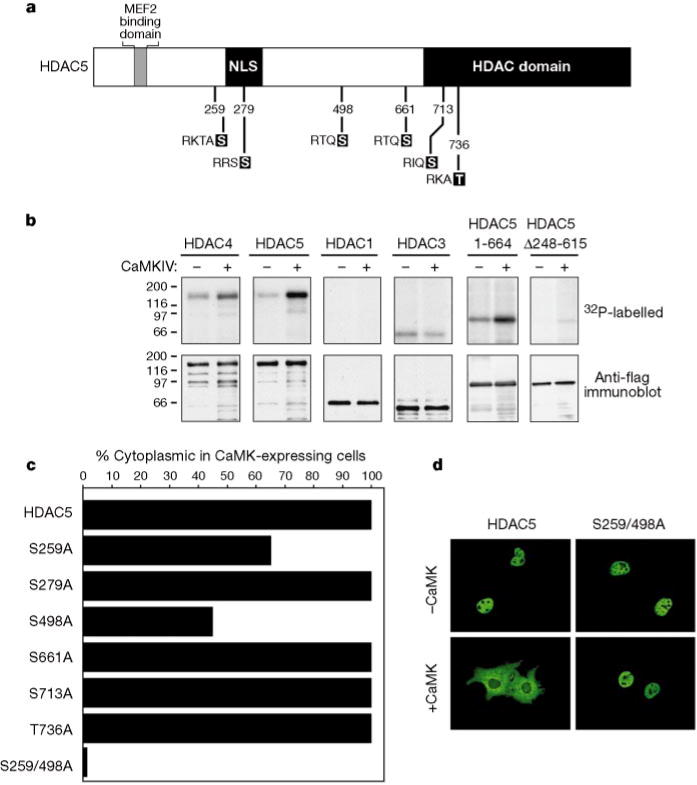
Identification of CaMK target sites in HDAC5. a, Six consensus CaMK sites in HDAC5 that are conserved in HDAC4 are shown. b, Cos cells were transfected with expression vectors encoding Flag-tagged forms of the indicated HDAC or a Flag-tagged derivative of activated CaMKIV and Flag-tagged proteins were immunoprecipitated and incorporated into in vitro phosphorylation reactions (see Methods). Proteins were resolved by SDS–PAGE, transferred to PVDF membranes, and visualized by autoradiography (top) followed by immunoblotting with anti-Flag antibodies (bottom). c, Cos cells were co-transfected with expression vectors for Flag-tagged forms of the indicated HDAC5 point mutants and an expression plasmid for an HA-tagged derivative of activated CaMKI. Cells were analysed by double immunofluorescence using a monoclonal anti-Flag antibody and polyclonal antibodies against the HA tag. Values represent the percentage of CaMKI-expressing cells in which HDAC5 exhibited cytoplasmic staining. In d, the immunofluorescence images reveal the loss of CaMK responsiveness of the HDAC5(S259/498A) double mutant.
If the subcellular distribution of HDAC5 is governed by its phosphorylation status, then disruption of the relevant phosphoacceptor sites would be predicted to block CaMK-mediated nuclear export. To test this hypothesis, each potential CaMK phosphorylation site in HDAC5 was substituted with alanine and the resultant mutants were examined for their ability to undergo nuclear export in response to CaMK signalling. In agreement with the in vitro phosphorylation data (Fig. 4b), mutation of Ser 661, Ser 713 or Thr 736 in HDAC5 had no effect on CaMK-dependent nuclear export (Fig. 4c). Likewise, disruption of Ser 279, which lies within the region of HDAC5 phosphorylated by CaMK (see Fig. 4b), did not prevent nuclear export in the presence of activated CaMK. In contrast, replacement of either Ser 259 or Ser 498 with alanine markedly reduced CaMK-mediated trafficking of HDAC5 from the nucleus to the cytoplasm, indicating that these sites may be involved in nuclear export. Indeed, simultaneous disruption of Ser 259 and Ser 498 completely blocked CaMK-mediated nuclear export of HDAC5 (Fig. 4c, d) and reduced the degree of phosphorylation by CaMK (data not shown). This mutant was still showed low-level phosphorylation, however, suggesting that phosphorylation at other sites in HDAC5 is not sufficient to induce nuclear export. These results show that phosphorylation of Ser 259 and Ser 498 is required for nuclear export of HDAC5.
To address the role of HDAC5 nuclear export in muscle differentiation, we tested the HDAC5(S259/498A) mutant for its effects on MyoD-dependent muscle gene activation in 10T1/2 fibroblasts, which requires MEF2 (ref. 1) and HAT activity18. Transfection of 10T1/2 cells with MyoD alone resulted in efficient conversion to the muscle cell lineage, assayed by myosin staining (Fig. 5a). Over-expression of wild-type HDAC5 interfered with this process. The HDAC5(S259/498A) mutant consistently blocked MyoD-dependent myogenic conversion of 10T1/2 cells even more potently than wild-type HDAC5. In contrast, a constitutively cytoplasmic mutant of HDAC5 lacking its nuclear localization signal (ΔNLS) was severely impaired in its ability to inhibit myogenesis. As described previously5, CaMK signalling completely rescued cells from the block to myogenesis imposed by HDAC5 (Fig. 5a). In cells expressing the HDAC5(S259/498A) mutant, however, the myogenic program remained largely blocked, despite the presence of activated CaMK. Together, these results suggest that nuclear HDAC5 represses myogenesis and that export of HDAC5 from the nucleus is required to execute the myogenic program.
Figure 5.
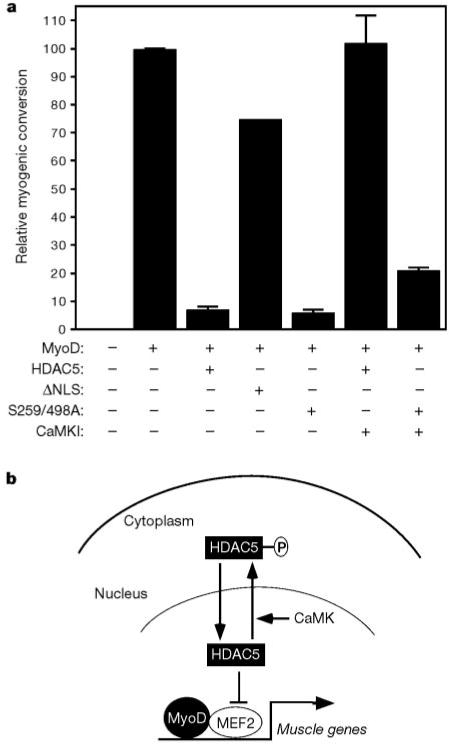
Regulation of myogenesis by HDAC5 nuclear export. a,10T1/2 fibroblasts were co-transfected with expression vectors for MyoD and the indicated HDAC5 protein in the absence or presence of a plasmid for activated CaMKI. Cells were transferred to differentiation medium 2 d post-transfection and stained with anti-myosin antibodies after 4d more in culture. Relative myogenic conversion represents the percentage of myosin-positive cells in HDAC transfectants relative to cells expressing MyoD alone (100 represents about 500 myosin-positive cells per 35-mm dish). Values represent the mean ± s.d. from at least two experiments. b, Model for signal-dependent regulation of myogenesis. HDAC5 blocks muscle differentiation by repressing the transcriptional activity of MEF2. CaMK phosphorylates HDAC5 and stimulates its nuclear export, freeing MEF2 to cooperate with MyoD to activate genes required for skeletal myogenesis.
A model for signal-dependent regulation of myogenesis through nuclear export of HDAC5 is depicted in Fig. 5b. Although our results show the potency and specificity of CaMK for promoting nuclear export of HDAC5, it remains possible that other signalling molecules with substrate specificities similar to that of CaMK can regulate HDAC5 nuclear export in certain cellular contexts. In addition to its apparent role in skeletal myogenesis, CaMK signalling regulates learning and memory19 as well as cardiac hypertrophy20. As class II HDACs and MEF2 are expressed primarily in skeletal muscle, heart and brain1,21, it is intriguing to speculate that CaMK controls these diverse biological processes by stimulating nuclear export of HDACs, resulting in activation of MEF2-dependent genes.
Methods
Cell culture
Cos, C2 and 10T1/2 cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with fetal bovine serum (10%), L-glutamine and penicillin/streptomycin. Lipid-mediated DNA transfections were performed using Fugene 6 reagent (Roche Molecular Biochemicals) and 0.5 μg of the indicated plasmids.
Myogenic conversion assays
For myogenic differentiation of confluent C2 and MyoD-transfected 10T1/2 cultures, growth medium was replaced with DMEM containing horse serum (2%), L-glutamine and penicillin/streptomycin. Transiently transfected 10T1/2 cells were exposed to differentiation medium 2 d after transfection and assessed by immunoperoxidase staining with the Vectastain Elite ABC kit (Vector Laboratories), and an anti-skeletal myosin antibody (Sigma) after a further 4 d.
Plasmids
Mammalian expression vectors for HDAC1, HDAC3 and HDAC5 (all with C-terminal Flag tags)22, constitutively active mutants of CaMKI23, CaMKIV (C-terminal Flag tag)24, calcineurin25, MKK626, AKT27, MEK528, ERK528 and wild-type forms of GSK-3β (ref. 29) and PKA (ref. 30) have been described. The cDNAs encoding activated CaMKI23 and CaMKIV24 contain stop codons in place of Ile 294 and Gln 318, respectively, thereby removing the C-terminal autoinhibitory domains. Both of these CaMK mutants function constitutively without the requirement for calcium and calmodulin for activation. We fused sequences encoding the indicated epitope tags to MEF2C (C-terminal Myc tag), HDAC5 (N-terminal Myc tag), calcineurin (N-terminal Flag tag), MKK6 (C-terminal Flag tag) and CaMKI (N-terminal 3×HA (haemagglutinin) tag), in the pCDNA3.1 expression vector (Invitrogen). HDAC5–GFP fusion proteins were constructed in the pEGFP-N1 expression plasmid (Clontech). Site-directed mutagenesis was performed using the QuikChange kit (Stratagene). For mutants S279A and S661A, the adjacent serines at positions 278 and 662, respectively, were also converted to alanine.
Indirect immunofluorescence
Cos cells were grown on glass coverslips, fixed in 10% paraformaldehyde, and stained in PBS containing 3% bovine serum albumin and 0.1% nonidet-P40. C2 cells were grown on gelatin-coated glass coverslips, fixed in methanol and stained in PBS containing 3% goat serum and 0.1% Nonidet-P40. Primary antibodies against Flag (mouse monoclonal; Sigma), Myc (rabbit polyclonal; Santa Cruz) and HA (rabbit polyclonal; Santa Cruz) were used at a dilution of 1:200. Secondary fluorochrome-conjugated antibodies (Vector Labs) were also used at a dilution of 1:200. Rabbit polyclonal antisera for HDAC5 were raised against a glutathione-_S_-transferase fusion protein containing amino acids 11–70 of rat HDAC5. For immunostaining with anti-HDAC5 antibodies, we purifed IgG fractions from crude serum using protein A-agarose beads (Pierce).
In vitro phosphorylation assays
For in vitro phosphorylation assays, Cos cells were transiently transfected with expression vectors encoding Flag-tagged HDACs or a Flag-tagged derivative of activated CaMKIV. Cells were collected in PBS containing 0.5% Triton X-100, 1 mM EDTA, 1 mM sodium pyrophosphate, 2 mM sodium fluoride, 10 mM β-glycerol phosphate, 1 mM sodium molybdate, 1 mM sodium orthovanadate, 1 mM PMSF and a protease inhibitor cocktail (Complete; Roche Molecular Biochemicals). Cells were sonicated briefly and cellular debris was removed by centrifugation. Flag-tagged proteins were immunoprecipitated using anti-Flag affinity resin (Sigma). Immunoprecipitates were washed two times in lysis buffer followed by three additional washes in a solution of 25 mM HEPES pH 7.6 and 10 mM MgCl2. Where indicated, CaMKIV-bound beads were mixed with those bound to HDAC immediately before the addition of reaction buffer (40 μl) consisting of 25 mM HEPES pH 7.6, 10 mM MgCl2, 2 mM CaCl2, 20 μg ml−1 calmodulin, 12.5 μM ATP and [γ-32P]ATP (10 μCi; 4,500 Ci mmol−1). Reaction mixtures were incubated at 25 °C for 20 min. Reactions were terminated by addition of SDS–PAGE loading buffer followed by boiling. Immunoprecipitated proteins were resolved by SDS-PAGE, transferred to PVDF membranes and visualized by autoradiography followed by immunoblotting with anti-Flag antibodies.
Acknowledgments
We thank M. Cobb, J. Han, R. Lin, G.S. McKnight, A. Means, S. O’Keefe, S. Schreiber, T. Chatila and J. Woodgett for expression plasmids; R. Prives for anti-MEF2 antisera; and M. Yoshida for leptomycin B. We are grateful to A. Tizenor for graphics, and J. Page and W. Simpson for editorial assistance. E.N.O. was supported by grants from NIH, the Robert A. Welch Foundation, the D. W. Reynolds Foundation and Myogen, Inc. T.A.M. is a Pfizer fellow of The Life Sciences Research Foundation.
References
- 1.Molkentin JD, Black BL, Martin JF, Olson EN. Cooperative activation of muscle gene expression by MEF2 and myogenic bHLH proteins. Cell. 1995;83:1125–1136. doi: 10.1016/0092-8674(95)90139-6. [DOI] [PubMed] [Google Scholar]
- 2.Sparrow DB, et al. MEF-2 function is modified by a novel co-repressor, MITR. EMBO J. 1999;18:5085–5098. doi: 10.1093/emboj/18.18.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miska EA, et al. HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J. 1999;18:5099–5107. doi: 10.1093/emboj/18.18.5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu J, McKinsey TA, Nicol RL, Olson EN. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc Natl Acad Sci USA. 2000;97:4070–4075. doi: 10.1073/pnas.080064097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu J, McKinsey TA, Zhang CL, Olson EN. Regulation of skeletal myogenesis by association of MEF2 with class II histone deacetylases. Mol Cell. 2000;6:233–244. doi: 10.1016/s1097-2765(00)00025-3. [DOI] [PubMed] [Google Scholar]
- 6.Kuo MH, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. BioEssays. 1998;20:615–626. doi: 10.1002/(SICI)1521-1878(199808)20:8<615::AID-BIES4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 7.Han J, Jiang Y, Li Z, Kravchenko VV, Ulevitch RJ. Activation of the transcription factor MEF2C by the MAP kinase p38 in inflammation. Nature. 1997;386:296–299. doi: 10.1038/386296a0. [DOI] [PubMed] [Google Scholar]
- 8.Kato Y, et al. BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. EMBO J. 1997;16:7054–7066. doi: 10.1093/emboj/16.23.7054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mao Z, Wiedmann M. Calcineurin enhances MEF2 DNA binding activity in calcium-dependent survival of cerebellar granule neurons. J Biol Chem. 1999;274:31102–31107. doi: 10.1074/jbc.274.43.31102. [DOI] [PubMed] [Google Scholar]
- 10.Wu H, et al. MEF2 responds to multiple calcium-regulated signals in the control of skeletal muscle fiber type. EMBO J. 2000;19:1–11. doi: 10.1093/emboj/19.9.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nishi K, et al. Leptomycin B targets a regulatory cascade of crm1, a fission yeast nuclear protein, involved in control of higher order chromosome structure and gene expression. J Biol Chem. 1994;269:6320–6324. [PubMed] [Google Scholar]
- 12.Fukuda M, et al. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature. 1997;390:308–311. doi: 10.1038/36894. [DOI] [PubMed] [Google Scholar]
- 13.Gorner W, et al. Nuclear localization of the C2H2 zinc finger protein Msn2p is regulated by stress and protein kinase A activity. Genes Dev. 1998;12:586–597. doi: 10.1101/gad.12.4.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beals CR, Sheridan CM, Turck CW, Gardner P, Crabtree GR. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science. 1997;275:1930–1934. doi: 10.1126/science.275.5308.1930. [DOI] [PubMed] [Google Scholar]
- 15.Brunet A, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 16.Pinna LA, Ruzzene M. How do protein kinases recognize their substrates? Biochem Biophys Acta. 1996;1314:191–225. doi: 10.1016/s0167-4889(96)00083-3. [DOI] [PubMed] [Google Scholar]
- 17.Grozinger CM, Schreiber SL. Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc Natl Acad Sci USA. 2000;97:7835–7840. doi: 10.1073/pnas.140199597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sartorelli V, Huang J, Hamamori Y, Kedes L. Molecular mechanisms of myogenic coactivation by p300: direct interaction with the activation domain of MyoD and with the MADS box of MEF2C. Mol Cell Biol. 1997;17:1010–1026. doi: 10.1128/mcb.17.2.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mayford M, et al. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- 20.Passier R, et al. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J Clin Invest. 2000;105:1395–1406. doi: 10.1172/JCI8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Black BL, Olson EN. Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu Rev Cell Dev Biol. 1998;14:167–196. doi: 10.1146/annurev.cellbio.14.1.167. [DOI] [PubMed] [Google Scholar]
- 22.Grozinger CM, Hassig CA, Schreiber SL. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc Natl Acad Sci. 1999;96:4868–4873. doi: 10.1073/pnas.96.9.4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haribabu B, et al. Human calcium-calmodulin dependent protein kinase I: cDNA cloning, domain structure and activation by phosphorylation at threonine-177 by calcium-calmodulin dependent protein kinase I kinase. EMBO J. 1995;14:3679–3686. doi: 10.1002/j.1460-2075.1995.tb00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chatila T, Anderson KA, Ho N, Means AR. A unique phosphorylation-dependent mechanism for the activation of Ca2+/calmodulin-dependent protein kinase type IV/GR. J Biol Chem. 1996;271:21542–21548. doi: 10.1074/jbc.271.35.21542. [DOI] [PubMed] [Google Scholar]
- 25.O’Keefe SJ, Tamura J, Kincaid RL, Tocci MJ, O’Neill EA. FK-506- and CsA-sensitive activation of the interleukin-2 promoter by calcineurin. Nature. 1992;357:692–694. doi: 10.1038/357692a0. [DOI] [PubMed] [Google Scholar]
- 26.Jiang Y, et al. Characterization of the structure and function of a new mitogen-activated protein kinase (p38beta) J Biol Chem. 1996;271:17920–17926. doi: 10.1074/jbc.271.30.17920. [DOI] [PubMed] [Google Scholar]
- 27.Rybkin II, Cross ME, McReynolds EM, Lin RZ, Ballou L. M alpha(1A) adrenergic receptor induces eukaryotic initiation factor 4E-binding protein 1 phosphorylation via a Ca(2+)-dependent pathway independent of phosphatidylinositol 3-kinase/Akt. J Biol Chem. 2000;275:5460–5465. doi: 10.1074/jbc.275.8.5460. [DOI] [PubMed] [Google Scholar]
- 28.English JM, et al. Contribution of the ERK5/MEK5 pathway to Ras/Raf signaling and growth control. J Biol Chem. 1999;274:31588–31592. doi: 10.1074/jbc.274.44.31588. [DOI] [PubMed] [Google Scholar]
- 29.Stambolic V, Woodgett JR. Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem J. 1994;303:701–704. doi: 10.1042/bj3030701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mellon PL, Clegg CH, Correll LA, McKnight GS. Regulation of transcription by cyclic AMP-dependent protein kinase. Proc Natl Acad Sci USA. 1989;86:4887–4891. doi: 10.1073/pnas.86.13.4887. [DOI] [PMC free article] [PubMed] [Google Scholar]