Rational development and characterization of humanized anti–EGFR variant III chimeric antigen receptor T cells for glioblastoma (original) (raw)
. Author manuscript; available in PMC: 2016 Feb 18.
Abstract
Chimeric antigen receptors (CARs) are synthetic molecules designed to redirect T cells to specific antigens. CAR-modified T cells can mediate long-term durable remissions in B cell malignancies, but expanding this platform to solid tumors requires the discovery of surface targets with limited expression in normal tissues. The variant III mutation of the epidermal growth factor receptor (EGFRvIII) results from an in-frame deletion of a portion of the extracellular domain, creating a neoepitope. We chose a vector backbone encoding a second-generation CAR based on efficacy of a murine scFv–based CAR in a xenograft model of glioblastoma. Next, we generated a panel of humanized scFvs and tested their specificity and function as soluble proteins and in the form of CAR-transduced T cells; a low-affinity scFv was selected on the basis of its specificity for EGFRvIII over wild-type EGFR. The lead candidate scFv was tested in vitro for its ability to direct CAR-transduced T cells to specifically lyse, proliferate, and secrete cytokines in response to antigen-bearing targets. We further evaluated the specificity of the lead CAR candidate in vitro against EGFR-expressing keratinocytes and in vivo in a model of mice grafted with normal human skin. EGFRvIII-directed CAR T cells were also able to control tumor growth in xenogeneic subcutaneous and orthotopic models of human EGFRvIII+ glioblastoma. On the basis of these results, we have designed a phase 1 clinical study of CAR T cells transduced with humanized scFv directed to EGFRvIII in patients with either residual or recurrent glioblastoma (NCT02209376).
Introduction
Immune therapies that engage T cells have the potential to induce long-term durable remissions of cancer. In hematologic malignancies, allogeneic hematopoietic stem cell transplant can be curative in part due to T cell–mediated antitumor immunity; in solid tumors, checkpoint blockade with anti–CTLA-4 or anti–PD-1 monoclonal antibodies (mAbs) can mediate long-term responses (1, 2) by releasing T cells from tightly controlled peripheral tolerance. Redirecting T cells with chimeric antigen receptors (CARs) is an alternative method of overcoming tolerance and can be performed in the autologous setting. In B cell malignancies, CAR T cells directed to CD19 can mediate long-term remissions without the need for an allogeneic human leukocyte antigen (HLA)–matched donor (3–5). However, CAR immunotherapy in solid tumors remains challenging, largely due to the lack of appropriate surface antigens whose expression is confined to malignant tissue. Off-tumor expression of the antigen target has potential to cause on-target toxicity with varying degrees of severity depending on the affected organ tissue (6–8).
Epidermal growth factor receptor variant III (EGFRvIII) is a putative tumor-specific oncogenic mutation and is the most common variant of the EGFR observed in human tumors (9–13). EGFRvIII results from the in-frame deletion of exons 2 to 7 and the generation of a novel glycine residue at the junction of exons 1 and 8; this novel juxtaposition within the extracellular domain (ECD) of the EGFR creates a tumor-specific and immunogenic epitope. The EGFRvIII mutation is most frequently observed in glioblastoma, where it occurs in about 30% of cases. Median survival for patients with newly diagnosed glioblastoma is less than 15 months, and expression of EGFRvIII is linked to poor long-term survival regardless of other factors such as extent of resection and age (14). The current standard of care for patients with newly diagnosed glioblastoma involves primary surgical resection, followed by concurrent temozolomide and radiation, followed by adjuvant temozolomide alone for six cycles at minimum (15). No current treatment is curative. Novel temozolomide agents (16, 17) and a variety of targeted kinase inhibitors (18) have limited efficacy when used as monotherapy, and there has been extensive interest in immunotherapeutic approaches.
Classically, the central nervous system (CNS) has been considered an “immunoprivileged” site where immune surveillance is minimal. Indeed, several cellular and molecular mechanisms underlying the unique immunosuppression of the CNS tumors have been delineated (19, 20). However, the presence of lymphocytes within malignant gliomas can be a positive prognostic indicator of survival (21, 22). Although such tumor-infiltrating lymphocytes are not potent enough to mediate regression of gliomas, the primary defect is not likely a result of a lack of immune surveillance. Indeed, naturally occurring autoimmune diseases, such as paraneoplastic cerebellar generation and multiple sclerosis, provide evidence that immune cells can traffic to the CNS and target the brain.
Immunotherapeutic approaches to glioblastoma, and EGFRvIII in particular, are currently in clinical development. Rindopepimut is a peptide vaccine strategy currently in phase 3 trials for EGFRvIII-expressing glioblastoma. Rindopepimut consists of the EGFRvIII-specific peptide sequence conjugated to the carrier protein keyhole limpet hemocyanin. Three phase 2 trials of rindopepimut have been completed in newly diagnosed EGFRvIII-positive glioblastoma patients with consistent results: across all studies, rindopepimut has been generally well tolerated with generation of robust, specific, and durable immune responses (23,24). Generation of T cell responses to peptide vaccine is nevertheless limited by the available repertoire of T cells, which have undergone selection in the thymus; in contrast, adoptive immunotherapy with redirected T cells obviates both the existing T cell repertoire and the need for antigen processing and presentation by professional antigen-presenting cells. Therefore, direct transfer of EGFRvIII-directed T cells is potentially more effective and may have more favorable kinetics compared to peptide- or cell-based vaccines. However, whereas in the vaccine setting, high-avidity T cells reactive to self-antigens are unlikely to be present or cause tissue damage, T cells that are redirected to a novel antigen require extensive validation to avoid on-target, off-tumor toxicity or cross-reactivity to self-antigens (8, 25, 26).
We have previously described CARs directed to EGFRvIII in human and murine systems based on single-chain variable fragments (scFvs) derived from two mAbs: human 139 and murine 3C10 (27–29). Here, after selection of a basic CAR design, we humanized the murine 3C10 scFv to avoid human anti-mouse antibody (HAMA) responses, which could limit the persistence of the CAR T cells or cause allergic sensitization (30). Because many antibodies to EGFRvIII cross-react with wild-type EGFR (EGFRwt), including cetuximab (31, 32), we focused much of our validation efforts on determining the degree of specificity to EGFRvIII and cross-reactivity with EGFRwt. The interaction between scFv and antigen was evaluated at the biophysical level, with either or both as soluble molecules, and at the biologic level as CARs expressed on human T cells. The initial panel of scFv candidates was tested in silico, and the panel was progressively narrowed on the basis of testing in vitro. The lead humanized CAR was further tested for safety in a mouse model grafted with normal human skin and for efficacy as a single agent in subcutaneous and orthotopic xenograft models of human EGFRvIII+ glioblastoma. On the basis of the validation data described here, we have opened a phase 1 clinical trial to treat patients with EGFRvIII+ glioblastoma either at recurrence or in conjunction with temozolomide in newly diagnosed patients with residual disease.
Results
Selection of the anti-EGFRvIII CAR design
As a general rule, the scFv dictates the new antigen specificity of the redirected CAR T cell, whereas the intracellular signaling domains of the CAR dictate the functional outcome of CAR-mediated T cell activation. Our institution has had success with a CD19-directed CAR designed to include a murine-derived scFv fused to a CD8a hinge and transmembrane domain, and the intracellular domains of human 4-1BB and CD3z (or “z”). However, it is possible that targeting antigens presented by solid tumors could be optimized with a different CAR design, such as a third-generation CAR that includes the intracellular domain of the CD28 costimulatory molecule (33). To determine an optimal CAR design for glioblastoma, a series of EGFRvIII-specific CARs were generated on the basis of either the murine 3C10 or the human 139 scFv (27) fused with either 4-1BB or CD28–4-1BB and CD3ζ (z) signaling domains (Fig. 1A). Human T cells were lentivirally transduced with each construct and tested in vitro and in vivo. A flow cytometry–based cytotoxicity assay using the human glioma cell line U87 and the transduced cell line U87-EGFRvIII as targets indicated no difference among three different types of CAR T cell products directed to EGFRvIII (Fig. 1B). We then compared the three anti-EGFRvIII CAR designs in an in vivo xenogeneic mouse model of intracranial glioblastoma, using orthotopically injected U87-EGFRvIII tumor cells (Fig. 1C). In this model, mice were treated with CART cells intravenously 1 week after tumor implantation, along with temozolomide (17 mg/kg) daily for 4 days (29, 34) starting the day of T cell injection. Tumor progression was evaluated by serial bioluminescent imaging. Mice receiving any of the anti-EGFRvIII CAR–transduced T cells demonstrated sustained tumor regression compared with mice receiving control CD19–specific CARs, temozolomide alone, or saline. However, the 3C10.BBz construct induced a tumor regression earlier than 3C10.28.BBz or 139.BBz (Fig. 1C). On the basis of these results, we decided to proceed with the 3C10.BBz vector design for clinical translation.
Fig. 1. Selection of a 4-1BB-endodomain–based anti-EGFRvIII CAR construct.

(A) Vector maps of tested anti-EGFRvIII CAR designs. (B) Flow-based cytotoxicity of CART-EGFRvIII cells against EGFRvIII+ glioma cell line. The glioblastoma cell line U87MG was stably transduced with EGFRvIII, fluorescently labeled with CFSE, and cocultured for 18 hours. Untransduced (UTD) or CD19.BBz CAR T cells were used as negative controls and compared to 3C10.BBz, 3C10.28BBz, or 139.BBz EGFRvIII CAR T cells. Lysis of parental cells (U87MG) and EGFRvIII-transduced U87 cells was analyzed at different effector/target (E:T) ratios by flow cytometry. One representative experiment is shown. Samples were performed in duplicate in two replicative experiments. (C) CART-EGFRvIII cells eradicate glioblastoma in an orthotopic xenogeneic mouse model. Seven days after 5 × 104 U87-EGFRvIII cells were orthotopically implanted into mouse brains, mice were injected intravenously with either phosphate-buffered saline (PBS) alone, temozolomide (TMZ) alone, or temozolomide with 1 × 106 transduced T cells expressing the indicated CAR constructs. Tumor burden was quantified as total flux in units of photons/second. Bars indicate means ± SD (n = 10 mice per group). Note that all mice in the PBS group died by day 15.
Generation and selection of a humanized scFv
Previous studies of CAR T cells using murine-based scFvs have elicited HAMA responses, resulting in elimination of the transferred cells over relatively short periods of time, particularly in solid tumors (6, 35, 36). Although the HAMA response itself is not clinically significant, it could potentially limit retreatment options. In one case at our institution, repeated dosing of murine scFv CAR T cells resulted in anaphylaxis (30), which was likely caused by an immunoglobulin E (IgE)–mediated response to the murine scFv. On the basis of the CD19 CAR data demonstrating that persistence of CAR T cells is required for optimal efficacy (5, 37), and to allow for the possibility of more than one infusion, we chose to humanize the murine 3C10 scFv. A panel of eight humanized scFv constructs was generated and produced as soluble fragments. Each fragment was biotinylated and tested for binding to EGFRvIII and EGFRwt-transfected cells over a wide range of concentrations (Fig. 2A). We noted that the original murine 3C10 scFv bound to EGFRwt at high concentrations. Two of the constructs (2173 and 2174) did not bind to EGFRwt in the concentration ranges tested, though they also appeared to have a lower affinity for EGFRvIII.
Fig. 2. Selection of a humanized scFv with high specificity for EGFRvIII.
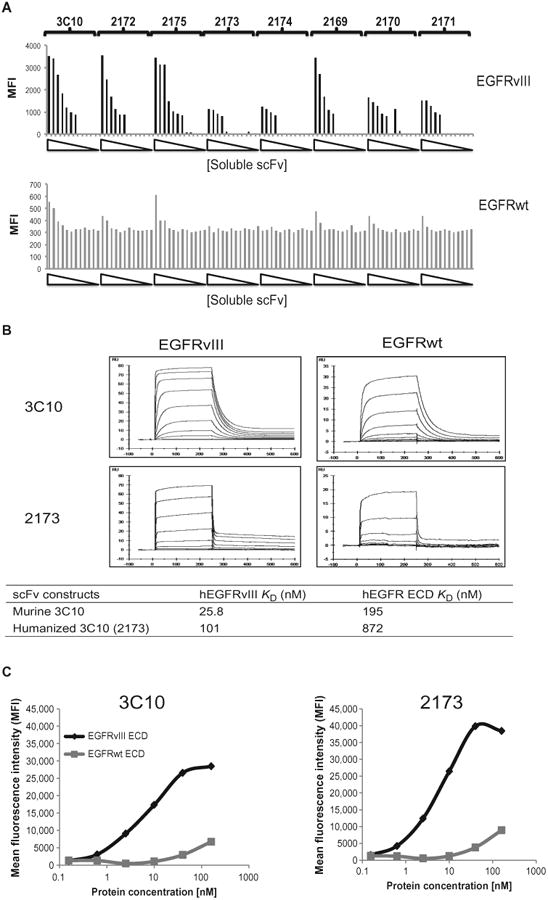
(A) Binding of soluble scFvs to 293 cells transiently transfected with EGFRvIII or EGFRwt. Eight his-tagged solubilized humanized scFvs, at increasing concentrations from 0.002 to 200 nM, were incubated with either EGFRvIII-or EGFRwt-transfected 293 cells. After detection with phycoerythrin (PE)–conjugated anti-his secondary antibodies, the mean fluorescence intensity (MFI) was quantified. (B) Affinity measurements of soluble scFv binding to EGFRvIII or EGFRwt. Sensograms of soluble versions of the lead humanized 2173 scFv and the original murine 3C10 scFv flowed at various concentrations over a sensor chip binding the ECDs of EGFRvIII or EGFRwt. Association (after 240 s) or dissociation (after 360 s) rates were measured. Calculated _K_D values for these two scFvs for affinity of binding to either EGFRvIII or EGFRwt are shown in the table. (C) Membrane-bound CAR recognition of soluble EGFRvIII or EGFRwt recombinant proteins. MFI of murine 3C10– or humanized 2173 CAR–transduced T cells stained with varying concentrations of soluble and biotinylated forms of the ECDs of EGFRvIII or EGFRwt.
We chose to pursue the lower-affinity scFv to minimize the potential binding to EGFRwt, given that EGFR is expressed in a wide variety of normal tissues, including keratinocytes and multiple epithelial tissues. In light of previous on-target, off-tumor toxicities observed in several cases of gene-engineered T cell therapy in melanoma (38), colon cancer (39), renal cell cancer (6), and toxicity due to recognition of HER2 and MAGE-A3 mimotopes on normal, nontumor cells (7, 8, 25, 26), minimizing the potential recognition of the EGFRwt protein was of paramount importance. Affinity constants were determined for the anti-EGFRvIII scFv constructs before and after humanization. Binding affinities were measured by Biacore (surface plasmon resonance) using soluble, biotinylated versions of the EGFRvIII and EGFRwt ECDs coupled to a streptavidin sensor chip; purified soluble scFv candidates were flowed over the chip in serial dilutions, and association and dissociation rates were monitored for 240 and 360 s, respectively. The sensograms for murine 3C10 and the humanized lead candidate (2173) are shown (Fig. 2B). The data were fitted to a steady-state affinity model, and _K_D values were calculated (Fig. 2B). The _K_D for the humanized 2173 scFv was 101 nM for EGFRvIII and 872 nM for EGFRwt. In contrast, the _K_D for the murine 3C10 scFv was 25.8 nM for EGFRvIII and 195 nM for EGFRwt. These values confirm the relatively lower affinity of the humanized 2173 scFv for both EGFRwt and EGFRvIII.
The affinity of a soluble scFv is a measure of the strength of its interaction with antigen as a monovalent protein. Because cell-anchored CAR constructs will dimerize and bind to antigen with the accumulated strength of multiple interactions (that is, avidity), we proceeded to determine the effect of CAR-mediated binding to EGFRwt and EGFRvIII by performing the converse experiment to Fig. 2A. Here, T cells transduced with murine 3C10 or humanized 2173 CAR were stained with varying concentrations of soluble proteins derived from the ECD of EGFRvIII or EGFRwt. We found that although there was a reduction in binding affinity of the humanized 2173–based CARs, the specificity toward EGFRvIII was maintained, if not enhanced, compared to the murine 3C10–based CAR (Fig. 2C). Thus, there was strong biophysical evidence for specific binding of the anti-EGFRvIII CAR to the EGFRvIII mutant protein.
In vitro characterization of CART-EGFRvIII T cells
The murine (3C10) and humanized (2173) EGFRvIII CARs were extensively evaluated in a series of in vitro immunologic assays. Cytotoxicity assays used to evaluate CART-EGFRvIII cells against U87 glioblastoma cell lines expressing EGFR (naturally) or transduced to express EGFRvIII demonstrated specific killing of EGFRvIII-expressing cells. Untransduced T cells and CAR T cells directed to CD19 served as negative controls (Fig. 3A). No cytolytic activity toward EGFR-expressing cells was noted. Because most cultured human tumor cell lines express some degree of EGFR, we generated a panel of baby hamster kidney (BHK) cell lines that lack human EGFR expression; these were main- tained as the parental line or transduced to express either EGFRwt or EGFRvIII. CART-EGFRvIII cells based on either the murine 3C10 scFv or the humanized 2173 scFv produced IFN-γ specifically in response to EGFRvIII-expressing cells such as BHK-EGFRvIII or U87-EGFRvIII (Fig. 3B). Similarly, T cells transduced with either the murine 3C10 or humanized 2173 CARs proliferated specifically in response to stimulation with EGFRvIII antigen, but not EGFRwt (Fig. 3C).
Fig. 3. In vitro function of murine 3C10 and humanized 2173 CART-EGFRvIII cells.

(A) Luciferase-based cytotoxicity assay of UTD, CART19, murine 3C10, or humanized 2173 CART-EGFRvIII cells cultured for 18 to 20 hours with U87 and U87-vIII target cells expressing luciferase. (B) Levels of interferon-γ (IFN-γ) as measured by enzyme-linked immunosorbent assay (ELISA) 24 hours after culturing UTD, CART19, murine 3C10, or humanized 2173 CART-EGFRvIII cells alone or with BHK, BHK-EGFRvIII (BHK-vIII), BHK-EGFRwt (BHK-wt), U87, or U87-EGFRvIII at a 1:1 E:T ratio. (C) Proliferation of UTD, CART19, 3C10, or 2173 CART-EGFRvIII cells after 6 days of coculture with either BHK-wt or BHK-vIII cells relative to proliferation over T cells cultured with parental BHK cells. (D) Long-term proliferation of T cells in response to repetitive stimulation with either antigen (U87-EGFRvIII cells) or anti-CD3/CD28 beads. T cells were either UTD or transduced to express either murine 3C10 or humanized EGFRvIII CAR, or an anti-mesothelin (SS1) CAR. All CARs are of the same vector design (4-1BB-ζ). (E) Enrichment of CAR-expressing T cells with repetitive antigen stimulation. CAR expression was quantified by flow cytometry just before each stimulation and normalized at each new stimulation as indicated by arrows.
To determine whether repeated antigen stimulation could specifically enhance CAR T cell expansion and persistence, we stimulated EGFRvIII-directed CAR T cells with U87-EGFRvIII cells or anti-CD3/CD28–coated beads over the course of 4 weeks in vitro. The timing of restimulation was based on return to resting cell size because cell size is a marker of lymphocyte activation state, and restimulation of resting lymphocytes reduces activation-induced cell death (40). Repeated stimulation with EGFRvIII-expressing cells allowed CART-EGFRvIII cells to continue to expand, whereas lack of antigen stimulation or repeated CD3/CD28 bead stimulation resulted in culture growth arrest (Fig. 3D). T cell cultures were normalized for CAR expression at each stimulation, and specific enrichment of CAR-expressing cells was noted after the first antigen-specific stimulation and was further enhanced after two encounters with antigen (Fig. 3E). We again noted modest antigen-independent proliferation of transduced T cells bearing the 4-1BB signaling domain (SS1 anti-mesothelin CAR T cells) (41). Thus, EGFRvIII-directed CAR T cells demonstrated cytolytic activity, cytokine production, and specific proliferation in response to EGFRvIII but not EGFRwt stimulation in vitro.
Extensive in vitro and in vivo testing against EGFRwt demonstrates lack of reactivity
Having confirmed the in vitro function of EGFRvIII-directed CAR T cells to EGFRvIII-expressing targets, we next sought to evaluate the humanized 2173 CAR candidate for potential cross-reactivity to EGFRwt in more biologically relevant assays. First, we generated a positive control CAR on the basis of the heavy and light chains from the mAb cetuximab, which does not distinguish EGFR from EGFRvIII. Cetuximab is U.S. Food and Drug Administration–approved for use in colon cancer and head and neck cancers, which overexpress EGFR; common side effects of cetuximab are rash and diarrhea due to endogenous EGFR expression in keratinocytes and epithelial tissues. A cetuximab-based CAR (cetux-CAR) constructed with the same design as the 2173 anti-EGFRvIII CAR was expressed in human T cells. Cetux-CAR bound equally well to soluble EGFRwt and EGFRvIII ECDs, whereas the 2173 CAR bound only to EGFRvIII (Fig. 4A). Similarly, cetux-CAR T cells were tested in vitro for their ability to proliferate in response to stimulation with either BHK-EGFRwt or BHK-EGFRvIII (Fig. 4B). Cetux-CAR T cells proliferated equally well in response to stimulation with BHK-EGFRwt as the they did to BHK-EGFRvIII, whereas 2173 CAR T cells proliferated only in response to BHK-EGFRvIII and equally as well as cetux-CAR T cells. Similarly, 2173 CAR T cells produced type I cytokines [IFN-γ, inter-leukin-2 (IL-2), and tumor necrosis factor–α (TNF-α)] only in response to EGFRvIII, whereas cetux-CARs responded to both EGFRwt and EGFRvIII. Again, the levels of cytokine production for 2173 and cetux-CAR T cells were similar in response to EGFRvIII, indicating that the relatively low affinity of 2173 for EGFRvIII did not result in lower in vitro activity against target cells expressing high levels of antigen.
Fig. 4. In vitro comparison of humanized 2173 EGFRvIII-specific CAR to cetux-CAR.
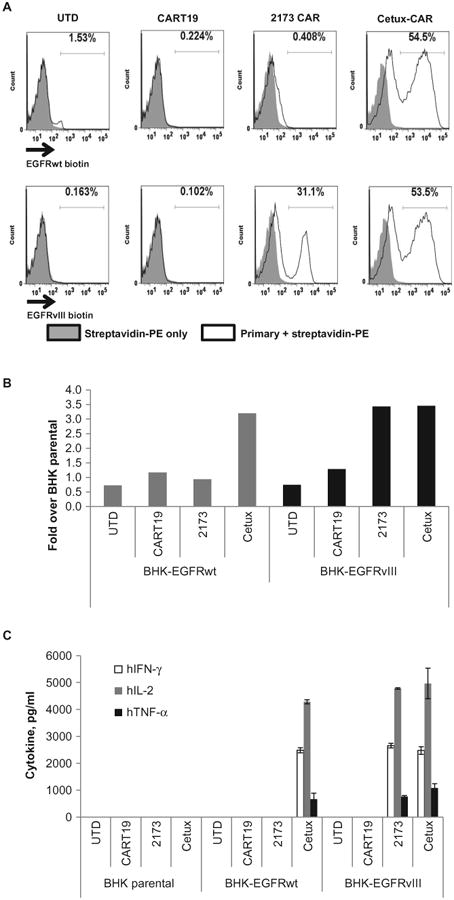
(A) Membrane-bound CAR recognition of soluble EGFRvIII or EGFR recombinant proteins. T cells that were either UTD or transduced with the indicated CARs were incubated with soluble biotinylated ECDs of EGFR or EGFRvIII. Histogram plots are shown with gates indicating the percentage of cells that stained with streptavidin-PE. (B) Proliferation of UTD or T cells transduced with the indicated CAR T cells after 4 days of coculture with BHK cells expressing EGFRwt or EGFRvIII. Y axis indicates fold proliferation over CAR T cells stimulated with parental BHK cells. (C) ELISA-based cytokine analysis on supernatants collected 24 hours after stimulation of T cells with BHK, BHK-EGFRwt, or BHK-EGFRvIII cells.
Having confirmed the function of the cetux-CAR, we next sought to compare reactivity of 2173 CAR and cetux-CAR T cells in response to endogenous levels of EGFR in normal tissues in vitro. We cultured primary human keratinocytes and used them to stimulate CAR T cells in vitro. We observed production of type I cytokines by cetux-CAR T cells in response to EGFR presented by either U87 or keratinocytes, as well as U87-EGFRvIII cells. In contrast, 2173 CAR T cells only produced type I cytokines in response to EGFRvIII antigen (Fig. 5A; gating strategy and controls are shown in the Supplementary Materials, fig. S1). Similarly, whereas cetux-CAR T cells lysed U87, U87-EGFRvIII, and keratinocytes, 2173 CAR T cells only lysed EGFRvIII-expressing target cells (Fig. 5B). There was no significant cytokine production or lysis of keratinocytes or U87 glioma cells by the humanized anti-EGFRvIII–directed 2173 CAR T cells.
Fig. 5. Evaluation of cross-reactivity of CAR T cells to human skin in vitro and in vivo.
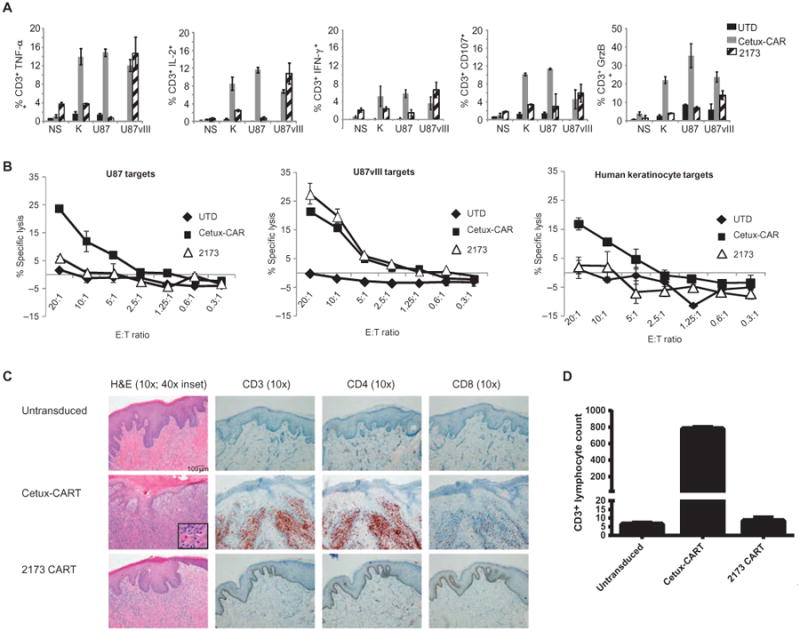
(A) Intracellular cytokine staining on untransduced (UTD), 2173 CAR, or cetux-CAR T cells after they were cocultured with no stimulation (NS), human keratinocytes (K), U87, or U87-EGFRvIII for 16 hours. Plots indicate percentage of T cells (±SD of two replicates) that stained positive for each cytokine as determined by flow cytometry. Data shown are representative of three independent experiments. GrzB, granzyme B. (B) Chromium release assays of cytotoxicity of UTD, 2173 CAR, or cetux-CAR T cells at ratios from 20:1 to 0.3:1 with either U87, U87-EGFRvIII, or human keratinocyte targets. Plots indicate means ± SEM of triplicate wells. (C) Hematoxylin and eosin (H&E)–stained human skin grafts excised from NSG mice 2 weeks after intravenous injection of either UTD, cetux-CAR–transduced, or humanized 2173 CAR–transduced T cells at ×10 magnification (degrees of magnification indicated at the top), along with an additional magnification inset in the cetux-CART–treated mice. On the right, immuno-histochemical stains for CD3, CD4, and CD8 of punch biopsy specimens obtained 3 days after T cell injection are shown. Scale bar, 100 mm (all 10× images). Results are representative of two experiments. (D) T cells infiltrating the epidermis were enumerated in a punch biopsy specimen. Bars show average and range of counts from two separate sections of adjacent cuts.
Finally, because cultured human keratinocytes may not express EGFR at endogenous levels, we sought to evaluate the reactivity of CART-EGFRvIII cells in human skin. We therefore tested CAR T cells in an in vivo mouse model of human skin grafted onto immunodeficient mice. NSG mice were surgically grafted with human foreskins and allowed to heal over 4 to 6 weeks. Skin-grafted mice were injected intravenously with either untransduced or cetux-CAR or 2173 anti-EGFRvIII CAR T cells. Two weeks after CAR T cell injection, skin grafts were excised and examined histologically (Fig. 5C). Mice treated with untransduced T cells had intact epidermis and dermis, with minimal infiltration of lymphocytes in the epidermis and dermis. No significant infiltration of CD3, CD4, or CD8 T cells was observed by immunohistochemistry of a skin biopsy specimen taken 3 days after T cell injection (Fig. 5C). In contrast, mice injected with cetux-CAR T cells had a prominent lymphocytic infiltrate of the epidermis and dermis. Lymphocyte-induced keratinocyte apoptosis was observed (inset), which is a typical histological feature of cutaneous graft-versus-host disease. Most of the infiltrate appeared to be composed of CD3+ CD4+ T cells. The injected CAR T cells were 75% CD4+ T cells and 25% CD8+ T cells. Mice injected with anti-EGFRvIII CAR T cells (2173) had mild immune infiltration of the dermis, but the basal cell layer and epider- mis and keratinocytes were intact. No significant CD3+ T cell infiltrate was observed by immunohistochemistry in the epidermis or dermis (Fig. 5C). Numbers of CD3+ T cells infiltrating the epidermis were quantified in two consecutive tissue sections taken from a punch biopsy specimen obtained 3 days after T cell injection (Fig. 5D); there were negligible numbers of T cells observed in the human skin of mice treated with untransduced and 2173 CAR T cells compared to the large numbers of T cells in the skin of cetux-CAR–treated mice.
CART-EGFRvIII efficacy in human tumors in subcutaneous and orthotopic mouse models
Having demonstrated both the specificity and effector functions of 2173 CAR T cells in response to EGFRvIII in vitro, we sought to confirm the in vivo antitumor activity in NSG mice bearing human glioma tumors established for 7 days. U87-EGFRvIII tumors were implanted subcutaneously, and engraftment was confirmed via bioluminescence imaging 5 days later. On day 7, human T cells expressing CD19 CAR, 3C10 CAR, or 2173 CAR at similar levels were injected intravenously as a single agent. Animals in all groups showed tumor progression; mice receiving control CD19 CARs demonstrated rapid tumor growth, with all mice reaching a humane experimental endpoint by 17 days after initial tumor engraftment. In contrast, mice treated with murine 3C10 or humanized 2173 EGFRvIII-specific CARs had reduced tumor growth, with both groups having mice surviving greater than 40 days. In the 3C10 and 2173 CAR treatment groups, median survival also increased from 14 to 21 days compared with mice receiving CD19 CAR control T cells (Fig. 6A; the raw data are shown in table S1).
Fig. 6. Antitumor activity of humanized 2173 CART-EGFRvIII cells in vivo.
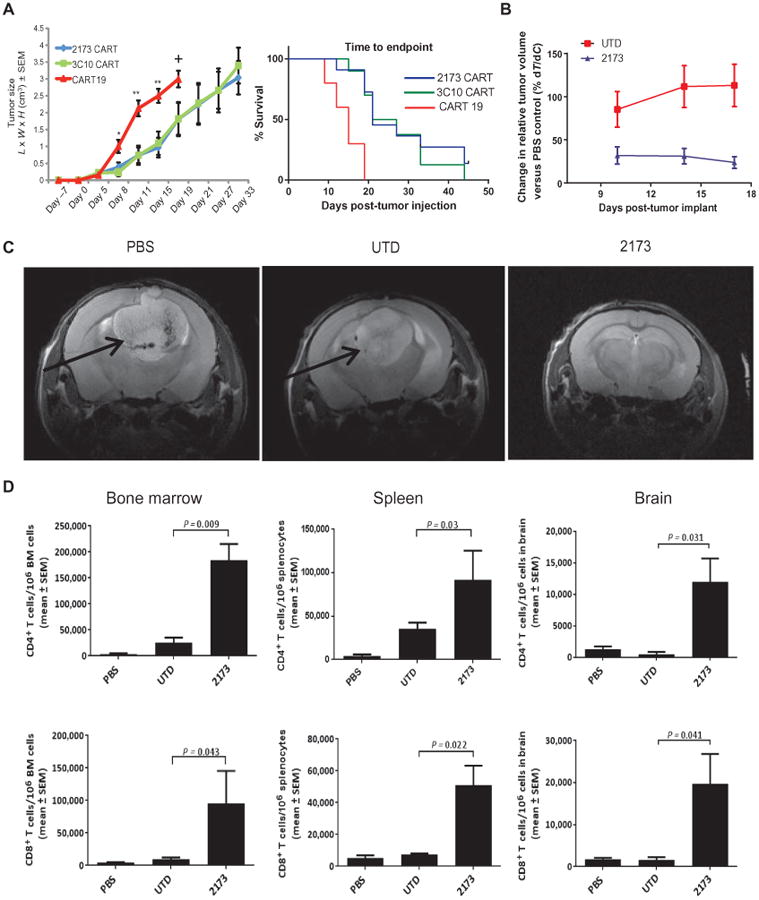
(A) NSG mice injected with U87-EGFRvIII tumors subcutaneously on day 0 and with T cells as indicated intravenously on day 7. Left plot indicates mean ± SEM of calculated tumor volume based on caliper measurements over time. Statistically significant differences between EGFRvIII CARs and CD19 CARs are marked by asterisks [*P = 0.01, **P = 0.003; n = 10 mice per group; one-way analysis of variance (ANOVA), Kruskal-Wallis test]. + indicates euthanasia. Survival based on time to endpoint was plotted using a Kaplan-Meier curve (Prism software); statistically significant differences between the experimental groups were determined using log-rank Mantel-Cox test (P = 0.0002). Endpoint was defined by tumor reaching 2 cm in any direction or a volume of 4 cm3, ulceration, more than 10% weight loss, or inability to ambulate. (B) Effect of 2173 CART-EGFRvIII cells and UTD T cells compared to PBS on intra cranial tumor volume over time. Mice were injected intracranially with 5 × 104 U87-EGFRvIII cells on day 0; on day 7, mice were injected intravenously with 4 × 106 CAR T cells or a matched number of total UTD T cells or PBS. Plots show means ± SEM of the calculated %T/Δ_C_ for the days indicated. The study was terminated, and all mice were euthanized on day 18 due to the disease burden and condition of the mice in the PBS and UTD groups. n = 10 mice per group. (C) Magnetic resonance imaging (MRI) of intracranial U87-EGFRvIII tumors in mice 11 days after CAR T cell injection (day 18 of study). Arrows indicate tumor mass. n = 2 mice per group imaged with one representative image shown. (D) Numbers of CD4+ and CD8+ T cells in bone marrow (BM), spleen, and brain of intracranial tumor-bearing mice on day 18. Tissues were harvested, processed into a single-cell suspension, and stained with antibodies for enumeration of T cells by flow cytometry. Plots indicate means ± SEM number of T cells per million cells as measured by flow cytometry of each homogenized organ. Statistically significant differences were calculated by one-way ANOVA, using Dunnett's test (n = 8 mice per group; P values as indicated).
In an orthotopic model of glioblastoma, U87-EGFRvIII tumors were intracranially implanted into NSG mice (day 0), and 7 days later, mice were injected intravenously with PBS alone, untransduced human T cells (UTD), or 2173 CAR–expressing human T cells (2173). In this model, CAR T cells were given as a single agent. Tumors in the 2173 CAR group had a rapid reduction in volume (∼80%) relative to the PBS group 3 days after receiving T cells (Fig. 6B; the raw data and calculations are shown in table S2). MRI performed 11 days after T cell injection (day 18) demonstrated a large mass in both the PBS and UTD groups compared to the 2173 group (Fig. 6C). Eighteen days after tumor implantation (11 days after T cell injection), mice were euthanized to determine persistence and trafficking of T cells within the bone marrow, spleen, or brain. A significant (P < 0.05) and large increase in both CD4+ and CD8+ T cells was found in all three organs in the 2173 group compared to the UTD group (Fig. 6D), indicating engraftment and trafficking of CAR T cells in mice bearing antigen-expressing tumor. The injected CAR T cells were 80% CD4+ T cells and 20% CD8+ T cells, and both types of T cells infiltrated the tumor-bearing brain, with a predominance of CD8+ T cells. This was in contrast to the skin graft infiltration of cetux-CAR T cells, where the predominant infiltrate was composed of CD4+ T cells. The significance of this finding is unclear, although it is possible that passenger human immune cells were present in the skin graft (such as dendritic cells) and that these favored infiltration of CD4+ T cells over CD8+ T cells. In both models where T cells were given as a single agent, CART-EGFRvIII cells significantly delayed tumor progression and appeared to control tumor effectively, as measured by the most commonly used clinical modality (MRI) for evaluation of glioblastoma.
Discussion
The application of gene-modified T cell immunotherapy as a potentially curative therapy for a multitude of cancer types has been met with abundant enthusiasm (42). Although most success has resulted from clinical trials targeting CD19 for hematological malignancies (3–5, 43, 44), promising responses have also been observed in solid tumors when targeting NY-ESO-1 on melanoma and synovial cell sarcoma (45). Additional ongoing trials and preclinical studies are targeting PSMA (prostate-specific membrane antigen) for prostate cancers (46), ganglioside GD2 for neuroblastoma (47), mesothelin for lung cancers (30), and others. The greatest impediment to widespread application of CAR T cell therapies remains the selection of appropriate target antigens. Phase 1 trials in the field have revealed toxicities when targeting molecules expressed at near undetectable levels on vital organs, such as Her2/neu (7) or CAIX (6). Furthermore, despite the strong selective pressure that targeted T cells can impose, targeting nondriver or nononcogenic proteins may nevertheless result in tumor recurrence by antigen escape variants (5). The ideal CAR target would be a surface-expressed protein that is specific to malignant cells, is homogeneously expressed, and plays a critical role in maintaining onco genesis. EGFRvIII appears to meet most of these criteria; although its expression in glioblastoma is heterogeneous, the association of its expression with the cancer stem cell and its role in oncogenesis make it an attractive target (27). Nevertheless, immunologic escape is possible and has already been proposed to be an issue with anti-EGFRvIII pep-tide vaccination (48).
In this relatively new field, developed for the most part in the academic setting, most scFvs have been derived from published sequences of the heavy and light chains of murine mAbs. The degree of specificity and potential for cross-reactivity has only been minimally examined. Here, we sought to develop a novel scFv to target EGFRvIII and validate the selection, by first generating a large panel of humanized scFvs, and then performing a deep and extensive characterization of their affinity as soluble molecules and their binding affinity under cellular conditions, directed both to the target antigen and to the most likely cross-reactive antigen, EGFRwt. Humanization of the scFv has the potential benefit of increasing the persistence of CAR-modified T cells, which has been shown to be a key factor in the efficacy of CAR T cells directed to CD19 (3–5, 37, 49). Furthermore, if persistence wanes before eradication of antigen-expressing tumor, retreatment with humanized scFv–based CARs is less likely to result in immediate rejection or cause allergic responses than murine scFv–based CARs.
Of the eight humanized scFvs that were generated and tested for affinity, avidity, and specificity in biophysical assays and in vitro, we ultimately selected a relatively low-affinity candidate as the lead for clinical translation. Although the lead candidate (2173) had a relatively lower affinity than most scFvs in clinical use as CARs, the gain in specificity to binding EGFRvIII over EGFRwt was prioritized. Although the affinity of a specific CAR construct is likely to play a role in directing the functionality of the redirected T cell, evidence from targeting the CD22 molecule indicates that affinity is of relatively lesser importance than the site of epitope binding (50). Here, we also noted that despite the relatively lower affinity of the 2173 scFv compared to cetuximab, both CARs were able to induce T cell proliferation and cytokine production to similar degrees in response to antigen stimulation by targets expressing high levels of antigen. One limitation of this study is that we were unable to test the 2173 CAR T cells against glioblastoma targets expressing endogenous levels of EGFRvIII, because patient-derived glioblastoma cells from EGFRvIII+ tumors frequently lose their endogenous expression of EGFRvIII in culture (51, 52).
Given the single amino acid epitope junction that defines the specificity of EGFRvIII compared to the ubiquitously expressed EGFRwt, we sought to ensure the specificity of the lead scFv to the greatest possible extent. To this end, we tested the lead CART-EGFRvIII construct for reactivity against primary human tissues in vitro and in vivo. To our knowledge, the skin graft assay used in this study is the first time normal human tissue has been used in vivo to test the potential toxicities of CAR T cells. Nevertheless, the potential for EGFR-directed toxicity will be carefully monitored in human studies; the monitoring period could be prolonged, depending on the persistence of the CAR T cells. On the basis of the toxicity of cetuximab, the most likely toxicities that could indicate cross-reactivity are skin rash and diarrhea.
Xenogeneic mouse models are commonly used to test the efficacy of novel constructs of CAR T cells against human tumors. In this regard, we tested three mouse models: two orthotopic models, where human glioma tumor is implanted in the brain and CAR T cells are administered with or without chemotherapy, and a subcutaneous tumor model. Measurements of tumor in mouse models are often quantified with bioluminescent imaging, which is quite convenient and sensitive, but cannot be used clinically to assess antitumor responses. We measured tumor responses by bioluminescent imaging, caliper measurements of subcutaneous tumors, and MRI in orthotopic models. Of these, MRI is the only modality that can be used clinically to measure disease response in glioblastoma. When administered as a single agent, CART-EGFRvIII cells were able to eliminate orthotopically injected glioma by MRI, but deeper regression was observed when CART-EGFRvIII cells were administered with adjuvant temozolomide. Encouragingly, CART-EGFRvIII cells also seemed to traffic to the site of antigen in the brain, indicating that intravenous administration is likely a suitable route to target CNS tumors with T cells.
The main limitation of our study is that preclinical testing does not necessarily predict safety or efficacy in human trials. Here, the target glioblastoma cells used to test efficacy were derived from a single cell line (U87), and the only form of EGFRvIII antigen expression tested was by overexpression of the gene via stable transduction of target cells. In contrast, glioblastoma is a heterogeneous disease, and each tumor is composed of heterogeneous cell populations. Unfortunately, primary cultured glioblastoma cells are very challenging to obtain, and often lose EGFRvIII expression in culture. The T cells used in all the experiments were derived from healthy donors, which may also be more functional and have more favorable characteristics than those derived from patients with glioblastoma, who are often treated with lymphotoxic agents including temozolomide, radiation, and/or cortico steroids. Nevertheless, we feel that these data indicate compelling rationale to proceed with clinical testing.
Two other clinical trials are currently open using CAR T cells to treat glioblastoma. The first trial, conducted by Baylor College of Medicine, is targeting the Her2 antigen using cytomegalovirus-specific CAR T cells; the CAR design uses the CD28 signaling domain instead of 4-1BB (NCT01109095). The second trial, conducted by the National Cancer Institute (NCI), is also targeting EGFRvIII but differs from ours in vector type and CAR design; the NCI is using a gammaretrovirus vector to express the CAR, and the CAR design incorporates the signaling domains of CD28, 4-1BB, and CD3ζ (NCT01454596). On the basis of the data described here, we have opened a clinical trial that will test the efficacy of 2173 CART-EGFRvIII cells in patients with EGFRvIII+ glioblastoma (NCT02209376). Patients will be enrolled on one of two cohorts, depending on whether they have recurrent disease after primary resection and adjuvant therapy (cohort 1), or residual disease after primary resection (cohort 2). In the second cohort, CART-EGFRvIII cells are administered after radiation and concurrent with adjuvant temozolomide.
The University of Pennsylvania has entered into an alliance with Novartis Pharmaceuticals to develop CAR T cells as a platform for multiple cancers. The degree of in vitro and in vivo characterization described here sets a new standard for preclinical validation of novel CAR T cell products as they enter phase 1 trials in patients with cancer.
Materials and Methods
Study design
The purpose of this study was to design and rigorously test an optimal and specific EGFRvIII-directed CAR T cell product for potential use as a therapeutic agent in patients with EGFRvIII-expressing tumors such as glioblastoma. We tested a panel of CAR designs with different signaling domains in vitro and in vivo, and then varied and tested a large panel of humanized scFvs as antigen recognition domains in silico and in vitro. We verified the specificity of the lead CAR, focusing on potential cross-reactivity to the native EGFRwt protein, by testing CAR T cells against cultured cell lines, primary tissues, and skin-grafted mice. We tested efficacy in three different xenogeneic models, including two orthotopic models with and without standard chemotherapy. We measured tumors by calipers, bioluminescent imaging, and MRI. Thus, multiple modalities including in silico, in vitro, and multiple mouse models were tested. Each experiment was performed multiple times, with T cells derived from various normal donors. Furthermore, experiments were performed in three different institutions, and overall results with regard to transduction efficiencies, in vitro testing, and in vivo animal models were consistent.
Cell lines and culture
The human cell lines U87 and U87-EGFRvIII were provided by S. Chang (Northwestern University, Chicago, IL). These cell lines were lentivi rally transduced to express the click beetle green luciferase and green fluorescent protein (GFP) under control of the EF-1a promoter. At 48 hours after transduction, cells were sorted on an Influx cell sorter (BD Biosciences) on the basis of GFP expression to be 100% GFP positive and subsequently expanded. BHK-21 parental cells were purchased from the American Type Culture Collection and cultured in Eagle's modified Eagle's medium (EMEM) with 10% fetal bovine serum (FBS). For the BHK cell line derivatives, lentivirus was produced to express EGFRvIII or EGFRwt under the control of the EF-1α promoter along with blasticidin selection marker. The produced viruses were used to transduce BHK cells. Blasticidin-resistant cells were selected and FACS (fluorescence-activated cell sorting)-sorted for EGFRvIII-or EGFRwt-positive cells. BHK-EGFRwt and BHK-EGFRvIII cells were maintained in EMEM with 10% FBS and blasticidin (2 μg/ml). Primary human keratinocytes were purchased from the Dermatology Core Facility at the University of Pennsylvania.
Vector constructs
The 3C10.28BBz CAR was constructed by ligating the huCD8 leader-3C10scFv coding sequence into the Nhe I/Nhe I sites of pELNS SS128BBz. The 3C10.BBz CAR was constructed by ligating the Nhe I/Nhe I fragment containing the huCD8 leader-3C10scFv from 3C10.28BBz into the Nhe I/Nhe I sites of pELNS SS1.BBz. The nucleotide coding sequences of 139 scFv with the huCD8 leader were synthesized with 5′ Nhe I and 3′ Nhe I sites and ligated into Nhe I/Nhe I sites of pELNS SS1.BBz. Likewise, DNA for humanized EGFRvIII scFv constructs as well as for the cetuximab-based scFv control including the huCD8 leader sequence and part of the huCD8 hinge region were synthesized and subcloned into the pELNS vector using 5′ Bam HI and 3′ Bsp EI sites. The pELNS vectors have been described (53).
Recombinant proteins
Plasmids encoding the amino acids for EGFRwt ECD (EGFR-ECD), EGFRvIII-ECD, 2173 scFv, and 3C10 scFv constructs were synthesized at DNA 2.0 Inc. All proteins were synthesized with a C-terminal 8×His tag to facilitate purification. EGFR-ECD and EGFRvIII-ECD included an additional Avi-tag on the C terminus for site-selective biotinylation of the proteins upon cotransfection with BirA biotin ligase to mimic orientation of the natural ECD on the cell surface. Proteins were produced transiently in human embryonic kidney (HEK) 293F cells. For each construct, HEK293F cells were transfected with 1:3 ratio of plasmid/polyethylenimine at a density at 3 × 106 cells/ml. The cells were incubated at 37°C with 8% CO2 for 6 days. Proteins were harvested by centrifugation, and the supernatant was purified by binding to either Ni-NTA agarose beads (Qiagen) or Ni-Sepharose 6FF (GE Healthcare). The resin was washed, and the protein was step-eluted with imidazole and dialyzed against PBS.
Surface plasmon resonance
Binding affinities of the 2173 and 3C10 scFvs were measured on the Biacore T200 molecular interaction system (GE Healthcare). Biotinylated EGFRvIII-ECD and biotinylated EGFR-ECD were coupled to a streptavidin sensor chip at a target density of about 150 relative units. Purified scFv was injected over the chip under constant flow rate in threefold serial dilutions. Association and dissociation rates were monitored for 240 and 360 s, respectively. Samples were run in duplicate, double referencing was performed against a blank immobilized flow cell and a buffer blank, and data were fit using a steady-state affinity model with the Biacore T200 evaluation software.
Flow cytometry
For measurement of scFv binding to target cells, 293 cells were transiently transfected with a plasmid encoding either full-length EGFRvIII or EGFRwt protein, using a 1:3 ratio of plasmid to polyethylenimine at a density at 3 × 106 cells/ml. Forty-eight hours after transfection, purified, his-tagged scFvs (0.002 to 200 nM) were incubated with 293-EGFRvIII– or 293-EGFRwt–expressing cells. Binding was detected by the addition of PE-conjugated anti-his antibody (R&D Systems). After a short incubation, cells were washed twice and resuspended in PBS with 2% paraformaldehyde. Fluorescence was assessed using a BD LSR II flow cytometer, and data were analyzed with FlowJo software.
For detection of CAR expression, cells were stained using either biotinylated EGFRvIII or EGFRwt protein. Cells alone or expressing EGFRvIII CAR were pelleted, washed once with PBS, and resuspended in PBS containing 0.2% bovine serum albumin (FACS buffer) at 1 × 106 cells/ml. Biotinylated EGFRvIII protein was added to 100 μl of cells at a concentration of 1 μg/ml and incubated at 4°C for 30 min. Cells were washed twice with FACS buffer, and secondary detection was carried out by the addition of streptavidin-coupled PE (Jackson ImmunoResearch). After a short incubation, cells were washed twice and resuspended in PBS with 2% paraformaldehyde. Fluorescence was assessed using a BD LSR II flow cytometer, and data were analyzed with FlowJo software.
In vitro T cell transduction and cultures
Isolated T cells were derived from leukapheresis products obtained from deidentified healthy donors under an institutional review board–approved protocol. T cells were stimulated with Dynabeads Human T-Activator CD3/CD28 (Life Technologies) at a bead to cell ratio of 3:1 (first stimulation). T cells were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum, Hepes buffer (20 mM), and penicillin and streptomycin (1%). The end of first stimulation was determined on the basis of decrease of log-phase growth and reduction of mean lymphocytic volume to 300 to 330 fl as measured on a Coulter Multisizer. This is usually reached 10 days after stimulation, at which point cells are frozen down for functional assays or used for restimulation. T cells from the first stimulation were normalized to 30% CAR+ and cocultured with either U87-EGFRvIII or Dynabeads Human T-Activator CD3/CD28 at a 1:1 ratio (second stimulation). Target cells were irradiated with 100 Gy of γ-radiation before coculture.
T cell expansions were monitored via mean lymphocytic volume and total cell counts as described in the first stimulation. Upon resting, T cells from the second stimulation were normalized to 52% CAR+ and cocultured and expanded as described in the second stimulation. T cells in all three rounds of stimulations were maintained at a concentration of 0.7 × 106 cells/ml throughout the culture period. In the second and third stimulation period, IL-2 (100 IU/ml) was added every other day.
Cytotoxicity assays
We used three different kinds of cytotoxicity assays based on luciferase, flow cytometry, and chromium release. The ability of EGFRvIII-specific CAR T cells to kill targets was tested in a 20-hour luciferase-based killing assay. Both U87 and U87-vIII cell lines were engineered to stably express firefly luciferase. These engineered cell lines served as targets for the killing assay. The effector CAR T cells used were transduced to express the EGFRvIII-specific CARs 3C10 or 2173 and the control CART19. Transduced T cells and UTD were thawed and rested for 24 hours at 37°C in a six-well plate in T cell medium. The effectors and targets were mixed together at the indicated E:T ratios and cultured in black-walled 96-well flat-bottom plates with 3 × 104 target cells in a total volume of 200 μl per well in T cell medium. Target cells alone were seeded at the same cell density to determine the maximal luciferase expression (relative light units; RLUmax). After 20 hours, 100 μl of supernatant per well was removed, and 100 μl of luciferase substrate (Bright-Glo, Promega) was added to the remaining supernatant and cells. Emitted light was measured after 10 min of incubation using the EnVision plate reader. Lysis was determined as [1 – (RLUsample)/(RLUmax)] × 100. Two replicate experiments were performed; each was done in duplicate.
Flow-based cytotoxicity assays were performed with T cells harvested at the end of the first stimulation; untransduced and CD19.BBz-transduced CAR cells served as control T cells. 3C10.BBz, 3C10.28BBz, and 139.BBz were tested for efficacy of the murine anti-EGFRvIII CARs. U87MG (parental) and U87-EGFRvIII were harvested and labeled with 5 μM carboxyfluorescein diacetate succinimidyl ester (CFSE) (Life Technologies), then plated in a 24-well plate, and left to adhere overnight at 37°C, 5% O2. The following day, CAR+ T cells were normalized by CAR expression and cocultured with the tumors at 0.1:1, 0.33:1, 1:1, 3:1, 10:1, and 20:1 E:T ratios for 18 hours. All cells were harvested and stained with CD45 allophycocyanin (APC)–H7 to identify T cells and 7-aminoactinomycin D (7-AAD) for live/dead discrimination. Tumor cells were CFSE-positive. Counting beads were added to quantify the number of live tumor cells. Samples were acquired on LSR II, and data were analyzed with FlowJo v8.8.7 (TreeStar). Lysis was determined as [(% lysis sample – % lysis minimum)/(% lysis max [100%] – % lysis minimum)] × 100%. Two replicate experiments were performed; each was done in duplicate.
Cytotoxicity of the CAR-expressing T cells was also tested in a 4-hour 51Cr release assay. Target cells were labeled with 51Cr for 1 hour at 37°C. Radioactive 51Cr (50 μCi) was used to label 1 × 106 target cells. After labeling, cells were washed with 10 ml of non–phenol red RPMI medium plus 5% FBS twice and resuspended at 10 × 105 cells/ml. One hundred microliters of labeled target cells (n = 5000) was plated in each well of a 96-well plate. Effector cells were added in a volume of 100 μl at different E:T ratios. Effector and targets were incubated together for 4 hours at 37°C. Supernatant (30 μl) from each well was collected and transferred onto the filter of a LumaPlate. The filter was allowed to dry overnight. Radioactivity released in the culture medium was measured using a β-emission reading liquid scintillation counter. Percentage specific lysis was calculated as follows: (sample counts – spontaneous counts)/(maximum counts – spontaneous counts) × 100.
Intracellular cytokine analysis
CAR-transduced or untransduced T cells were cocultured with target cells (tumors, cell lines, or human primary cells) in a 1:1 ratio at 2 × 106/ml in 96-well round bottom tissue culture plates at 37°C, 5% CO2 for 16 hours in RPMI 1640 plus 10% FBS in the presence of CD107a mAb and Golgi inhibitors monensin and brefeldin A. Cells were washed, stained with live/dead viability stain, followed by surface staining for CD3 and CD8, then fixed and permeabilized, and intracellularly stained for IFN-γ, TNF-α, IL-2, granzyme B, and CD154. Cells were analyzed by nine-color flow cytometry (Becton Dickinson Fortessa or LSR II) and gated on live, single-cell lymphocytes and CD3-positive lymphocytes (supplementary data for Fig. 5A).
Enzyme-linked immunosorbent assay
Thawed and rested (see Cytotoxicity assays) CAR T cells were cocultured with target-expressing cells for 24 hours at an E:T ratio of 1:1. The following effector cells were used: T cells transduced to express the EGFRvIII-specific CARs 3C10 or 2173, the EGFR-specific cetux-CAR, or the control CART19. Target cells were BHK parental cells, BHK cells overexpressing human EGFRwt protein (BHK-wt), and BHK cells overexpressing human EGFRvIII protein (BHK-vIII), as well as the human glioblastoma cell line U87MG (U87), which endogenously expresses the EGFRwt protein, and U87MG cells overexpres sing EGFRvIII (U87-vIII). Cells were cultured in 96-well U-bottom plates with 3 × 104 (BHK lines) or 1.7 × 104 (U87 lines) target cells in a total volume of 200 μl per well in T cell medium. After 24 hours, supernatants were removed from the cultures and IFN-γ secretion was quantified by ELISA (eBioscience). Alternatively, IFN-γ, IL-2, and TNF-α were quantified by CBA (BD Biosciences).
Proliferation assay
Assessment of antigen-mediated proliferation was performed in 96-well microtiter plates by mixing 2.5 × 104 washed T cells with 2.5 × 104 parental BHK cells or BHK cells expressing EGFRvIII (BHK-vIII) or EGFRwt (BHK-wt) for a final T cell/BHK cell ratio of 1:1. CAR T cell populations were normalized to equivalent percentages of CAR+ cells before plating. BHK cells were irradiated at 10,000 rads before use. T cells were enumerated in cultures using CountBright fluorescent beads (Invitrogen) and analyzed on a BD LSR II as described by the manufacturer.
Mouse models
All mouse experiments were conducted according to Institutional Animal Care and Use Committee (IACUC)–approved protocols. For orthotopic models, 1 × 10 U87-luc-EGFRvIII cells were implanted intracranially into 6- to 8-week-old female NSG mice (Jackson), with 10 mice per group. The surgical implants were done using a stereotactic surgical setup with tumor cells implanted 1 mm right and 1 mm anterior to the bregma and 3 mm into the brain. Before surgery and for 3 days after surgery, mice were treated with an analgesic and monitored for adverse symptoms in accordance with the IACUC. Mice were injected with CAR T cells in 100 μl of PBS intravenously via the tail vein. Bioluminescent measurements are used as a surrogate for tumor volume. %T/Δ_C_ was calculated for each measurement after CART cells were dosed. Percent treatment/control (T/C) values were calculated using the following formula:
%Regression=100×T/TinitialifT<0
where T is the individual tumor volumes of the mice in the drug-treated group on the final day of the study; T_initial is the individual tumor volumes of the mice in the drug-treated group on initial day of dosing; Δ_T is the individual tumor volumes of the mice in the drug-treated group on the final day of the study minus the individual tumor volumes of the mice in the drug-treated group on the initial day of dosing C is the mean tumor volume of the control group on the final day of the study; and ΔC is the mean tumor volume of the control group on the final day of the study minus the mean tumor volume of the control group on the initial day of dosing. T/C values in the range of 100 to 42% are interpreted to have no or minimal antitumor activity. T/C values that are ≤42% and >10% are interpreted to have antitumor activity or tumor growth inhibition. T/C values ≤10% or regression values ≥–10% are interpreted to be tumor stasis. Regression values <–10% are reported as regression.
In subcutaneous models, NSG mice were injected with 5.0 × 105 U87-v III/Luc+ tumors subcutaneously in 100 μ l of PBS on day 0. Tumor progression was evaluated by luminescence emission on a Xenogen IVIS Spectrum after intraperitoneal d-luciferin injection according to the manufacturer's directions (GoldBio). Additionally, tumor size was measured by calipers in three dimensions, L × W × H, for the duration of the experiment. Seven days later, CAR T cells were stained with anti-idiotype (goat anti-mouse or goat anti-human) antibodies, and CAR expression was normalized by addition of donor-matched untransduced T cells. Mice were treated with 1.0 × 10 CAR T cells or a matched number of untransduced T cells intravenously via tail vein in 100 μl of PBS. Survival was followed over time until predetermined IACUC-approved endpoint was reached (n = 8 to 10 mice per group). The experiment was repeated twice.
In skin-grafted models, 8- to 10-week-old male NSG mice (Jackson) were surgically grafted with human neonatal foreskin obtained from the Penn Skin Disease Research Center. After complete healing of the skin graft (about 6 weeks), mice were injected with 2 × 10 CAR T cells intravenously. Skin grafts were monitored daily for 2 weeks and surgically excised at day 15. Skin punch biopsies were taken 3 days after T cell injection. Formalin-fixed paraffin-embedded tissue was cut and stained by standard H&E and by immunohistochemistry with antibodies to CD3, CD4, and CD8.
MRI of mice
In orthotopic mouse models, tumors were evaluated with a 7-T MRI machine at a single time point 18 days after tumor implantation. For each animal imaged, 20 to 30 slices were taken in the coronal orientation of the brain. Each slice was 0.5 mm thick and was T2-weighted. The images were used to estimate the solid tumor volume in the brain.
Statistical methods
Data are presented as means ± SD or SEM as stated in the figure legends. Statistically significant differences were tested by specific tests as indicated in the figure legends. Survival curves were analyzed with log-rank test as stated in the figure legends. The number of animals per group in each experiment was determined on the basis of previous statistical analyses by our group and by the Novartis Pharmacology team. All exact p values are provided in the figures or their legends. All statistical analyses were performed with Prism software version 6.0 (GraphPad).
Supplementary Material
supplement figure
Fig. S1. Untransduced T cells stimulated with U87vIII tumor cells for 16 hours.
supplement table 1
Table S1. Source data for Fig. 6A (excel).
supplement table 2
Table S2. Source data for Fig. 6B (excel).
Acknowledgments
We thank S. Chang for providing the U87 and U87-EGFRvIII cell lines.
Funding: This work was supported by funding from the NIH (DP2CA174502; L.A.J.), NCI (K08-166039; M.V.M.), American Society of Clinical Oncology Conquer Cancer Foundation Young Investigator Award (M.V.M.), and the Novartis Institutes for Biomedical Research as part of an alliance with the University of Pennsylvania.
Footnotes
Author contributions: L.A.J., J.S., A.L., R.S., G.P., J.T.S., H.O., J.L.B., and M.V.M. designed the experiments. L.A.J., J.S., T.O., A.K., P.R.P., S.E.M., A.K.N., P.T., A.L., A.C.B., A.P.C., T.C., J.A.F., C.C.K., A.D.P., B.E., R.S., T.E., N.I., M.H.R., N.L., and L.Z. performed the experiments. L.A.J., C.H.J., and M.V.M. provided funding. All the authors contributed to the writing and editing of the manuscript.
Competing interests: The University of Pennsylvania and Novartis hold a patent in the use of CAR T cells in oncology. L.A.J., C.H.J., H.O., J.L.B., J. Scholler, A.L., and M.V.M. are inventors on a patent related to these data.
References
- 1.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor–modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, Milone MC, Levine BL, June CH. Chimeric antigen receptor–modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, Gratama JW, Stoter G, Oosterwijk E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: First clinical experience. J Clin Oncol. 2006;24:e20–e22. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 7.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM, Phan GQ, Hughes MS, Kammula US, Miller AD, Hessman CJ, Stewart AA, Restifo NP, Quezado MM, Alimchandani M, Rosenberg AZ, Nath A, Wang T, Bielekova B, Wuest SC, Akula N, McMahon FJ, Wilde S, Mosetter B, Schendel DJ, Laurencot CM, Rosenberg SA. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36:133–151. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hatanpaa KJ, Burma S, Zhao D, Habib AA. Epidermal growth factor receptor in glioma: Signal transduction, neuropathology, imaging, and radioresistance. Neoplasia. 2010;12:675–684. doi: 10.1593/neo.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gupta P, Han SY, Holgado-Madruga M, Mitra S, Li G, Nitta R, Wong A. Development of an EGFRvIII specific recombinant antibody. BMC Biotechnol. 2010;10:72. doi: 10.1186/1472-6750-10-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong A, Bigner S, Bigner D, Kinzler K, Hamilton S, Vogelstein B. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc Natl Acad Sci U S A. 1987;84:6899–6903. doi: 10.1073/pnas.84.19.6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friguet B, Chaffotte A, Djavadi-Ohaniance L, Goldberg M. Measurements of the true affinity constant in solution of antigen-antibody complexes by enzyme-linked immunosorbent assay. J Immunol Methods. 1985;77:305–319. doi: 10.1016/0022-1759(85)90044-4. [DOI] [PubMed] [Google Scholar]
- 13.Mukasa A, Wykosky J, Ligon K, Chin L, Cavenee W, Furnari F. Mutant EGFR is required for maintenance of glioma growth in vivo, and its ablation leads to escape from receptor dependence. Proc Natl Acad Sci U S A. 2010;107:2616–2621. doi: 10.1073/pnas.0914356107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feldkamp MM, Lala P, Lau N, Roncari L, Guha A. Expression of activated epidermal growth factor receptors, Ras-guanosine triphosphate, and mitogen-activated protein kinase in human glioblastoma multiforme specimens. Neurosurgery. 1999;45:1442–1453. doi: 10.1097/00006123-199912000-00034. [DOI] [PubMed] [Google Scholar]
- 15.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 16.Chi AS, Norden AD, Wen PY. Antiangiogenic strategies for treatment of malignant gliomas. Neurotherapeutics. 2009;6:513–526. doi: 10.1016/j.nurt.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verhoeff J, van Tellingen O, Claes A, Stalpers L, van Linde M, Richel D, Leenders W, van Furth W. Concerns about anti-angiogenic treatment in patients with glioblastoma multiforme. BMC Cancer. 2009;9:444. doi: 10.1186/1471-2407-9-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sathornsumetee S, Reardon DA. Targeting multiple kinases in glioblastoma multiforme. Expert Opin Investig Drugs. 2009;18:277–292. doi: 10.1517/13543780802692603. [DOI] [PubMed] [Google Scholar]
- 19.Okada H, Kohanbash G, Zhu X, Kastenhuber ER, Hoji A, Ueda R, Fujita M. Immunotherapeutic approaches for glioma. Crit Rev Immunol. 2009;29:1–42. doi: 10.1615/critrevimmunol.v29.i1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujita M, Okada H. In: Brain Tumors—Current and Emerging Therapeutic Strategies. Abujamra AL, editor. InTech; Rijeka, Croatia: 2011. [Google Scholar]
- 21.Palma L, Di Lorenzo N, Guidetti B. Lymphocytic infiltrates in primary glioblastomas and recidivous gliomas. Incidence, fate, and relevance to prognosis in 228 operated cases. J Neurosurg. 1978;49:854–861. doi: 10.3171/jns.1978.49.6.0854. [DOI] [PubMed] [Google Scholar]
- 22.Brooks WH, Markesbery WR, Gupta GD, Roszman TL. Relationship of lymphocyte invasion and survival of brain tumor patients. Ann Neurol. 1978;4:219–224. doi: 10.1002/ana.410040305. [DOI] [PubMed] [Google Scholar]
- 23.Swartz AM, Li QJ, Sampson JH. Rindopepimut: A promising immunotherapeutic for the treatment of glioblastoma multiforme. Immunotherapy. 2014;6:679–690. doi: 10.2217/imt.14.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dixit S. Immunotherapy for high-grade glioma. Future Oncol. 2014;10:911–915. doi: 10.2217/fon.14.20. [DOI] [PubMed] [Google Scholar]
- 25.Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ, Binder-Scholl GK, Smethurst DP, Gerry AB, Pumphrey NJ, Bennett AD, Brewer JE, Dukes J, Harper J, Tayton-Martin HK, Jakobsen BK, Hassan NJ, Kalos M, June CH. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122:863–871. doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, Grand F, Brewer JE, Gupta M, Plesa G, Bossi G, Vuidepot A, Powlesland AS, Legg A, Adams KJ, Bennett AD, Pumphrey NJ, Williams DD, Binder-Scholl G, Kulikovskaya I, Levine BL, Riley JL, Varela-Rohena A, Stadtmauer EA, Rapoport AP, Linette GP, June CH, Hassan NJ, Kalos M, Jakobsen BK. Identification of a Titin-derived HLA-A1–presented peptide as a cross-reactive target for engineered MAGE A3–directed T cells. Sci Transl Med. 2013;5:197ra103. doi: 10.1126/scitranslmed.3006034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, Feldman SA, Chinnasamy N, Kuan CT, Song H, Zhang W, Fine HA, Rosenberg SA. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther. 2012;23:1043–1053. doi: 10.1089/hum.2012.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sampson JH, Choi BD, Sanchez-Perez L, Suryadevara CM, Snyder DJ, Flores CT, Schmittling RJ, Nair SK, Reap EA, Norberg PK, Herndon JE, II, Kuan CT, Morgan RA, Rosenberg SA, Johnson LA. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Cancer Res. 2014;20:972–984. doi: 10.1158/1078-0432.CCR-13-0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohno M, Ohkuri T, Kosaka A, Tanahashi K, June CH, Natsume A, Okada H. Expression of miR-17-92 enhances anti-tumor activity of T-cells transduced with the anti-EGFRvIII chimeric antigen receptor in mice bearing human GBM xenografts. J Immunother Cancer. 2013;1:21. doi: 10.1186/2051-1426-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maus MV, Haas AR, Beatty GL, Albelda SM, Levine BL, Liu X, Zhao Y, Kalos M, June CH. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res. 2013;1:26–31. doi: 10.1158/2326-6066.CIR-13-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jungbluth AA, Stockert E, Huang HJ, Collins VP, Coplan K, Iversen K, Kolb D, Johns TJ, Scott AM, Gullick WJ, Ritter G, Cohen L, Scanlan MJ, Cavenee WK, Old LJ. A monoclonal antibody recognizing human cancers with amplification/overexpression of the human epidermal growth factor receptor. Proc Natl Acad Sci U S A. 2003;100:639–644. doi: 10.1073/pnas.232686499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patel D, Lahiji A, Patel S, Franklin M, Jimenez X, Hicklin DJ, Kang X. Monoclonal antibody cetuximab binds to and down-regulates constitutively activated epidermal growth factor receptor vIII on the cell surface. Anticancer Res. 2007;27:3355–3366. [PubMed] [Google Scholar]
- 33.Maus MV, Fraietta JA, Levine BL, Kalos M, Zhao Y, June CH. Adoptive immunotherapy for cancer or viruses. Annu Rev Immunol. 2014;32:189–225. doi: 10.1146/annurev-immunol-032713-120136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanchez-Perez LA, Choi BD, Archer GE, Cui X, Flores C, Johnson LA, Schmittling RJ, Snyder D, Herndon JE, II, Bigner DD, Mitchell DA, Sampson JH. Myeloablative temozolomide enhances CD8+ T-cell responses to vaccine and is required for efficacy against brain tumors in mice. PLOS One. 2013;8:e59082. doi: 10.1371/journal.pone.0059082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, White DE, Wunderlich JR, Canevari S, Rogers-Freezer L, Chen CC, Yang JC, Rosenberg SA, Hwu P. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12:6106–6115. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, Qian X, James SE, Raubitschek A, Forman SJ, Gopal AK, Pagel JM, Lindgren CG, Greenbergt PD, Riddell SR, Press OW. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 2008;12:2261–2271. doi: 10.1182/blood-2007-12-128843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR, Lee CC, Restifo NP, Schwarz SL, Cogdill AP, Bishop RJ, Kim H, Brewer CC, Rudy SF, VanWaes C, Davis JL, Mathur A, Ripley RT, Nathan DA, Laurencot CM, Rosenberg SA. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM, Hughes MS, Kammula US, Phan GQ, Lim RM, Wank SA, Restifo NP, Robbins PF, Laurencot CM, Rosenberg SA. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19:620–626. doi: 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levine BL, Bernstein WB, Connors M, Craighead N, Lindsten T, Thompson CB, June CH. Effects of CD28 costimulation on long-term proliferation of CD4+ T cells in the absence of exogenous feeder cells. J Immunol. 1997;159:5921–5930. [PubMed] [Google Scholar]
- 41.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, Campana D, Riley JL, Grupp SA, June CH. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17:1453–1464. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. 2013;342:1432–1433. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 43.Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, Olszewska M, Bernal Y, Pegram H, Przybylowski M, Hollyman D, Usachenko Y, Pirraglia D, Hosey J, Santos E, Halton E, Maslak P, Scheinberg D, Jurcic J, Heaney M, Heller G, Frattini M, Sadelain M. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, Qu J, Wasielewska T, He Q, Fink M, Shinglot H, Youssif M, Satter M, Wang Y, Hosey J, Quintanilla H, Halton E, Bernal Y, Bouhassira DC, Arcila ME, Gonen M, Roboz GJ, Maslak P, Douer D, Frattini MG, Giralt S, Sadelain M, Brentjens R. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra225. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL, Kammula US, Hughes MS, Restifo NP, Raffeld M, Lee CC, Levy CL, Li YF, El-Gamil M, Schwarz SL, Laurencot C, Rosenberg SA. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Santoro S, Kim S, Motz G, Alatzoglou D, Li C, Irving M, Powell D, Jr, Coukos G. T cells bearing a chimeric antigen receptor against prostate-specific membrane antigen mediate vascular disruption and result in tumor regression. Cancer Immunol Res. 2015;3:68–84. doi: 10.1158/2326-6066.CIR-14-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, Liu H, Wu MF, Gee AP, Mei Z, Rooney CM, Heslop HE, Brenner MK. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6056. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, Gilbert MR, Herndon JE, II, Mc Lendon RE, Mitchell DA, Reardon DA, Sawaya R, Schmittling RJ, Shi W, Vredenburgh JJ, Bigner DD. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28:4722–4729. doi: 10.1200/JCO.2010.28.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, Kamble RT, Bollard CM, Gee AP, Mei Z, Liu H, Grilley B, Rooney CM, Heslop HE, Brenner MK, Dotti G. CD28 costimulation improves expansion and persistence of chimeric antigen receptor–modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, Dimitrov DS, Morgan RA, Fitz Gerald DJ, Barrett DM, Wayne AS, Mackall CL, Orentas RJ. Anti-CD22–chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121:1165–1174. doi: 10.1182/blood-2012-06-438002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pandita A, Aldape KD, Zadeh G, Guha A, James CD. Contrasting in vivo and in vitro fates of glioblastoma cell subpopulations with amplified EGFR. Genes Chromosomes Cancer. 2004;39:29–36. doi: 10.1002/gcc.10300. [DOI] [PubMed] [Google Scholar]
- 52.Schulte A, Gunther HS, Martens T, Zapf S, Riethdorf S, Wulfing C, Stoupiec M, Westphal M, Lamszus K. Glioblastoma stem–like cell lines with either maintenance or loss of high-level EGFR amplification, generated via modulation of ligand concentration. Clin Cancer Res. 2012;18:1901–1913. doi: 10.1158/1078-0432.CCR-11-3084. [DOI] [PubMed] [Google Scholar]
- 53.Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, Varela-Rohena A, Haines KM, Heitjan DF, Albelda SM, Carroll RG, Riley JL, Pastan I, June CH. Control large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. 2009;106:3360–3365. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplement figure
Fig. S1. Untransduced T cells stimulated with U87vIII tumor cells for 16 hours.
supplement table 1
Table S1. Source data for Fig. 6A (excel).
supplement table 2
Table S2. Source data for Fig. 6B (excel).