Integrative clinical genomics of advanced prostate cancer (original) (raw)
. Author manuscript; available in PMC: 2016 May 21.
SUMMARY
Toward development of a precision medicine framework for metastatic, castration resistant prostate cancer (mCRPC), we established a multi-institutional clinical sequencing infrastructure to conduct prospective whole exome and transcriptome sequencing of bone or soft tissue tumor biopsies from a cohort of 150 mCRPC affected individuals. Aberrations of AR, ETS genes, TP53 and PTEN were frequent (40–60% of cases), with TP53 and AR alterations enriched in mCRPC compared to primary prostate cancer. We identified novel genomic alterations in PIK3CA/B, R-spondin, BRAF/RAF1, APC, β-catenin and ZBTB16/PLZF. Aberrations of BRCA2, BRCA1 and ATM were observed at substantially higher frequencies (19.3% overall) than seen in primary prostate cancers. 89% of affected individuals harbored a clinically actionable aberration including 62.7% with aberrations in AR, 65% in other cancer-related genes, and 8% with actionable pathogenic germline alterations. This cohort study provides evidence that clinical sequencing in mCRPC is feasible and could impact treatment decisions in significant numbers of affected individuals.
Keywords: precision oncology, whole exome sequencing, transcriptome sequencing, prostate cancer genomics
INTRODUCTION
Prostate cancer is among the most common adult malignancies, with an estimated 220,000 US men diagnosed yearly (ACS, 2015). Some men will develop metastatic prostate cancer and receive primary androgen deprivation therapy (ADT). However, nearly all men with metastatic prostate cancer develop resistance to primary ADT, a state known as metastatic castration resistant prostate cancer (mCRPC). Multiple “second generation” ADT treatments, like abiraterone acetate (de Bono et al., 2011; Ryan et al., 2013) and enzalutamide (Beer et al., 2014; Scher et al., 2012), have emerged for mCRPC affected individuals; however, nearly all affected individuals will also develop resistance to these agents. In the US, an estimated 30,000 men die of prostate cancer yearly.
Multiple studies have identified recurrent somatic mutations, copy number alterations, and oncogenic structural DNA rearrangements (chromoplexy) in primary prostate cancer (Baca et al., 2013; Barbieri et al., 2012; Berger et al., 2011; Cooper et al., 2015; Pflueger et al., 2011; Taylor et al., 2010; Tomlins et al., 2007; Wang et al., 2011). These include point mutations in SPOP, FOXA1, and TP53; copy number alterations involving MYC, RB1, PTEN, and CHD1; and E26 transformation-specific (ETS) fusions, among other biologically relevant genes. While certain primary prostate cancer alterations or signatures have prognostic clinical significance (Hieronymus et al., 2014; Lalonde et al., 2014), the therapeutic impact of primary prostate cancer genomic events has not yet been realized.
Genomic studies of metastatic prostate cancers demonstrated additional alterations in AR (Taplin et al., 1995) and in the androgen signaling pathway (Beltran et al., 2013; Grasso et al., 2012; Gundem et al., 2015; Hong et al., 2015), although these studies were performed predominantly using autopsy samples or preclinical models with limited cohort sizes. Prospective genomic characterization of fresh biopsy samples from living mCRPC affected individuals has been limited due to challenges in obtaining adequate tumor tissue, especially from bone biopsies (Mehra et al., 2011; Van Allen et al., 2014a), which is the most common site of metastatic disease. Thus, the landscape of genomic alterations in mCRPC disease remains incompletely characterized. Moreover, the low frequency of actionable genomic alterations in primary prostate cancer has limited the inclusion of mCRPC among cohorts wherein precision cancer medicine approaches have been piloted to guide treatment or clinical trial enrollment.
We conducted a systematic and multi-institutional study of mCRPC tumors obtained from living affected individuals to determine the landscape of somatic genomic alterations in this cohort, dissect genomic differences between primary prostate cancer and mCRPC, and discover the potential relevance of these findings from a biological and clinical perspective.
RESULTS
Clinical, biopsy, and pathology parameters
An international consortium consisting of eight academic medical center clinical sites was established to capture fresh clinical mCRPC affected individual samples as part of standard-of-care approaches or through a cohort of prospective clinical trials (Fig. 1A, B). Standard-of-care approaches for mCRPC included abiraterone acetate or enzalutamide. Clinical trials included in this study focused on combination strategies involving abiraterone acetate or enzalutamide, inhibitors of poly ADP ribose polymerase (PARP), or inhibitors of aurora kinase. Here we report the results of genomic profiling from mCRPC biopsy samples obtained at time of entry into the cohort study. Future reports will include longitudinal clinical data such as treatment response. The consortium utilized two sequencing and analysis centers, one centralized digital pathology review center, and one centralized data visualization portal (Cerami et al., 2012; Gao et al., 2013; Robinson et al., 2011; Thorvaldsdottir et al., 2013). Cross-validation of sequencing data from the two original sequencing sites demonstrated comparable variant calls for adequately powered genetic loci (Van Allen et al, In preparation).
Figure 1. Overview of the SU2C-PCF IDT multi-institutional clinical sequencing of mCRPC project.
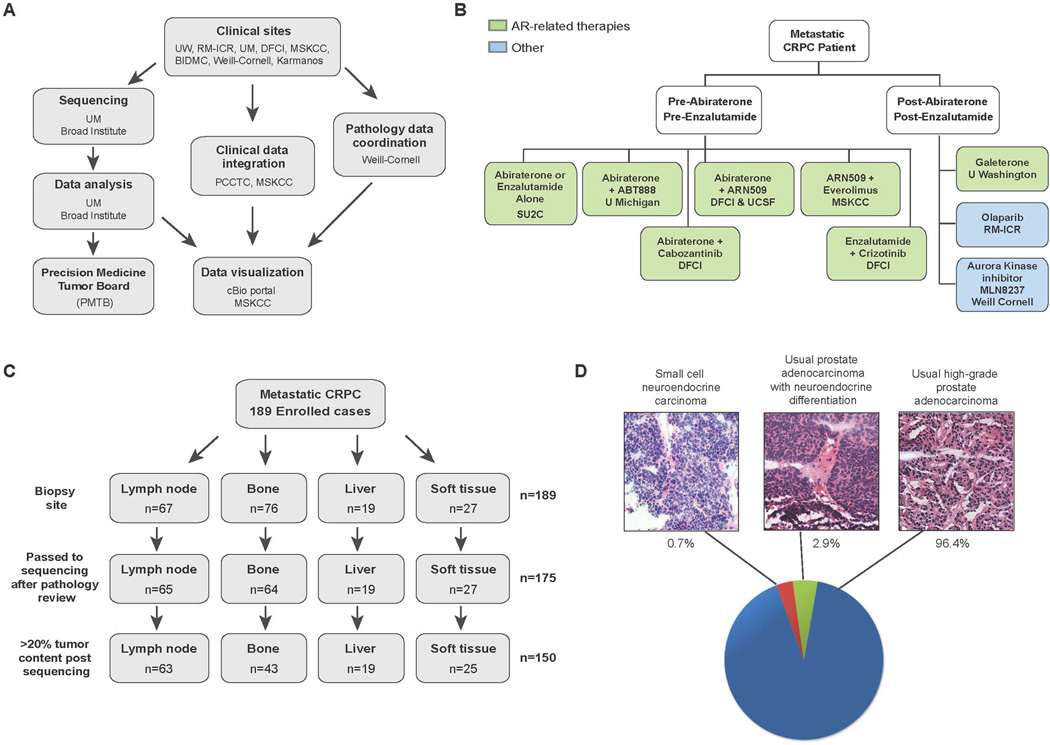
A, Schema of multi-institutional clinical sequencing project work flow. B, Clinical trials associated with the SU2C-PCF mCRPC project. C, Biopsy sites of the samples used for clinical sequencing. D, Histopathology of the cohort. Representative images of morphological analysis of mCRPC are shown along with prevalence in our cohort.
Here we describe 150 affected individuals with metastatic disease with complete integrative clinical sequencing results (whole exome, matched germline, and transcriptome data) (Fig. 1C), and summarized in Supp. Table S1. One hundred and eighty-nine affected individuals were enrolled in the study and 175 cases were sequenced after pathology review and assessment of tumor content. Of these, 150 biopsies had >20% tumor content as defined by computational analysis, based on mutant allele variant fractions and zygosity shifts. The biopsies sequenced were from lymph node (42%), bone (28.7%), liver (12.7%) and other soft tissues (16.7%). Baseline clinical information is available in Supp. Table S2. A majority of cases (96.4%) displayed typical high-grade prostate adenocarcinoma features while 2.9% of cases showed neuroendocrine differentiation. One case (0.7%) exhibited small cell neuroendocrine features (Epstein et al., 2014) (Fig. 1D).
Landscape of mCRPC alterations
Somatic aberrations in a panel of 38 statistically or clinically significant genes are illustrated in Fig. 2. Mean target coverage for tumor exomes was 160X and for matched normal exomes was 100X. While the average mutation rate for mCRPC was 4.4 mutations/Mb, there were four cases that exhibited a mutation rate of nearly 50 per Mb, three of which are likely due to alterations in the mismatch repair genes MLH1 and MSH2 as discussed later.
Figure 2. Integrative landscape analysis of somatic and germline aberrations in metastatic CRPC obtained through DNA and RNA sequencing of clinically obtained biopsies.

Columns represent individual patients and rows represent specific genes grouped in pathways. Mutations per Mb shown in the upper histogram while incidence of aberrations in the cohort is in the right histogram. Copy number variations (CNVs) common to mCRPC are shown in in the lower matrix with pink representing gain and light blue representing loss. Color legend of the aberrations represented including amplification, 2 copy loss, 1 copy loss, copy neutral loss of heterozygosity (LOH), splice site mutation, frameshift mutation, missense mutation, in-frame indel, and gene fusion. Cases with more aberration in a gene are represented by split colors.
Frequent copy number gains of 8q as well as copy number losses of 8p, 13q, 16q, and 18q were also observed. The mean number of identified biologically relevant genetic aberrations per case was 7.8 (Fig. 2). All mutations identified are presented in Supp. Table S3. The landscape of copy number alterations demonstrated expected recurrent amplification peaks (frequent AR, 8q gain) and deletion peaks (CHD1, PTEN, RB1, TP53) (Fig. 3A). Additional frequent focal amplifications were observed in regions encompassing CCND1 and PIK3CA and PIK3CB. A novel recurrent focal homozygous deletion event was observed in chr11q23, encompassing the transcriptional repressor ZBTB16.
Figure 3. Classes of genomic aberrations seen in mCPRC.
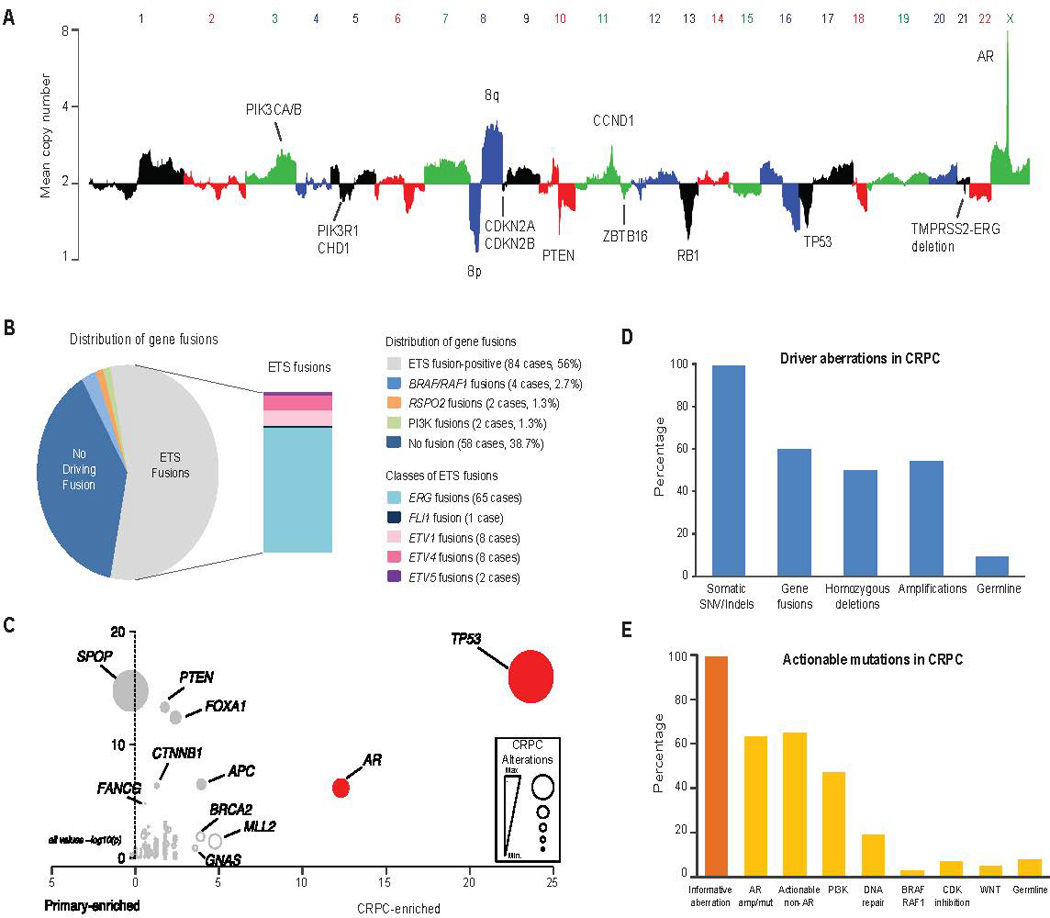
A, Copy number landscape of the SU2C-PCF mCRPC cohort. Individual chromosomes are represented by alternating colors and key aberrant genes are indicated. B, The gene fusion landscape of mCRPC. Pie chart of all driver fusions identified and the box plot represents specific ETS fusions. C, Mutations enriched in mCRPC relative to hormone naïve primary prostate cancer. Primary prostate cancer data derived from published studies (Barbieri et al., 2012) (TCGA, Provisional 2015). Level of CRPC enrichment represented by the×axis and MutSig CRPC significance analysis provided by the y axis. Diameters are proportional to the number of cases with the specific aberration. Genes of interest are highlighted. D, Classes of driver aberrations identified in mCRPC. E, Classes of clinically actionable mutations identified in mCRPC.
To identify gene fusions, analysis of 215 transcriptome libraries derived from the 150 tumor RNAs was performed and identified 4122 chimeras with at least 4 reads spanning the fusion junction. These fusion junctions resulted from 2247 unique gene pairs, an average of 15 gene fusions per tumor (Supplemental Table S4). Among chimeric fusion transcripts identified, recurrent ETS fusions (Tomlins et al., 2005) were observed in 84 cases (56%), with a majority of these fusions to ERG and also novel fusions to FLI1, ETV4, and ETV5 (Fig. 3B). In addition, potential clinically actionable fusions (involving BRAF, RAF1, PIK3CA/B, or RSPO2) were seen in eight cases (Fig. S1 and covered subsequently).
To place the mCRPC mutation landscape in the context of primary prostate cancer somatic genomics, we performed a selective enrichment analysis to compare somatic point mutations and short insertion/deletions observed in this cohort with those observed in somatic whole exome mutation data from 440 primary prostate cancer exomes (Barbieri et al., 2012)(TCGA Provisional Data, 2015) (Fig. 3C, Supp. Table S5). Focusing on genes previously implicated in cancer (n = 550), somatic TP53 mutations were the most selectively mutated (q < 0.001; Benjamini-Hochberg), followed by AR, KMT2D, APC, BRCA2, and GNAS (q < 0.1; Benjamini-Hochberg; Supp. Table S6). Both AR and GNAS were mutated exclusively in mCRPC. We found no genes selectively mutated in primary prostate cancer compared to mCRPC.
We identified an established biological “driver” aberration in a cancer-related gene (i.e., known oncogene or tumor suppressor; Supp. Table S7) in nearly all the cases (Fig. 3D). While 99% of the mCPRC cases harbored a potential driver single nucleotide variant (SNV) or indel, other classes of driver aberrations were also highly prevalent. These include driver gene fusions in 60%, driver homozygous deletions in 50% and driver amplifications in 54%. While informative mutations were present in virtually all mCRPC cases, 63% harbored aberrations in AR, an expected finding in castrate resistant disease but with higher frequency than in prior reports (Fig. 3E). Interestingly, even when AR was not considered, 65% of cases harbored a putatively clinically actionable alteration (defined as predicting response or resistance to a therapy, having diagnostic or prognostic utility across tumor types) (Supp. Table S8) (Roychowdhury et al., 2011; Van Allen et al., 2014c). Non-AR related clinically actionable alterations included aberrations in the PI3K pathway (49%), DNA repair pathway (19%), RAF kinases (3%), CDK inhibitors (7%) and the WNT pathway (5%). In addition to somatic alterations, clinically actionable pathogenic germline variants were seen in 8% of mCRPC affected individuals, potentially emphasizing the need for genetic counseling in affected individuals with prostate cancer.
Genomically aberrant pathways in mCRPC
Integrative analysis using both biological and statistical frameworks (Lawrence et al., 2014; Lawrence et al., 2013) of somatic point mutations, short insertion/deletions, copy number alterations, fusion transcripts, and focused germline variant analysis identified discrete molecular subtypes of mCRPC (Fig. 2). These subtypes were classified based on alteration clustering and existing biological pathway knowledge, and implicated the AR signaling pathway, phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K), WNT, DNA repair, cell cycle, and chromatin modifier gene sets, among others. The most frequently aberrant genes in mCRPC included AR (62.7%), ETS family (56.7%), TP53 (53.3%) and PTEN (40.7%) (Fig. 2).
AR signaling pathway
In aggregate 107/150 (71.3%) of cases harbored AR pathway aberrations, the majority of which were direct alterations affecting AR through amplification and mutation (Fig. 4A). Figure 4B summarizes the key genes altered in AR signaling including AR itself, FOXA1 as a pioneer transcription factor, NCOR1/2 as negative regulators of AR, SPOP as a putative androgen receptor transcriptional regulator (Geng et al., 2013), and ZBTB16 as an AR inducible target gene that may also negatively regulate AR. Recurrent hotspot mutations in AR were observed at residues previously reported to confer agonism to AR antagonists such as flutamide (T878A) and bicalutamide (W742C), as well as to glucocorticoids (L702H). Some but not all of these affected individuals had documented prior exposures that could explain enrichment for these mutations. Additional clinical data collection is ongoing (Fig. 4C). Rare AR mutations not previously described were seen in our cohort, although these are of unclear functional significance. Furthermore, one affected individual (Case 89) harbored two putatively functional AR mutations (T878A and Q903H), which may further suggest intra-tumor heterogeneity emerging in the CRPC setting (Carreira et al., 2014). Analysis of AR splice variants from RNA-seq data demonstrated a distribution of splice variants observed throughout these mCRPC tumor cases (Fig. 4D). Analysis of the TCGA prostate dataset revealed that many of these variants were also present at varying levels in primary prostate cancer and benign prostate tissue. AR-V7, which has been implicated in abiraterone acetate and enzalutamide resistance (Antonarakis et al., 2014), was observed in a majority of pre-abiraterone/enzalutamide cases but at very low ratios relative to full length AR. Implications for treatment response are unknown at this time.
Figure 4. Aberrations in the AR pathway found in mCRPC.

A, Cases with aberrations in the AR pathway. Case numbering as in Fig. 2. B, Key genes found altered in the AR pathway of mCRPC. DHT, dihydrotestosterone. C, Point mutations identified in AR. Amino acids altered are indicated. NTAD, N-terminal activation. DBD, DNA-binding. LBD, ligand binding. D, Splicing landscape of AR in mCRPC. Specific splice variants are indicated by exon boundaries and junction read level provided. SU2C, this mCRPC cohort. PRAD tumor, primary prostate cancer from the TCGA. PRAD normal, benign prostate from the TCGA. E, Homozygous deletion of ZBTB16. Copy number plots with×axis representing chromosomal location and the y axis referring to copy number level. Red outline indicates region of ZBTB16 homozygous loss.
In addition to AR mutations itself, we observed alterations in AR pathway members (Fig. 4A). These included known alterations in NCOR1, NCOR2, and FOXA1 that have been previously reported in primary prostate cancers and mCRPC (Barbieri et al., 2012; Grasso et al., 2012). In this cohort, truncating and missense mutations in FOXA1 form a cluster near the end of the Forkhead DNA binding domain (Fig. S2).
Recurrent homozygous deletions of the androgen-regulated gene ZBTB16 (also known as PLZF) were seen in 8 (5%) cases (Fig. 4E), not previously reported in clinical mCRPC biopsies. Analysis of the minimally deleted region seen in this cohort narrowed the candidate genes in the chr11q23 region to ZBTB16 (Fig. S3). ZBTB16 has been previously implicated in prostate cancer tumorigenesis and androgen resistance in preclinical models (Cao et al., 2013; Kikugawa et al., 2006), with loss of ZBTB16 upregulating the MAPK signaling pathway (Hsieh et al., 2015).
New PI3K pathway discoveries
The PI3K pathway was also commonly altered, with somatic alterations in 73/150 (49%) of mCRPC affected individuals (Fig. 5A). This included biallelic loss of PTEN, as well as hotspot mutations, amplifications and activating fusions in PIK3CA and p.E17K activating mutations in AKT1 (Fig. S2). Of note, PIK3CA amplifications resulted in overexpression compared to the remaining cohort (Fig. S3).
Figure 5. Aberrations in the PI(3)K pathway found in mCRPC.

A, Cases with aberrations in the PIK3 pathway. Case numbering as in Fig. 2. B, Point mutations identified in PIK3CB. Amino acids altered are indicated. Analogous, recurrent COSMIC mutations in PIK3CA are shown as expansion views. C, Outlier expression of PK3CA in CRPC case harboring the TBL1XR1-PIK3CA gene fusion. Structure of the gene fusion is inset. UTR, untranslated region. CDS, coding sequence. D. As in C, except for PIK3CB and the ACPP-PIK3CB gene fusion.
Interestingly, mutations in another member of the PI3K catalytic subunit, PIK3CB, were observed in this cohort for the first time, at equivalent positions to canonical activating mutations in PIK3CA (Fig. 5B). PIK3CB mutations appeared in the context of PTEN deficient cases, consistent with a previous report demonstrating some PTEN deficient cancers are dependent on PIK3CB, rather than PIK3CA (Wee et al., 2008). Furthermore, two affected individuals harbored fusions involving PIK3CA/B, with these events resulting in overexpression of the gene relative to other tumors in the cohort (Fig. 5C–D).
New Wnt pathway discoveries
27/150 (18%) of our cases harbored alterations in the Wnt signaling pathway (Fig. 6A). Hotspot activating mutations in CTNNB1 were seen (Fig. 6B), as previously described (Voeller et al., 1998). Notably, recurrent alterations in APC were also observed, which have not been previously described in clinical mCRPC affected individuals. This prompted a broader examination of Wnt signaling genes (Fig. 6B). Through integrative analysis, we identified alterations in RNF43 and ZNRF3, which were recently described in colorectal, endometrial and adrenocortical cancers (Assie et al., 2014; Giannakis et al., 2014) and were mutually exclusive with APC alterations (Fig. 6A). Moreover, we also discovered R-spondin fusions involving RSPO2, as previously observed in colorectal carcinoma (Seshagiri et al., 2012) in association with RSPO2 overexpression in these cases (Fig. 6C). RSPO2 is a key factor in prostate cancer organoid methodology (Gao et al., 2014). Affected individuals with aberrations in RNF43, ZNRF3, or RSPO2 (overall 6% of affected individuals) are predicted to respond to porcupine inhibitors (Liu et al., 2013).
Figure 6. Aberrations in the WNT pathway found in mCRPC.

A, Cases with aberrations in the WNT pathway. Case numbering as in Fig. 2. B, Aberrations identified in APC and CTNNB1. Amino acids altered are indicated. ARM, armadillo repeat. Phos, phosphorylation domain. TAD, trans-activating domain. EB1, end binding protein-1 domain. CC, coiled coil. C, Outlier expression of RSPO2 in CRPC and the GRHL2-RSPO2 gene fusion. RNA-seq expression across our CRPC cohort. Structure of the gene fusion is inset. UTR, untranslated region. CDS, coding sequence.
Cell cycle pathway
We observed RB1 loss in 21% of cases (Fig. S4). Expanding the scope of cell cycle genes implicated in mCRPC, we noted focal amplifications involving CCND1 in 9% of cases, as well as less common (< 5%) events in CDKN2A/B, CDKN1B, and CDK4 (Fig S4). Cell cycle derangement, such as through CCND1 amplification or CDKN2A/B loss, may result in enhanced response to CDK4 inhibitors in other tumor types (Finn et al., 2015), and preclinical mCRPC models predict similar activity in prostate cancer (Comstock et al., 2013).
DNA Repair pathway
Integrative analysis of both the somatic and pathogenic germline alterations in BRCA2 identified 19/150 (12.7%) of cases with loss of BRCA2, of which approximately 90% exhibited biallelic loss (Fig. 7A). This was commonly a result of somatic point mutation and loss of heterozygosity, as well as homozygous deletion. One of the clinical trials in our consortium is evaluating poly(ADP-ribose) polymerase (PARP) inhibition in unselected mCRPC affected individuals. Importantly, multiple affected individuals in this trial who experienced clinical benefit harbored biallelic BRCA2 loss, providing further evidence of clinical actionability (Mateo et al., 2014). Eight affected individuals (5.3%) harbored pathogenic germline BRCA2 mutations (Fig. 7B) with a subsequent somatic event that resulted in biallelic loss, revealing a surprisingly high frequency relative to primary prostate cancer.
Figure 7. Aberrations in the DNA repair pathway found in mCRPC.

A, Cases with aberrations in the DNA repair pathway. Case numbering as in Fig. 2. B, Aberrations identified in BRCA2, ATM and BRCA1. Amino acids altered are indicated. HELC, helical domain. OB, oligonucleotide binding fold. FAT, FRAP-ATM-TRRAP domain. PIK3c, PI3 kinase domain. CC, coiled coil. BRC, Brca repeat. C, Microsatellite instability analysis of representative hypermutated CRPC cases and non-hypermutated cases.
We therefore expanded the focus to other DNA repair/recombination genes and identified alterations in at least 34/150 (22.7%) of cases. These include recurrent biallelic loss of ATM (Fig. 7B), including multiple cases with germline pathogenic alterations. ATM mutations were also observed inaffected individuals who achieved clinical responses to PARP inhibition (Mateo et al., 2014). In addition, we noted events in BRCA1, CDK12, FANCA, RAD51B, and RAD51C. If aberrations of BRCA2, BRCA1 and ATM all confer enhanced sensitivity to PARP inhibitors, 29/150 (19.3%) of mCRPC affected individuals would be predicted to benefit from this therapy. Interestingly, 3 out of 4 mCRPC tumors exhibited hypermutation and harbored alterations in the mismatch repair pathway genes MLH1 or MSH2 (Fig. 2, 7C), corroborating a recent report identifying structural alterations in MSH2 and MSH6 mismatch repair genes in hypermutated prostate cancers (Pritchard et al., 2014).
DISCUSSION
To effectively implement precision cancer medicine, prospective identification of predictive biomarkers should be performed with information derived from the most contemporary tumor assessments that reflect the affected individual’s prior therapies and treatment opportunities. In mCRPC, precision cancer medicine activities have been limited by difficulties obtaining clinical samples from mCRPC affected individuals, and a lack of comprehensive genomic data for potentially actionable alterations. By demonstrating the feasibility of prospective genomics in mCRPC and defining the mutational landscape in a focused metastatic clinical cohort, this report may inform multiple genomically driven clinical trials, and biological investigations into key mediators of mCRPC. In nearly all of the mCRPC analyzed in this study, we identified biologically informative alterations; almost all harbored at least one driver SNV/indel and approximately half harbored a driver gene fusion, amplification or homozygous deletion. Remarkably, in nearly 90% of mCRPC affected individuals we identified a potentially actionable somatic or germline event.
The high frequency of AR pathway alterations in this cohort strongly implies that the vast majority of mCRPC affected individuals remain dependent on AR signaling for viability. Newer “second generation” AR-directed therapies (e.g. abiraterone acetate, enzalutamide) may select for distinct phenotypes that may be indifferent to AR signaling, and prospective characterization of such cases will be of particular interest. We hypothesize that affected individuals with acquired AR mutations, including novel AR mutations discovered in this cohort, will harbor differential responses to these second-generation ADT therapies. As the number of affected individuals in this cohort with AR mutations increases, we will subsequently be able to link specific AR mutations with clinical phenotypes to determine which mutations confer selective response or resistance to subsequent AR-directed therapy.
Moreover, these data identify multiple therapeutic avenues warranting clinical investigation in the CRPC population. Excluding AR aberrations, 65% of mCRPC have a potentially actionable aberration that may suggest an investigational drug or approved therapy. For example, focusing on the PI3K pathway, PIK3CB-specific inhibitors may have utility in affected individuals with mutation, amplification and/or fusion of this gene (Schwartz et al., 2015); multiple affected individuals who achieved durable (> 1 year) responses to PIK3CB-specificin inhibition harbored activating mutation or amplification in PIK3CB (de Bono et al., 2015). RAF kinase fusions in 3% of mCPRC affected individuals would suggest the use of pan-RAF inhibitors or MEK inhibitors (Palanisamy et al., 2010). In addition, the emergence of porcupine inhibitors (Liu et al., 2013) and R-spondin antibodies may warrant investigation in mCRPC tumors harboring Wnt pathway alterations or specifically R-spondin fusions, respectively. These observations will need to be prospectively assessed in the clinical trials.
Additionally, biallelic inactivation of BRCA2, BRCA1 or ATM was observed in nearly 20% of affected individuals. Previous work in other cancer types suggest that these affected individuals may benefit from PARP inhibitors (Fong et al., 2009; Kaufman et al., 2015; Weston et al., 2010) or platinum based chemotherapy, and prior reports have implicated the presence of germline BRCA2 alterations in primary prostate cancer with poor survival outcomes (Castro et al., 2013). Given the incidence of pathogenic germline BRCA2 mutations in this cohort with subsequent somatic events (5%), along with enrichment for somatic BRCA2 alterations in mCRPC (13%), germline genetic testing in mCRPC affected individuals warrants clinical consideration.
The ability to molecularly characterize mCRPC biopsy samples from affected individuals actively receiving therapy will also enable focused studies of resistance to secondary ADT therapies, including neuroendocrine-like phenotypes. This will require iterative sampling of pre-treatment and resistant tumors from matching affected individuals and may warrant multiregional biopsies from affected individuals (if feasible) given heterogeneity in mCRPC (Carreira et al., 2014; Gundem et al., 2015). Towards that end, in some affected individuals we observed multiple AR mutations emerging in the same biopsy, which may indicate clonal heterogeneity within these mCRPC tumor samples. Additional genomic alterations discovered in this cohort (e.g. ZBTB16) warrant exploration in prostate cancer model systems, including organoid cultures (Gao et al., 2014).
Broadly, our effort demonstrates the utility of applying comprehensive genomic principles developed for primary malignancies (e.g., TCGA) to a clinically relevant metastatic tumor cohort. Our effort may also catalyze multi-institutional efforts to profile tumors from cohorts of affected individuals with metastatic, treated tumors in other clinical contexts, since our results demonstrate multiple new discoveries within this advanced disease stage that have not been observed in primary tumor profiling. Moreover, this study sets the stage for epigenetic and other profiling efforts in mCRPC not taken in this study, which may enable biological discovery and have immediate therapeutic relevance in mCRPC (Asangani et al., 2014). Overall, our efforts demonstrate the feasibility of comprehensive and integrative genomics on prospective biopsies from individual mCRPC affected individuals to enable precision cancer medicine activities in this large affected individual population.
METHODS
Affected individual enrollment
Affected individuals with clinical evidence of mCRPC who were being considered for abiraterone acetate or enzalutamide as standard of care, or as part of a clinical trial, were considered for enrollment. Affected individuals with metastatic disease accessible by image-guided biopsy were eligible for inclusion. All affected individuals provided written informed consent to obtain fresh tumor biopsies, and to perform comprehensive molecular profiling of tumor and germline samples.
Biopsies and pathology review
Biopsies of soft tissue or bone metastases were obtained under radiographic guidance. Digital images of biopsy slides were centrally reviewed using schema established to distinguish usual adenocarcinoma from neuroendocrine prostate cancer (Epstein et al., 2014). All images were reviewed by genitourinary oncology pathologists (M.R., J.M.M., L.P.K., S.A.T., R.M., V.R., A.G., M.L., R.L., M.B.)
Sequencing and Analysis
Normal DNAs from buccal swabs, buffy coats, or whole blood were isolated using the Qiagen DNeasy Blood & Tissue Kit. Flash frozen needle biopsies with highest tumor content for each case, as determined by pathology review, were extracted for nucleic acids. Tumor genomic DNA and total RNA were purified from the same sample using the AllPrep DNA/RNA/miRNA kit (QIAGEN) with disruption on a Tissuelyser II (Qiagen). RNA integrity was verified on an Agilent 2100 Bioanalyzer using RNA Nano reagents (Agilent Technologies).
Whole exome capture libraries were constructed from 100ng to 1 µg of DNA from tumor and normal tissue after sample shearing, end repair, and phosphorylation and ligation to barcoded sequencing adaptors. Ligated DNA was size selected for lengths between 200–350 bp and subjected to hybrid capture using SureSelect Exome v4 baits (Agilent). Exome sequence data processing and analysis were performed using pipelines at the Broad Institute and the University of Michigan. A BAM file aligned to the hg19 human genome build was produced using Illumina sequencing reads for the tumor and normal sample and the Picard pipeline. Somatic mutation analysis was performed as described previously (Cibulskis et al., 2013; Van Allen et al., 2014c) and reviewed with Integrated Genomics Viewer (IGV) (Robinson et al., 2011).
Copy number aberrations were quantified and reported for each gene as the segmented normalized log2-transformed exon coverage ratios between each tumor sample and matched normal sample (Lonigro et al., 2011). To account for observed associations between coverage ratios and GC content across the genome, lowess normalization was used to correct per-exon coverage ratios prior to segmentation analysis. Mean GC percentage was computed for each targeted region, and a lowess curve was fit to the scatterplot of log2-coverage ratios vs. mean GC content across the targeted exome using the lowess function in R (version 2.13.1) with smoothing parameter f=0.05. The resulting copy ratios were segmented using the circular binary segmentation algorithm (Olshen et al., 2004).
Statistical analysis of recurrently mutated genes was performed using MutSig (Lawrence et al., 2013). Selective enrichment analysis (Van Allen et al., 2014b) of mutations observed in mCRPC compared to primary prostate cancer was performed by tabulating the frequency affected individual-normalized mutations observed in either CRPC or primary prostate cancer and performing a two-sided Fisher’s Exact Test using allelic fraction cut off of 0.1 or greater and a set of biologically relevant cancer genes (n = 550 genes) (Futreal et al., 2004). Multiple hypothesis test correction was performed using Benjamini-Hochberg method.
Transcriptome libraries were prepared using 200–1000 ng of total RNA. PolyA+ RNA isolation, cDNA synthesis, end-repair, A-base addition, and ligation of the Illumina indexed adapters were performed according to the TruSeq RNA protocol (Illumina). Libraries were size-selected for 250–300 bp cDNA fragments on a 3% Nusieve 3:1 (Lonza) gel, recovered using QIAEX II reagents (Qiagen), and PCR-amplified using Phusion DNA polymerase (New England Biolabs). Total transcriptome libraries were prepared as above, omitting the poly A selection step and captured using Agilent SureSelect Human All Exon V4 reagents and protocols. Library quality was measured on an Agilent 2100 Bioanalyzer for product size and concentration. Paired-end libraries were sequenced with the Illumina HiSeq 2500, (2×100 nucleotide read length), with sequence coverage to 50M paired reads and 100M total reads
Paired-end transcriptome sequencing reads were aligned to the human reference genome (GRCh37/hg19) using a RNA-Seq spliced read mapper Tophat2(Kim and Salzberg, 2011) (Tophat 2.0.4), with ‘--fusion-search’ option turned on to detect potential gene fusion transcripts. Potential false positive fusion candidates were filtered out using ‘Tophat-Post-Fusion’ module. Further, the fusion candidates were manually examined for annotation and ligation artifacts. Gene expression, as fragments per kilobase of exon per million fragments mapped (FPKM; normalized measure of gene expression), was calculated using Cufflinks (Trapnell et al., 2012).
Supplementary Material
Figure S1
Table S5
Table S6
Table S7
Table S8
Figure S2
Figure S3
Figure S4
Supplemental Table of Contents
Table S1
Table S2
Table S3
Table S4
ACKNOWLEDGMENTS
We thank the affected individuals that participated in this study to better understand the feasibility and utility of precision medicine approaches for advanced prostate cancer. Individuals at our respective institutions who helped with this study are listed by institution. University of Michigan: Karen Giles, Lynda Hodges, Erica Rabban, Ning Yu, Fengyun Su, Rui Wang, Brendan Veeneman, Moshe Talpaz. MSKCC: Brett Carver, Kristen Curtis, Julie Filipenko. DFCI/Broad: Zhenwei Zhang, Daniele Depalo, Joseph Kramkowski. University of Washington: Jina Taub, Hiep Nguyen, Colm Morrissey, Robert Vessella. ICR/Royal Marsden: Suzanne Carreira, Ines Figueiredo, and Daniel Nava Rodrigues.
FUNDING
This work was supported by a Stand Up To Cancer-Prostate Cancer Foundation Prostate Dream Team Translational Cancer Research Grant. Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research (SU2C-AACR-DT0712). The project was also supported by NIH awards: Clinical Sequencing Exploratory Research (CSER) UM1HG006508 (A.M.C.), Early Detection Research Network grant UO1 CA111275(A.M.C.), Prostate SPORE grants P50 CA186786 (A.M.C.), P50 CA092629 (H.S., C.L.S., Y.C.), and P50 CA097186 (P.S.N., B.M., E.M., L.T.), P01 CA163227 (P.S.N., S.P., R.B., H.D.), R01 CA116337 (M.A.R., H.B., F.D.), R01 CA155169 (C.L.S.), R01 CA092629, P50 CA092629 (H.S., C.L.S., Y.C.), and R01 CA155169 (C.L.S.) DoD awards: W81XWH-09-1-0147 (PCCTC), DOD PC121341(H.B.). Starr Cancer Consortium (N.S., M.R., C.S., Y.C., L.A.G.) A.M.C. is an A. Alfred Taubman Scholar, and American Cancer Society Professor. H.B. is supported by Damon Runyon Cancer Research Foundation CI-67-13. C.P. is supported by a PCF Young Investigator Award and DoD PC131820. E.M.V is supported by a NIH 1K08CA188615. E.M.V., N.S. and F.Y.F. are supported by Prostate Cancer Foundation Young Investigator Awards. The RM and ICR team is supported by the Movember Foundation and Prostate Cancer UK, PCF, the ECMC network from Cancer Research UK and the Department of Health in the UK, and BRC grant funding.
Footnotes
Supplemental information includes Extended Experimental Protocols, four figures and eight tables. All data will be available in the SU2C-PCF IDT cBio portal and through dbGAP.
AUTHOR CONTRIBUTIONS
Y-M.W., N.S., R.J.L., J-M.M., R.M., M.E.T., C.C.P., G.A., H.B., and W.M.A. made equal contributions. D.R.R., E.M.V.A. and R.J.L. coordinated overall sequencing and bioinformatics analysis. Y.W., D.R.R., E.M.V.A., R.J.L., W.M.A., and J.V. coordinated figures and tables. N.S. developed the SU2C-PCF IDT cBio portal. R.J.L. coordinated copy number analyses. J-M.M. coordinated central pathology review and L.P.K coordinated UM pathology analysis. C.C.P. coordinated hypermutation analysis and clinical germline interpretations, R.M., M.E.T, G.A., H.B., M.H., H.I.S., and E.H. coordinated clinical enrollment at their specific sites. R.B., S.P., and H.D. carried out AR splice variant analysis. J.V. managed the clinical data portal. X.C., J.S., P.F., S.M. and C.S. were involved in project management. R.M., S.A.T., V.R., A.G., M.L., S.B., R.L., M.B., B.R., and L.D.T. were involved in pathology review. C.B., P.V., J.G., M.G., F.D., O.E., A.S., A.S., and K.E. contributed to bioinformatics analysis. K.A.C., D.C.S., F.Y.F., H.S., D.R.R., S.S., M.M., R.F., Z.Z., N.T., G.G., J.D., J.M., D.N., S.T.T., E.Y., Y.C., E.M., H.C., F.C., M.S., and K.J.P. are clinical contributors. E.M.V.A., D.R.R., C.L.S. and A.M.C. wrote the manuscript, which all authors reviewed. P.K., J.S.B, M.A.R., P.S.N., and L.A.G. are SU2C-PCF Dream Team Principals while C.L.S. and A.M.C. are Dream Team co-Leaders.
REFERENCES
- ACS. Cancer Facts and Figures 2015. 2015 [Google Scholar]
- Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. The New England journal of medicine. 2014;371:1028–1038. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, Escara-Wilke J, Wilder-Romans K, Dhanireddy S, Engelke C, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510:278–282. doi: 10.1038/nature13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assie G, Letouze E, Fassnacht M, Jouinot A, Luscap W, Barreau O, Omeiri H, Rodriguez S, Perlemoine K, Rene-Corail F, et al. Integrated genomic characterization of adrenocortical carcinoma. Nature genetics. 2014;46:607–612. doi: 10.1038/ng.2953. [DOI] [PubMed] [Google Scholar]
- Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, Park K, Kitabayashi N, Macdonald TY, Ghandi M, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013;153:666–677. doi: 10.1016/j.cell.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nature genetics. 2012;44:685–689. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, Iversen P, Bhattacharya S, Carles J, Chowdhury S, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. The New England journal of medicine. 2014;371:424–433. doi: 10.1056/NEJMoa1405095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran H, Yelensky R, Frampton GM, Park K, Downing SR, MacDonald TY, Jarosz M, Lipson D, Tagawa ST, Nanus DM, et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. European urology. 2013;63:920–926. doi: 10.1016/j.eururo.2012.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, Sboner A, Esgueva R, Pflueger D, Sougnez C, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Zhu S, Zhou W, Li J, Liu C, Xuan H, Yan J, Zheng L, Zhou L, Yu J, et al. PLZF mediates the PTEN/AKT/FOXO3a signaling in suppression of prostate tumorigenesis. PloS one. 2013;8:e77922. doi: 10.1371/journal.pone.0077922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira S, Romanel A, Goodall J, Grist E, Ferraldeschi R, Miranda S, Prandi D, Lorente D, Frenel JS, Pezaro C, et al. Tumor clone dynamics in lethal prostate cancer. Science translational medicine. 2014;6:254ra125. doi: 10.1126/scitranslmed.3009448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro E, Goh C, Olmos D, Saunders E, Leongamornlert D, Tymrakiewicz M, Mahmud N, Dadaev T, Govindasami K, Guy M, et al. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31:1748–1757. doi: 10.1200/JCO.2012.43.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature biotechnology. 2013 doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comstock CE, Augello MA, Goodwin JF, de Leeuw R, Schiewer MJ, Ostrander WF, Jr, Burkhart RA, McClendon AK, McCue PA, Trabulsi EJ, et al. Targeting cell cycle and hormone receptor pathways in cancer. Oncogene. 2013;32:5481–5491. doi: 10.1038/onc.2013.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper CS, Eeles R, Wedge DC, Van Loo P, Gundem G, Alexandrov LB, Kremeyer B, Butler A, Lynch AG, Camacho N, et al. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nature genetics. 2015;47:367–372. doi: 10.1038/ng.3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bono JS, Arkenau HT, Mateo J, Infante J, Burris H, Bang Y, Eder JP, Sharma S, Chung CH, Decordova S, et al. Paper presented at: 106th Annual Meeting of the American Association for Cancer Research. Philadelphia (PA): Cancer Res; 2015. Exploratory genetic analysis of tumors from a phase-I/II dose escalation study of GSK2636771 in patients (pts) with PTEN deficient advanced tumors. [Google Scholar]
- de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB, Jr, Saad F, et al. Abiraterone and increased survival in metastatic prostate cancer. The New England journal of medicine. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein JI, Amin MB, Beltran H, Lotan TL, Mosquera JM, Reuter VE, Robinson BD, Troncoso P, Rubin MA. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. The American journal of surgical pathology. 2014;38:756–767. doi: 10.1097/PAS.0000000000000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, Ettl J, Patel R, Pinter T, Schmidt M, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. The lancet oncology. 2015;16:25–35. doi: 10.1016/S1470-2045(14)71159-3. [DOI] [PubMed] [Google Scholar]
- Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. The New England journal of medicine. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nature reviews Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, Dowling C, Wanjala JN, Undvall EA, Arora VK, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159:176–187. doi: 10.1016/j.cell.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng C, He B, Xu L, Barbieri CE, Eedunuri VK, Chew SA, Zimmermann M, Bond R, Shou J, Li C, et al. Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:6997–7002. doi: 10.1073/pnas.1304502110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakis M, Hodis E, Jasmine Mu X, Yamauchi M, Rosenbluh J, Cibulskis K, Saksena G, Lawrence MS, Qian ZR, Nishihara R, et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nature genetics. 2014;46:1264–1266. doi: 10.1038/ng.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JM, Papaemmanuil E, Brewer DS, Kallio HM, Hognas G, Annala M, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015 doi: 10.1038/nature14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hieronymus H, Schultz N, Gopalan A, Carver BS, Chang MT, Xiao Y, Heguy A, Huberman K, Bernstein M, Assel M, et al. Copy number alteration burden predicts prostate cancer relapse. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:11139–11144. doi: 10.1073/pnas.1411446111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong MK, Macintyre G, Wedge DC, Van Loo P, Patel K, Lunke S, Alexandrov LB, Sloggett C, Cmero M, Marass F, et al. Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nature communications. 2015;6:6605. doi: 10.1038/ncomms7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh CL, Botta G, Gao S, Li T, Van Allen EM, Treacy DJ, Cai C, He HH, Sweeney CJ, Brown M, et al. PLZF, a Tumor Suppressor Genetically Lost in Metastatic Castration Resistant Prostate Cancer, is a Mediator of Resistance to Androgen Deprivation Therapy. Cancer research. 2015 doi: 10.1158/0008-5472.CAN-14-3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, Mitchell G, Fried G, Stemmer SM, Hubert A, et al. Olaparib Monotherapy in Patients With Advanced Cancer and a Germline BRCA1/2 Mutation. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015;33:244–250. doi: 10.1200/JCO.2014.56.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikugawa T, Kinugasa Y, Shiraishi K, Nanba D, Nakashiro K, Tanji N, Yokoyama M, Higashiyama S. PLZF regulates Pbx1 transcription and Pbx1-HoxC8 complex leads to androgen-independent prostate cancer proliferation. The Prostate. 2006;66:1092–1099. doi: 10.1002/pros.20443. [DOI] [PubMed] [Google Scholar]
- Kim D, Salzberg SL. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome biology. 2011;12:R72. doi: 10.1186/gb-2011-12-8-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde E, Ishkanian AS, Sykes J, Fraser M, Ross-Adams H, Erho N, Dunning MJ, Halim S, Lamb AD, Moon NC, et al. Tumour genomic and microenvironmental heterogeneity for integrated prediction of 5-year biochemical recurrence of prostate cancer: a retrospective cohort study. The lancet oncology. 2014;15:1521–1532. doi: 10.1016/S1470-2045(14)71021-6. [DOI] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014 doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Pan S, Hsieh MH, Ng N, Sun F, Wang T, Kasibhatla S, Schuller AG, Li AG, Cheng D, et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:20224–20229. doi: 10.1073/pnas.1314239110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonigro RJ, Grasso CS, Robinson DR, Jing X, Wu YM, Cao X, Quist MJ, Tomlins SA, Pienta KJ, Chinnaiyan AM. Detection of somatic copy number alterations in cancer using targeted exome capture sequencing. Neoplasia. 2011;13:1019–1025. doi: 10.1593/neo.111252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateo J, Hall E, Sandhu S, Omlin A, Miranda S, Carreira S, Goodall J, Gillman A, Mossop H, Ralph C, et al. Antitumour activity of the PARP inhibitor olaparib in unselected sporadic castration-resistant prostate cancer (CRPC) in the TOPARP trial. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2014;25:1–41. [Google Scholar]
- Mehra R, Kumar-Sinha C, Shankar S, Lonigro RJ, Jing X, Philips NE, Siddiqui J, Han B, Cao X, Smith DC, et al. Characterization of bone metastases from rapid autopsies of prostate cancer patients. Clin Cancer Res. 2011;17:3924–3932. doi: 10.1158/1078-0432.CCR-10-3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- Palanisamy N, Ateeq B, Kalyana-Sundaram S, Pflueger D, Ramnarayanan K, Shankar S, Han B, Cao Q, Cao X, Suleman K, et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nature medicine. 2010;16:793–798. doi: 10.1038/nm.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflueger D, Terry S, Sboner A, Habegger L, Esgueva R, Lin PC, Svensson MA, Kitabayashi N, Moss BJ, MacDonald TY, et al. Discovery of non-ETS gene fusions in human prostate cancer using next-generation RNA sequencing. Genome research. 2011;21:56–67. doi: 10.1101/gr.110684.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard CC, Morrissey C, Kumar A, Zhang X, Smith C, Coleman I, Salipante SJ, Milbank J, Yu M, Grady WM, et al. Complex MSH2 and MSH6 mutations in hypermutated microsatellite unstable advanced prostate cancer. Nature communications. 2014;5:4988. doi: 10.1038/ncomms5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nature biotechnology. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roychowdhury S, Iyer MK, Robinson DR, Lonigro RJ, Wu YM, Cao X, Kalyana-Sundaram S, Sam L, Balbin OA, Quist MJ, et al. Personalized oncology through integrative high-throughput sequencing: a pilot study. Science translational medicine. 2011;3:111ra121. doi: 10.1126/scitranslmed.3003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, Fizazi K, Mainwaring P, Piulats JM, Ng S, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. The New England journal of medicine. 2013;368:138–148. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. The New England journal of medicine. 2012;367:1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- Schwartz S, Wongvipat J, Trigwell CB, Hancox U, Carver BS, Rodrik-Outmezguine V, Will M, Yellen P, de Stanchina E, Baselga J, et al. Feedback suppression of PI3Kalpha signaling in PTEN-mutated tumors is relieved by selective inhibition of PI3Kbeta. Cancer cell. 2015;27:109–122. doi: 10.1016/j.ccell.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, Chaudhuri S, Guan Y, Janakiraman V, Jaiswal BS, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488:660–664. doi: 10.1038/nature11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, Keer HN, Balk SP. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. The New England journal of medicine. 1995;332:1393–1398. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. Integrative genomic profiling of human prostate cancer. Cancer cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in bioinformatics. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, Morris DS, Menon A, Jing X, Cao Q, Han B, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–599. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Foye A, Wagle N, Kim W, Carter SL, McKenna A, Simko JP, Garraway LA, Febbo PG. Successful whole-exome sequencing from a prostate cancer bone metastasis biopsy. Prostate cancer and prostatic diseases. 2014a;17:23–27. doi: 10.1038/pcan.2013.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Mouw KW, Kim P, Iyer G, Wagle N, Al-Ahmadie H, Zhu C, Ostrovnaya I, Kryukov GV, O'Connor KW, et al. Somatic ERCC2 mutations correlate with cisplatin sensitivity in muscle-invasive urothelial carcinoma. Cancer discovery. 2014b doi: 10.1158/2159-8290.CD-14-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Wagle N, Stojanov P, Perrin DL, Cibulskis K, Marlow S, Jane-Valbuena J, Friedrich DC, Kryukov G, Carter SL, et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nature medicine. 2014c;20:682–688. doi: 10.1038/nm.3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voeller HJ, Truica CI, Gelmann EP. Beta-catenin mutations in human prostate cancer. Cancer research. 1998;58:2520–2523. [PubMed] [Google Scholar]
- Wang XS, Shankar S, Dhanasekaran SM, Ateeq B, Sasaki AT, Jing X, Robinson D, Cao Q, Prensner JR, Yocum AK, et al. Characterization of KRAS rearrangements in metastatic prostate cancer. Cancer discovery. 2011;1:35–43. doi: 10.1158/2159-8274.CD-10-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee S, Wiederschain D, Maira SM, Loo A, Miller C, deBeaumont R, Stegmeier F, Yao YM, Lengauer C. PTEN-deficient cancers depend on PIK3CB. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:13057–13062. doi: 10.1073/pnas.0802655105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston VJ, Oldreive CE, Skowronska A, Oscier DG, Pratt G, Dyer MJ, Smith G, Powell JE, Rudzki Z, Kearns P, et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood. 2010;116:4578–4587. doi: 10.1182/blood-2010-01-265769. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Table S5
Table S6
Table S7
Table S8
Figure S2
Figure S3
Figure S4
Supplemental Table of Contents
Table S1
Table S2
Table S3
Table S4