The SHH/Gli pathway is reactivated in reactive glia and drives proliferation in response to neurodegeneration-induced lesions (original) (raw)
. Author manuscript; available in PMC: 2015 Oct 1.
Published in final edited form as: Glia. 2014 Jun 4;62(10):1595–1607. doi: 10.1002/glia.22702
Abstract
In response to neurodegeneration, the adult mammalian brain activates a cellular cascade that results in reactive astro- and microgliosis. The mechanism through which astrocytes become reactive and the physiological consequences of their activation in response to neurodegeneration is complex. While the activation and proliferation of astrocytes has been shown to occur during massive neuronal cell death, the functional relationship between these two events has not been clearly elucidated. Here we show that in response to Kainic acid- (KA) induced neurodegeneration, the mitogen sonic hedgehog (SHH) is upregulated in reactive astrocytes. SHH activity peaks at 7 days and is accompanied by increased Gli activity and elevated proliferation in several cell types. To determine the functional role of SHH-Gli signaling following KA lesions, we used a pharmacological approach to show that SHH secreted by astrocytes drives the activation and proliferation of astrocytes and microglia. The consequences of SHH-Gli signaling in KA-induced lesions appear to be independent of the severity of neurodegeneration.
Keywords: Sonic Hedgehog, neurodegeneration, Gli, astrogliosis, microgliosis
Introduction
Excessive glutamate levels cause neuronal dysfunction and degeneration. Glutamate excitotoxicity plays a key role in a number of neurodegenerative disorders including amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), and Parkinson’s disease (Zheng et al. 2011). Kainic acid (KA) is an agonist of the a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)/kainite class of glutamate receptors and induces neurotoxicity with 30-fold more potency than glutamate (Olney et al. 1974). Administration of KA to rodents results in hippocampal damage, neuronal death and seizures, and is a well-characterized model to study human neurodegeneration (Pollard et al. 1994). In the hippocampus, CA3 pyramidal neurons and interneurons in the hilus of dentate gyrus (DG) are the most vulnerable, followed by CA1 pyramidal neurons. In response to neuronal damage, microglia and astrocytes are activated by KA exposure (Miettinen et al. 1998). While the role of these cell populations in KA-induced neurodegeneration is still unclear, some studies suggest that pro-inflammatory cytokines and other molecules may play an important role in KA-induced neurodegeneration (Wang et al. 2005).
In response to diverse forms of CNS injury, microglia and astrocytes commonly undergo a phenotypic transformation, with changes in both morphology and in the expression of a wide range of signaling molecules. These pathologic processes are referred to as microgliosis and astrogliosis. Reactive microgliosis is characterized by a massive but transient proliferative expansion of the microglial cell population and a change in cell shape with elevated expression of CD11b and Iba1 (Kettenmann et al. 2011). The hallmark of reactive astrogliosis is increased expression of glial fibrillary acidic protein (GFAP) and vimentin in astrocytes. The process of astrocyte activation is triggered and controlled in different and selective ways in different neurodegenerative diseases, and consequences largely depend on the type of injury to the CNS (Burda and Sofroniew 2014; Sofroniew 2009). Astrocytes are activated in all CNS injuries, but their proliferation is observed only in certain types of injury (Cui et al. 2011; Gangoso et al. 2012). This activation can be regulated by a number of molecules including endothelin 1, FGF2, bone morphogenic proteins (BMPs), SHH and others (for more detail, see the review by (Burda and Sofroniew 2014))
In response to a KA-induced insult, first microglia are activated, followed by astrocytes (Mitchell et al. 1993; Serrano-Perez et al. 2011). While some studies have examined the role of microglial activation in the context of pyramidal neuronal death (Hong et al. 2010; Zhu et al. 2010), the role of astrocytes remains not well understood. Additionally, little is known about the potential role played by microglia or astrocytes in proliferation after KA-induced neurodegeneration.
Recently, we have shown that in response to a cortical freeze injury, reactive astrocytes express the developmental morphogen molecule sonic hedgehog (SHH) (Amankulor et al. 2009). We have also shown that astrocytes activate downstream targets of SHH-Gli in the context of gliomagenesis (Becher et al. 2008). Further, SHH-expressing astrocytes activated in response to injury and inflammation can induce proliferation of Olig2-positive cells (Amankulor et al. 2009). SHH is a secreted protein that exhibits both morphogenic and mitogenic activities during development. SHH ligand transmits its signal by binding to the transmembrane receptor Patched (Ptc), which results in de-repression of Smoothened and activation of the Gli1 and Gli2 transcription factors. One downstream effect of SHH signaling is to induce expression of the basic helix-loop-helix transcription factor Olig2, driving proliferation and differentiation of Olig2-expressing cells in the developing mammalian ventral neural tube (Danesin et al. 2006). SHH has been studied mainly in the context of development, but there is increasing evidence that the mitogenic properties of SHH may be re-activated during adulthood, where SHH can promote oncogenesis or tissue repair in different organs (Garcia et al. 2010; Mimeault and Batra 2010; Rao et al. 2003). Several recent studies have shown that SHH regulates proliferation of neural progenitor cells during ischemia/hypoxia (Sims et al. 2009), improves neurological recovery after cerebral ischemia (Bambakidis et al. 2012), and mediates neural stem cell differentiation in experimental autoimmune encephalomyelitis (EAE) and MS (Wang et al. 2008). Interestingly, the consequences of SHH activation and the source of SHH vary depending on the context of the injury. For example, in the case of ischemia/hypoxia, neurons produce SHH (Sims et al. 2009) whereas in EAE, TBI or LPS models, SHH is secreted from reactive astrocytes (Amankulor et al. 2009; Wang et al. 2008). One study documented increased expression of SHH by neurons in temporal lobe epileptic foci in human and experimental rats, although consequences of elevated SHH were not studied (Fang et al. 2011).
Here, we demonstrate that KA-induced neurodegeneration in mice leads to reactivation of SHH and its downstream target transcription factor, Gli. We also demonstrate that SHH secreted by reactive astrocytes increases the proliferation of astrocytes, microglia and Olig2-positive progenitors. Using a pharmacological approach, we show that both activation and proliferation of astrocytes and microglia in response to neurodegeneration are SHH-dependent. This study for the first time identifies the signaling pathway responsible for astrocyte activation and proliferation as well as microglia proliferation during KA-induced neurodegeneration.
Materials and Methods
Mice
All experiments were performed in accordance with the Institutional Animal Care and Use Committee guidelines at Memorial Sloan-Kettering Cancer Center (MSKCC) and Cleveland Clinic. Mice 6-8 weeks old were used in all experiments. In all experiments age, sex and strain were used as criteria to equally distribute mice into KA- and saline-injected groups. Gli-luciferase (Gli-Luc) mice in FVB/N background were generated as previously described (Becher et al. 2008).
KA and cyclopamine administration
Kainic acid (Sigma #K0250) was dissolved in physiological saline (0.9% NaCl solution) and was slowly injected intraperitoneally at a dose of 30mg/kg to produce systemic onset of seizures. Control mice were injected with physiological saline only. Cyclopamine treatment was administered orally starting 3 days before KA injection, at a twice-daily dose of 30 mg/kg. On the fourth day of KA injection and for the subsequent 5 days, cyclopamine was again used at a single daily dose of 30 mg/kg (Palma et al. 2005). Cyclopamine was a kind gift from Dale Gardner (United States Department of Agriculture).
Seizure scoring
2- to 3-month-old mice were injected i.p. at a dose of 30 mg/kg body weight with KA (Sigma). KA+Cyclopamine mice were pretreated for 3 days with twice-daily oral cyclopamine (30 mg/kg). On the fourth day, KA was injected. Mice were monitored for 360 min and maximum seizure severity was scored every 5 min. Mortality was determined as the time when mice ceased breathing and did not respond to a noxious stimulus. A modified version of Racine’s scale (Racine et al. 1972), previously reported for other Alzheimer’s disease (AD) mouse models (Ghosal et al. 2009), (Vogt et al. 2011), was used to score seizure severity, where:
- (0)
normal activity, - (1)
freezing/immobility, - (2)
mild twitch, - (3)
tail extension, - (4)
forelimb clonus/repeated forelimb extensions, - (5)
consistent loss of balance/tonic-clonal seizures, - (6)
hyperactivity with jumping, - (7)
full body extension, - (8)
death
Bioluminescence imaging (BLI)
Mice were dosed with ketamine/xylazine followed by a retro-orbital injection of D-luciferin (at a dose of 75mg/kg body weight, 30mg luciferin/ml dH2O and given 2.5μl per gram mouse weight) into the right eye as a bolus injection. D-luciferin was synthesized at MSKCC. Mice were sacrificed 2 minutes after injection. Their brains were placed in a 6-well dish with 1ml of PBS and 3mg of luciferin and images were acquired for 5 minutes in the IVIS 100 machine (Xenogen) at imaging level A. A circular region of interest (ROI) was used to cover the whole brain, with the same background mouse with no Gli light as an internal control. All images were analyzed using the same maximum and minimum threshold parameters. BLI values were defined as photon per second (photon/sec).
Immunohistochemistry (IHC) and Immunofluorescence (IF)
After BLI imaging, brains were washed with ice cold PBS and post-fixed in 10% formalin for 72 hours and embedded in paraffin blocks. 5μm coronal sections were used for all histological studies. All IHC procedures were performed as previously described (Becher et al. 2008). The following antibodies were used at the stated dilutions: rabbit polyclonal anti-Iba1, 1:100 (Wako Pure Chemicals, Osaka Japan); rabbit polyclonal anti-GFAP, 1:8000 (DAKO, Denmark); rabbit polyclonal anti-Olig2, 1:250 (Chemicon, Temecula CA); mouse monoclonal anti-PCNA, 1:2000 (DAKO, Denmark), mouse monoclonal anti-NeuN, 1:50 (Millipore) and goat polyclonal anti-SHH, 1:100 (Santa Cruz N-19). For IHC detection, an automated staining processor was used (Discovery, Ventana Medical Systems, Inc.) with protocols developed in the Holland and Hambardzumyan laboratories. For immunofluorescence (IF) staining, secondary antibodies conjugated to different Alexa-Fluor dyes (488, 555, 647 from Invitrogen) at a dilution of 1:500 in PBS/2%BSA were applied. For nuclear counterstaining, DAPI was used (Sigma).
Quantification of IF and IHC
IF images were quantified using the Volocity program (PerkinElmer). Eight to ten images (20X) were taken per mouse brain and the total number of PCNA-positive cells, as well as the total number of PCNA/Olig2; PCNA/Iba1 and PCNA/GFAP double-positive cells were quantified. For quantification of IHC, whole mounts of images containing the hippocampus were scanned using a Mirax scanner and the DAB-positive area was quantified using ImageJ and Volocity and plotted as percentage of total area or number of positive cells. For quantification of neuronal loss in the hippocampus, slides were stained with NeuN and TUNEL and scanned at 20x magnification using a Leica SCN400F slide scanner. Images were analyzed in ImageJ; a selection including both CA1 and CA3 was drawn over each side of the hippocampus and the number of NeuN-positive cells was determined through the automated counting of single color images. The total number of counted cells was then divided by the area of the selection and plotted as the number of NeuN-positive cells/area (mm2). Selections of the whole hippocampus were analyzed in ImageJ using the automated counting of single color images. The number of TUNEL-positive cells was divided by the area and plotted as the number of TUNEL-positive cells/area (mm2).
TUNEL assay
The terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay was performed on 5 μm sections by using a Terminal Transferase, recombinant kit (Roche, #3333566) in accordance with the manufacturer’s protocol.
Statistical analysis
Graphs were made using GraphPad Prism 4 (GraphPad Software) and were analyzed using an unpaired parametric two-tailed t-test, assuming equal standard deviations. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001, (****) _P<_0.0001; no asterisk= not significant.
Results
KA-induced neurodegeneration induces astrocyte and microglial activation
Age and sex-matched mice were injected with KA (30 mg/kg) and were sacrificed 7 days after injury in all experiments. This is the time point when we observed maximum GFAP expression (Fig. S1). Despite using age and sex-matched mice, we observed large variability in the KA-induced hippocampal neurodegenerative response, as determined by neuronal loss in the CA3 and CA1 regions (Fig 1B). Based on histological examination of the hippocampus (areas of pyknotic neurons), we classified KA-treated mice into high, medium and low responders (Fig. 1B). The responders were assigned to the “high” category if they had between 10 and 30 TUNEL-positive cells per 20X view, “medium” for 3-10 cells, and “low” for 1-3 cells per 20X view. Control animals had 0-0.5 TUNEL-positive cells per 20X view. The quantification of TUNEL-positive apoptotic nuclei confirmed the observations from H&E sections and demonstrated that despite large variability, KA treatment induced statistically significant cell death in the hippocampus (8.93 cells/mm2 vs 0.1688 cells/mm2; p = 0.035; Fig. 1C). Next we examined the number of neurons in the hippocampus and showed that there was a significant decrease in the number of NeuN-positive neurons in the hippocampus of KA-treated mice compared to saline-treated mice (185.1 cells/mm2 vs 386.0 cells/mm2; p = 0.0024). Despite some variability, KA treatment induced statistically significant neurodegeneration, which was more pronounced in the samples that had higher positivity for TUNEL in hippocampus (Fig. 1D).
Figure 1.

KA-treatment induces neural loss in hippocampus. A, High and low magnification H&E sections of the hippocampus in vehicle-treated animals. B, H&E sections of low, medium and high KA responders. Loss of pyramidal neurons is apparent in CA3 (white arrowheads) and also in CA1 area of high responders (white arrowheads). C. Representative images of TUNEL staining in KA- and saline-treated animals with corresponding quantification graphs showing a significant increase in the number of TUNEL-positive nuclei in KA-treated animals compared to saline-treated animals. D) IF images of NeuN staining in saline- and KA-treated animals showing a significant decrease in the number of NeuN-positive neurons in response to KA treatment. Scale bars for A and B are 50μm. *,**, and *** represent significance determined by unpaired _t_-test analysis for p<0.05, p<0.01, and p<0.001.
Since KA-induced neuronal death has been previously shown to result in activation of both microglia and astrocytes (Chen et al. 2005), we looked at microglial activation via Iba1 expression seven days after KA administration. While Iba1 can stain both activated and resting microglia, its expression is increased upon microglial activation (Ito et al. 1998). Staining for Iba1 revealed that microglia are activated in all mice treated with KA, regardless of response status (low, medium or high) (Fig. 2A), but the activation was more pronounced in high responders. On aggregate, KA-injected mice displayed more Iba1 expression, as quantified by immunohistochemistry (17.47% vs. 8.8% positive area; p = 0.0232, Fig. 2C). We observed a more pronounced activation of astrocytes in KA high responders compared to medium and low responders. Quantification of the areas of reactive gliosis showed significant upregulation of GFAP expression in KA-treated mice compare to the saline-treated group (15.29% vs. 5% positivity, p = 0.0103, Fig. 2_B,D_).
Figure 2.

Neural loss followed by activation of microglia and astrocytes in KA-treated animals. A, Images of Iba1 IHC of saline- and KA-treated animals showing that the highest up-regulation of Iba1 (increased brown intensity and size of cells) is in high KA responders. B, IHC images of GFAP in saline- and KA-treated animals showing that the highest level of reactive astrogliosis is in the high responders. C–D, graphs of quantification of Iba1 and GFAP staining showing significant up-regulation of reactive microgliosis and astrogliosis in KA-treated animals compared to saline-treated animals 7 days post-KA injection. Scale bar for the top right image of A is 500 μm, for the rest of the images in A, B, C and D scale bars are 50μm. *,**, and *** represent significance determined by unpaired _t_-test analysis for p<0.05, p<0.01, and p<0.001
Sonic Hedgehog is expressed by reactive astrocytes in KA-treated mouse brains
Upon activation, astrocytes produce pro- and anti-inflammatory cytokines, chemokines, and neurotrophic factors that regulate neuron-glia communication (Zhang and Zhu 2011). For example, upregulation of SHH in astrocytes has previously been reported in EAE, TBI and LPS injury models (Amankulor et al. 2009; Wang et al. 2008). However, the response in a seizure-induced neurodegeneration model has not been described. To this end we next investigated the SHH expression pattern in KA-injected mice. Saline-injected animals had low levels of SHH staining (Fig. 3A). By contrast, KA-injected mice showed elevated SHH protein immunostaining, especially near the hippocampus (Fig. 3B). Double-staining for SHH and GFAP demonstrated almost completely overlapping expression patterns, demonstrating that the source of upregulation of SHH is the reactive astrocytes (Fig. 3C). We also observed staining around the astrocytes, which is typical for secreted molecules like SHH (Fig. 3C).
Figure 3.
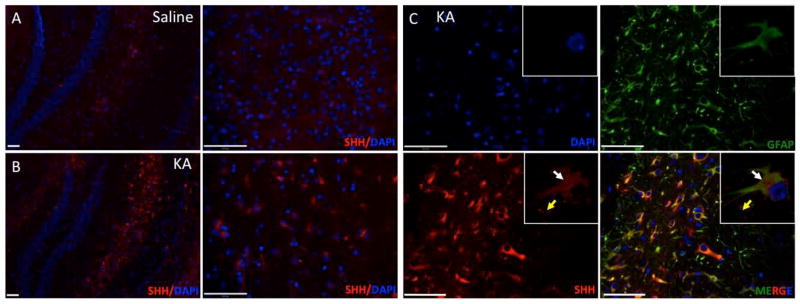
SHH is expressed at 7 days post-KA injection in the mouse brain. A–B, Brains from KA-treated or saline-treated mice 7 days after KA injections were used for immunofluorescence with anti-SHH antibody and nuclei were stained with DAPI (hippocampus area). C, SHH and GFAP immunofluorescence of post- KA-lesions in hippocampus. White arrowheads indicate cells that are double-positive for SHH and GFAP and yellow arrowhead indicate secreted SHH around astrocytes 7 days after the KA-injection, inserted confocal images of single astrocyte. Scale bars for A are 150 and for B, C are 50μm.
The Sonic Hedgehog effector Gli is activated in response to KA-induced neurodegeneration
The observed reactive gliosis and up-regulation of SHH expression by reactive astrocytes in KA-induced lesions led us to further investigate the role of the SHH/Gli signaling pathway. In canonical SHH pathway signaling, SHH binds to its receptor Patched (Ptc) and releases the inhibition of Smoothened (Smo), causing a series of downstream events that ultimately results in Gli translocation to the nucleus (referred to as Gli activation) and the transcription of Gli target genes (Becher et al. 2008; Stone et al. 1996). To investigate whether upregulation of SHH expression by reactive astrocytes results in activation of this signaling pathway; we utilized the Gli-Luciferase (Gli-Luc) reporter mouse. In this reporter mouse, eight contiguous Gli binding sites are positioned upstream of the firefly luciferase gene, allowing the use of bioluminescence imaging to quantify SHH pathway activation (Becher et al. 2008; Sasaki et al. 1997). Administration of KA in Gli-Luc mice resulted in upregulation of Gli activation as determined by bioluminescence imaging (BLI) (Fig. 4A). As was the case with hippocampal neurodegeneration, KA-induced Gli activation was variable and we divided the responses into low, medium and high responders as before. Despite the large variability in BLI intensity in KA-treated mice, quantification of BLI showed a statistically significant increase in Gli signaling in these mice compared to controls (62.95 x 103 vs. 50.26 x 103 photons/sec, p = 0.0114, Fig. 4B).
Figure 4.

Proliferation is increased and the Gli pathway is activated following injection of KA. A, Gli bioluminescence imaging of KA- and saline-injected brains imaged 7 days post-injection. A variable increase in BLI intensity was noted in KA-injected mice. B, Quantification and statistical analysis of the BLI intensity in saline and KA-treated animals 7 days post-injection. C, Images of PCNA immunohistochemistry of dentate gyrus and cortex of KA- and saline-treated brains 7 days post-injection. D, Quantification of PCNA immunohistochemistry for KA- and saline-treated brains 7 days after injection (p < 0.001). E, Simple linear regression showing a positive correlation (_r_2) between GFAP intensity and PCNA expression in KA-injected brains and the corresponding p value. Scale bars for C are 50μm.
KA administration is associated with increased proliferation in the hippocampal dentate gyrus (DG), which peaks at 7 days; however, the underlying mechanism of this proliferation is largely unknown (Kralic et al. 2005). We looked at the effect of KA-induced neurodegeneration on proliferation in the brain, focusing on the hippocampus and cortex of four high responders to further determine what populations of cells are proliferating. Proliferation was not limited to the DG but was also induced in the cortex (Fig. 4C). Quantification of PCNA staining (in both the cortex and hippocampus) demonstrated a statistically significant increase in the proliferation index seven days after KA injection (p = 0.0041, Fig 4D). We identified a strong positive correlation between proliferation (as determined by PCNA staining) and reactive gliosis (GFAP staining) (R2 = 0.590, p >0.0001, Fig. 4E). To determine which cell types proliferate in response to KA, we double-stained brains with the proliferation marker PCNA and three cell-type specific markers: GFAP (astrocytes), Olig2 (mostly OPCs), and Iba1 (microglia/macrophages) (representative images Fig. 5A, quantification Fig 5B-E). Saline-injected brains displayed very few proliferating cells in the cortex and hippocampus (Fig. 5B). We observed a significant increase in proliferating microglia in both the hippocampus and cortex of KA-treated mice (Fig. 5C), whereas astrocyte proliferation was significantly increased only in hippocampus (Fig. 5D). There was also a trend towards an increase in proliferation of Olig2-positive progenitors in both hippocampus and cortex (Fig. 5_E_).
Figure 5.

KA-induced neurodegeneration increases glial cell proliferation. A, Representative images of triple immunostaining with Iba1 or GFAP or Olig2 (green), PCNA (red) and DAPI counterstaining (blue) showing proliferating cells in cortex and hippocampus of high KA responders. B, Quantification of immunofluorescence staining for total PCNA+ cells C, Iba1/PCNA double positive cells D, GFAP/PCNA double-positive cells E, Olig2/PCNA double-positive cells. Scale bars for A are 50μm. *, **, *** and **** represent significance determined by unpaired _t_-test analysis for p<0.05, p<0.01, p<0.001 and p<0.0001.
Pharmacological inhibition of SHH/Gli signaling blocks proliferation in KA-treated mice
Given the observed activation of the SHH/Gli pathway and the concomitant increase in cellular proliferation, we wondered whether SHH expression was responsible for driving the proliferation in response to KA-induced neurodegeneration. In order to uncouple SHH from its downstream effector Gli, we employed cyclopamine to pharmacologically inhibit Smo, thereby blocking SHH activation of Gli (Taipale et al. 2000). We choose two cyclopamine treatment paradigms: a) mice were pretreated for 3 days with twice-daily oral cyclopamine (30 mg/kg). On the fourth day, KA was injected and mice were monitored for seizure severity up to 360 minutes. This allowed us to determine whether cyclopamine pretreatment has an impact on seizure severity; Fig. 6A demonstrates that cyclopamine pre-treatment does not significantly affect seizure severity. b) In the second paradigm mice were pretreated for 3 days with twice-daily oral cyclopamine (30 mg/kg). On the fourth day, KA was injected and cyclopamine treatment was continued once daily for 5 more days (30 mg/kg). Cyclopamine treatment had no statistically significant impact on mouse survival, but there was a higher tendency towards death in cyclopamine-treated mice compared to vehicle-treated mice (58% vs. 82%, p=0.5680, n=12 and n=11 correspondingly).
Figure 6.

Cyclopamine reduces reactive microgliosis, astrogliosis and the proliferative capacity of KA-treated mouse brain. A, Time course of seizure-like behavior in response to KA in saline- and cyclopamine-pre-treated animals. Higher scores correspond to more severe seizures as described in the Methods section. No significant difference was observed between the groups. B, Quantification graphs showing no significant difference in the number of TUNEL-positive nuclei or the number of NeuN-positive cells in hippocampus in KA+saline versus KA+cyclopamine animals. C, H&E and Iba1 images of KA+vehicle and KA+cyclopamine mouse brains. While cyclopamine treatment had no effect on cell death in the hippocampus, it decreased microglial activation. D, Quantification graph and representative whole mount images of GFAP immunohistochemistry for KA+vehicle and Ka+cyclopamine brains 7 days after injection. E, Quantification graph and representative whole mount images of PCNA immunohistochemistry for KA+vehicle and KA+cyclopamine brains 7 days after injection. Scale bars for C and E are 50μm. *,**, and *** represent significance determined by unpaired _t_-test analysis for p<0.05, p<0.01, and p<0.001.
We did not observe significant differences in neuronal loss in the hippocampus of cyclopamine+KA-treated mice determined by H&E, TUNEL staining (14.77 cells/mm2 vs 14.55 cells/mm2; p = 0.9716, Fig. 6B) or by the number of NeuN-positive cells (neuronal density, 171.6 cells/mm2 vs. 167.0 cells/mm2; p = 0.9235, Fig. 6B), suggesting that SHH does not affect neurogenesis in the context of KA-induced neuronal loss. We did observe a significant decrease in reactive microgliosis in the group that received cyclopamine (Fig. 6C). Quantification of reactive astrogliosis by immunohistochemistry revealed a statistically significant decrease in the activation of astrocytes in the cyclopamine+KA-treated group compared to the saline+KA controls (35.85% vs. 11.37%, p = 0.0215, Fig. 6D). Similarly, cyclopamine also significantly attenuated proliferation in response to neurodegeneration (p = 0.0069, Fig. 6E). Previously, using lineage-tracing experiments, we demonstrated that both reactive astrocytes and Olig2-positive glioma cells express Ptc, suggesting the potential for autocrine hedgehog signaling in astrocytes (Becher et al. 2008). These data demonstrate that abrogating SHH signaling inhibits astrocyte activation, suggesting that SHH regulates the activation of astrocytes by KA-induced lesions (Fig. 6B). Next we examined the effect of Smo inhibition by cyclopamine on Gli activation, using BLI in Gli-Luc mice. Gli signaling was identical in the cyclopamine+KA and saline+KA groups, as determined by BLI (Fig. S2B). The unchanged BLI is explained by the known effect of Smo inhibitors on the ABCG2/BCRP transporter, which modulates D-luciferin-based BLI (Zhang et al. 2009). ABCG2 is a direct transcriptional target of hedgehog signaling (Singh et al. 2011). When Smo is inhibited by cyclopamine, we also expect to inhibit expression of ABCG2, thereby influencing the efflux of D-luciferin via the cell membrane. BLI should therefore be considered an inaccurate readout of SHH signaling when cyclopamine, or any other Smo inhibitor, is used (Singh et al. 2011; Zhang et al. 2009). Histological analysis of cyclopamine-treated KA-injected mice versus only KA-injected mice showed a statistically significant decrease of proliferation in both the hippocampus and the whole brain (Fig. 5C).
Inhibition of SHH-GLI signaling reduces the proliferating Olig2-positive progenitor pool, GFAP-positive reactive astrocytes and Iba1-positive microglia in KA-induced lesions
Several studies have looked at the impact of stromal cells and their signals on KA-induced neurodegeneration in the hippocampus. The main focus of these studies was the role of astrocytes and microglia (or signals coming from these cells) in neuronal death after injection of KA. For example, one such study showed that in mice with microglia lacking Ikkβ, neuronal death was significantly reduced in response to KA (Cho et al. 2008). Some studies also characterized changes in the cellular composition and proliferative capacity of different cell populations, mainly in rats. These studies yielded different outcomes; some suggest that reactive astrocytes do not proliferate in response to KA, while others suggested they do. The same conclusion also holds true for microglia (Gangoso et al. 2012; Mitchell et al. 1993). The differences in these studies can be partially explained by variations in the dose of KA as well as differences in the assays used and the time points chosen for data collection. Nevertheless, the mechanism driving proliferation in KA-induced lesions is largely unknown. To characterize a potential role for SHH-regulated cell fate-specific proliferation following KA-induced neurodegeneration, we immunostained brains for a selection of markers at seven days post-KA injection in mice treated concomitantly with vehicle alone or with cyclopamine. Brains were stained with the proliferation marker PCNA and three cell-type specific markers: GFAP (astrocytes), Olig2 (mostly oligodendrocyte precursors), and Iba1 (microglia/macrophages). Saline-injected brains displayed very few proliferating cells in the cortex and hippocampus (data not shown, Fig. 5B-E). The cyclopamine pretreatment significantly decreased proliferation of all three cell populations, providing evidence that the proliferation of all three cell types may be driven by SHH signaling (Fig. 7A). The intriguing effect of cyclopamine on microglial proliferation requires further investigation because our knowledge regarding the factors driving microglial proliferation is extremely limited, especially in a disease context. To determine which proliferating cell types responded to cyclopamine pre-treatment, we stained brains treated with KA or KA+cyclopamine with the proliferation marker PCNA and three cell-type specific markers: GFAP (astrocytes), Olig2 (mostly oligodendrocyte precursors), and Iba1 (microglia/macrophages). We observed a significant decrease in proliferation of microglia in both the hippocampus and cortex of KA-treated mice (Fig. 7B). While astrocyte and Olig2-positive cell proliferation trended lower throughout the brain, proliferation was significantly decreased only in hippocampus (Fig. 7C,D). these data suggest that SHH most likely has an autocrine effect on astrocytes, and a paracrine effect on the other cell types.
Figure 7.

Cyclopamine pre-treatment attenuates seizure-induced glial proliferation. Quantification of immunofluorescence staining for total PCNA+ cells A, Iba1/PCNA double-positive cells B, GFAP/PCNA double-positive cells C, and Olig2/PCNA double-positive cells D. Scale bars for A are 50μm. *, **, *** and **** represent significance determined by unpaired _t_-test analysis for p<0.05, p<0.01, p<0.001 and p<0.0001.
Discussion
In the mammalian brain, where injury-related cell proliferation is transient, knowledge of factors that promote cell proliferation and an understanding of what cell types proliferate would be tremendously useful for clinical applications. This is especially true in the context of neurodegeneration, where cell loss is a major obstacle to recovery. The importance of these questions was highlighted recently by work from the Gotz laboratory demonstrating that astrocytes respond differently depending on the context of injury. Only acute injuries such as stab wounds or cerebral ischemia result in increased plasticity, in contrast to degenerative insults like amyloidosis or induced neuronal death (Sirko et al. 2013a).
The main focus of this current study was to investigate the role of glial proliferation in KA-induced neurodegeneration, a model that is known to cause massive neuronal death and associated cellular proliferation. We wanted to delineate potential correlations between astrocytic activation, proliferation and function in relation to the severity of disease within the same sensitive mouse strain. One of the challenges in using this model is the large degree of variability in the physiological response to KA-induced seizures. The hippocampal pyramidal neurons of certain commonly used mouse strains, specifically C57Bl/6 and BALB/c, are known to be resistant to KA-induced treatment as compared to the highly susceptible FVB/N and 129/svEms strains (Liu et al. 2012). However, despite the variability in neuronal cell death, all of these strains showed similar levels of seizure activity (Schauwecker 2002). In our experiments, which used high doses of KA (30mg/kg) and a sensitive mouse strain (FVB/N), we observed variable neuronal loss in the hippocampus even when equally distributing mice by age and sex into treatment and control groups.
Microglia and astrocytes are activated in response to neurodegeneration and their activation correlates with the severity of neuronal loss in hippocampus and cortex
We show here that both microglia and astrocytes are significantly activated in response to KA-induced neurodegeneration and that the levels of activation correlate with the degree of KA-induced neurodegeneration. Microglial activation was more pronounced in the areas of neuronal death, whereas reactive astrocytes were observed in the whole brain but were more pronounced in high KA responders. Several studies have provided evidence that microglia enhance hippocampal neuronal death following injection of KA (Zhang and Zhu 2011; Zhu et al. 2010). However, it is entirely unknown whether astrocytes enhance or decrease hippocampal neuronal death following KA-induced injury. Similarly, the role of astrocytes and microglia in the regenerative process after KA-induced neurodegeneration is not well understood. The role of reactive astrocytes in response to different models of CNS injury has been controversial, with some groups arguing that reactive astrocytosis can impede neurogenesis and axonal repair (Larsson et al. 2004; Silver and Miller 2004; Williams et al. 2007), while others claim that reactive and cytokine-stimulated astrocytes are able to restrict the volume of injured tissue by promoting cell proliferation, generating a number of growth factors, and by providing structural support to the damaged CNS tissue (Bush et al. 1999; Li et al. 2008; Liberto et al. 2004; Myer et al. 2006).
Reactive astrocytes up-regulate the SHH-Gli axis, serving as the main source of SHH in KA-treated brains
Our study shows that the SHH-Gli axis drives proliferation in response to KA-induced neurodegeneration. Our previous work in a stab-freeze injury model showed that the SHH-Gli axis was reactivated; resulting in proliferation of only Olig2-positive cells (Amankulor et al. 2009). In the current study, we observed significantly elevated levels of SHH in KA-treated mice seven days post-injection. Elevated SHH levels were mainly observed in reactive astrocytes in the hippocampus. Pharmacological inhibition of SHH/Gli signaling blocked astrocyte activation. Whether SHH is necessary for maintenance after astrocyte have already been activated is still an open question. We further observed significant up-regulation of proliferation seven days post-KA injection, especially in the hippocampus and cortex of high and medium responders. The levels of reactive astrogliosis were correlated with the levels of proliferation in KA-induced lesions. In last several years, many studies have shown elevated levels of SHH in different CNS disease contexts in mice, including brain tumors (Becher et al. 2008), ischemia/hypoxia (Sims et al. 2009), and autoimmune, traumatic or neuroinflammatory pathologies (Amankulor et al. 2009; Wang et al. 2008). One recent study demonstrated positive correlation between the levels of SHH induced by CNS injuries and the likelihood that astrocytes will behave like stem cells. In other words, astrocytes show stem cell-like behaviors (such as neurosphere formation in vitro) only when the injury induces a high level of SHH expression. The same study also demonstrated that not all CNS injuries induce high levels of SHH (Sirko et al. 2013b). Beneficial effects of SHH activity have been implicated in regeneration in many murine CNS models, including spinal cord injury (Bambakidis et al. 2012) and stroke (Ding et al. 2013), in contrast to the negative effects of SHH on repair observed during more chronic injuries such as EAE and MS (Wang et al. 2008).
SHH signaling regulates activation and proliferation of astrocytes and Olig2-positive cells in KA-induced lesions
In the current study, we used a pharmacological approach to demonstrate that SHH is both necessary and sufficient for astrocyte activation and proliferation in KA-induced lesions in vivo. This enhanced SHH production has been shown to correlate with the ability of astrocytes to form neurospheres in vitro (Sirko et al. 2013a). The significance of the subset of reactive astrocytes that are particularly plastic and immature is important in the context of novel therapies for CNS diseases for several reasons. First, these cells could potentially be manipulated to adopt a desirable fate in vivo. Additionally, these cells might serve as a source of SHH that, as we show here, can drive proliferation of Olig2-positive precursor cells. While in the current study we did not further pursue the fate of progeny of proliferating reactive astrocytes or SHH-driven newly formed Olig2-positive cells and their ability to differentiate into functional oligodendrocytes, these are promising avenues for future research. However, it is worth mentioning that there is emerging evidence to suggest that proliferating Olig2-positive cells in the injured mammalian brain can go on to form mature, myelinating oligodendrocytes (Dimou et al. 2008; Ligon et al. 2006). We therefore hypothesize that re-activation of the SHH pathway in astrocytes can be not only a source of stem cells but also can provide signaling that leads to the expansion of a crucial oligodendrocyte population. While SHH signaling resulted in the activation and proliferation of reactive astrocytes and Iba1-positive progenitors, it showed a trend but did not reach statistical significance on proliferation of Olig2-positive cells in KA-induced lesions, which can be explained by the low amount of proliferation in these cells and the high variability of the response to KA. Further investigation of the mechanism by which cyclopamine regulates microglial proliferation is valuable considering our limited knowledge about signals that regulate their proliferation (Meyers-Needham et al. 2012). The development of new mouse models that would allow SHH gene deletion in all astrocytes would provide a valuable tool to study the genetic validations of our observations determined by pharmacological inhibition of SHH/Gli signaling in KA-induced lesions.
Supplementary Material
Supp FigureS1-S2
Supplemental Figure 1. Reactive gliosis reached maximum levels at 7 days post KA-injection. Whole mount GFAP IHC images of KA-treated mice at days 0, 3, 5 and 7 demonstrate maximum activation at day 7.
Supplemental Figure 2. Cyclopamine pre-treatment and subsequent treatment had no significant impact on mouse survival or Gli-light. A, Cyclopamine pre-treatment did not show a statistically significant impact on mouse survival after KA injection but showed some tendency towards worsening symptoms. n=11 for Vehicle+KA and n=12 for Cyclopamine+KA groups. B, There was no significant difference in Gli-light determined by BLI in Vehicle+KA compared to Cyclopamine+KA.
Main Points.
- The mitogen SHH is upregulated in reactive astrocytes in response to neurodegeneration, and it regulates astrocyte activation and proliferation.
- Downstream targets of SHH-Gli are activated in several cell types, and these targets are responsible for proliferation in post-neurodegenerative lesions.
Acknowledgments
We thank Shannon Donnola for excellent technical support, members of the Hambardzumyan and Holland labs for technical advice and reagents. We also would like to thank Dr. Trapp for his continuous support, Drs. Rebecca Bish-Cornelissen and Chris Nelson for critical reading of manuscript. Cleveland Clinic Foundation and U01-CA160882 to D.H. and ECH and F31 NS076028 to KLP supported this research).
Footnotes
Contributions
K.L.P., I.T., X.F., N.A and D.H. performed and analyzed experiments. E.C.H. provided reagents, E.C.H and K.L.P., contributed to design of studies. D.H. conceived, designed the study and wrote the manuscript. All authors edited or commented on the manuscript.
References
- Amankulor NM, Hambardzumyan D, Pyonteck SM, Becher OJ, Joyce JA, Holland EC. Sonic hedgehog pathway activation is induced by acute brain injury and regulated by injury-related inflammation. J Neurosci. 2009;29(33):10299–308. doi: 10.1523/JNEUROSCI.2500-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bambakidis NC, Petrullis M, Kui X, Rothstein B, Karampelas I, Kuang Y, Selman WR, LaManna JC, Miller RH. Improvement of neurological recovery and stimulation of neural progenitor cell proliferation by intrathecal administration of Sonic hedgehog. J Neurosurg. 2012;116(5):1114–20. doi: 10.3171/2012.1.JNS111285. [DOI] [PubMed] [Google Scholar]
- Becher OJ, Hambardzumyan D, Fomchenko EI, Momota H, Mainwaring L, Bleau AM, Katz AM, Edgar M, Kenney AM, Cordon-Cardo C, et al. Gli activity correlates with tumor grade in platelet-derived growth factor-induced gliomas. Cancer Res. 2008;68(7):2241–9. doi: 10.1158/0008-5472.CAN-07-6350. [DOI] [PubMed] [Google Scholar]
- Burda JE, Sofroniew MV. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron. 2014;81(2):229–48. doi: 10.1016/j.neuron.2013.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron. 1999;23(2):297–308. doi: 10.1016/s0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- Chen Z, Duan RS, Quezada HC, Mix E, Nennesmo I, Adem A, Winblad B, Zhu J. Increased microglial activation and astrogliosis after intranasal administration of kainic acid in C57BL/6 mice. J Neurobiol. 2005;62(2):207–18. doi: 10.1002/neu.20099. [DOI] [PubMed] [Google Scholar]
- Cho IH, Hong J, Suh EC, Kim JH, Lee H, Lee JE, Lee S, Kim CH, Kim DW, Jo EK, et al. Role of microglial IKKbeta in kainic acid-induced hippocampal neuronal cell death. Brain. 2008;131(Pt 11):3019–33. doi: 10.1093/brain/awn230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui M, Huang Y, Tian C, Zhao Y, Zheng J. FOXO3a inhibits TNF-alpha- and IL-1beta-induced astrocyte proliferation:Implication for reactive astrogliosis. Glia. 2011;59(4):641–54. doi: 10.1002/glia.21134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danesin C, Agius E, Escalas N, Ai X, Emerson C, Cochard P, Soula C. Ventral neural progenitors switch toward an oligodendroglial fate in response to increased Sonic hedgehog (Shh) activity: involvement of Sulfatase 1 in modulating Shh signaling in the ventral spinal cord. J Neurosci. 2006;26(19):5037–48. doi: 10.1523/JNEUROSCI.0715-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimou L, Simon C, Kirchhoff F, Takebayashi H, Gotz M. Progeny of Olig2-expressing progenitors in the gray and white matter of the adult mouse cerebral cortex. J Neurosci. 2008;28(41):10434–42. doi: 10.1523/JNEUROSCI.2831-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X, Li Y, Liu Z, Zhang J, Cui Y, Chen X, Chopp M. The sonic hedgehog pathway mediates brain plasticity and subsequent functional recovery after bone marrow stromal cell treatment of stroke in mice. J Cereb Blood Flow Metab. 2013 doi: 10.1038/jcbfm.2013.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang M, Lu Y, Chen GJ, Shen L, Pan YM, Wang XF. Increased expression of sonic hedgehog in temporal lobe epileptic foci in humans and experimental rats. Neuroscience. 2011;182:62–70. doi: 10.1016/j.neuroscience.2011.02.060. [DOI] [PubMed] [Google Scholar]
- Gangoso E, Ezan P, Valle-Casuso JC, Herrero-Gonzalez S, Koulakoff A, Medina JM, Giaume C, Tabernero A. Reduced connexin43 expression correlates with c-Src activation, proliferation, and glucose uptake in reactive astrocytes after an excitotoxic insult. Glia. 2012;60(12):2040–9. doi: 10.1002/glia.22418. [DOI] [PubMed] [Google Scholar]
- Garcia AD, Petrova R, Eng L, Joyner AL. Sonic hedgehog regulates discrete populations of astrocytes in the adult mouse forebrain. J Neurosci. 2010;30(41):13597–608. doi: 10.1523/JNEUROSCI.0830-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosal K, Vogt DL, Liang M, Shen Y, Lamb BT, Pimplikar SW. Alzheimer’s disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc Natl Acad Sci U S A. 2009;106(43):18367–72. doi: 10.1073/pnas.0907652106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J, Cho IH, Kwak KI, Suh EC, Seo J, Min HJ, Choi SY, Kim CH, Park SH, Jo EK, et al. Microglial Toll-like receptor 2 contributes to kainic acid-induced glial activation and hippocampal neuronal cell death. J Biol Chem. 2010;285(50):39447–57. doi: 10.1074/jbc.M110.132522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res. 1998;57(1):1–9. doi: 10.1016/s0169-328x(98)00040-0. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91(2):461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- Kralic JE, Ledergerber DA, Fritschy JM. Disruption of the neurogenic potential of the dentate gyrus in a mouse model of temporal lobe epilepsy with focal seizures. Eur J Neurosci. 2005;22(8):1916–27. doi: 10.1111/j.1460-9568.2005.04386.x. [DOI] [PubMed] [Google Scholar]
- Larsson A, Wilhelmsson U, Pekna M, Pekny M. Increased cell proliferation and neurogenesis in the hippocampal dentate gyrus of old GFAP(−/−)Vim(−/−) mice. Neurochem Res. 2004;29(11):2069–73. doi: 10.1007/s11064-004-6880-2. [DOI] [PubMed] [Google Scholar]
- Li L, Lundkvist A, Andersson D, Wilhelmsson U, Nagai N, Pardo AC, Nodin C, Stahlberg A, Aprico K, Larsson K, et al. Protective role of reactive astrocytes in brain ischemia. J Cereb Blood Flow Metab. 2008;28(3):468–81. doi: 10.1038/sj.jcbfm.9600546. [DOI] [PubMed] [Google Scholar]
- Liberto CM, Albrecht PJ, Herx LM, Yong VW, Levison SW. Pro-regenerative properties of cytokine-activated astrocytes. J Neurochem. 2004;89(5):1092–100. doi: 10.1111/j.1471-4159.2004.02420.x. [DOI] [PubMed] [Google Scholar]
- Ligon KL, Kesari S, Kitada M, Sun T, Arnett HA, Alberta JA, Anderson DJ, Stiles CD, Rowitch DH. Development of NG2 neural progenitor cells requires Olig gene function. Proc Natl Acad Sci U S A. 2006;103(20):7853–8. doi: 10.1073/pnas.0511001103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Hamre KM, Goldowitz D. Kainic acid-induced neuronal degeneration in hippocampal pyramidal neurons is driven by both intrinsic and extrinsic factors: analysis of FVB/N<-->C57BL/6 chimeras. J Neurosci. 2012;32(35):12093–101. doi: 10.1523/JNEUROSCI.6478-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers-Needham M, Lewis JA, Gencer S, Sentelle RD, Saddoughi SA, Clarke CJ, Hannun YA, Norell H, da Palma TM, Nishimura M, et al. Off-target function of the Sonic hedgehog inhibitor cyclopamine in mediating apoptosis via nitric oxide-dependent neutral sphingomyelinase 2/ceramide induction. Mol Cancer Ther. 2012;11(5):1092–102. doi: 10.1158/1535-7163.MCT-11-0705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettinen R, Kotti T, Tuunanen J, Toppinen A, Riekkinen P, Sr, Halonen T. Hippocampal damage after injection of kainic acid into the rat entorhinal cortex. Brain Res. 1998;813(1):9–17. doi: 10.1016/s0006-8993(98)00915-9. [DOI] [PubMed] [Google Scholar]
- Mimeault M, Batra SK. Frequent deregulations in the hedgehog signaling network and cross-talks with the epidermal growth factor receptor pathway involved in cancer progression and targeted therapies. Pharmacol Rev. 2010;62(3):497–524. doi: 10.1124/pr.109.002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell J, Sundstrom LE, Wheal HV. Microglial and astrocytic cell responses in the rat hippocampus after an intracerebroventricular kainic acid injection. Exp Neurol. 1993;121(2):224–30. doi: 10.1006/exnr.1993.1089. [DOI] [PubMed] [Google Scholar]
- Myer DJ, Gurkoff GG, Lee SM, Hovda DA, Sofroniew MV. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain. 2006;129(Pt 10):2761–72. doi: 10.1093/brain/awl165. [DOI] [PubMed] [Google Scholar]
- Olney JW, Rhee V, Ho OL. Kainic acid: a powerful neurotoxic analogue of glutamate. Brain Res. 1974;77(3):507–12. doi: 10.1016/0006-8993(74)90640-4. [DOI] [PubMed] [Google Scholar]
- Palma V, Lim DA, Dahmane N, Sanchez P, Brionne TC, Herzberg CD, Gitton Y, Carleton A, Alvarez-Buylla A, Ruiz i Altaba A. Sonic hedgehog controls stem cell behavior in the postnatal and adult brain. Development. 2005;132(2):335–44. doi: 10.1242/dev.01567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard H, Charriaut-Marlangue C, Cantagrel S, Represa A, Robain O, Moreau J, Ben-Ari Y. Kainate-induced apoptotic cell death in hippocampal neurons. Neuroscience. 1994;63(1):7–18. doi: 10.1016/0306-4522(94)90003-5. [DOI] [PubMed] [Google Scholar]
- Racine R, Okujava V, Chipashvili S. Modification of seizure activity by electrical stimulation. 3. Mechanisms. Electroencephalogr Clin Neurophysiol. 1972;32(3):295–9. doi: 10.1016/0013-4694(72)90178-2. [DOI] [PubMed] [Google Scholar]
- Rao G, Pedone CA, Coffin CM, Holland EC, Fults DW. c-Myc enhances sonic hedgehog-induced medulloblastoma formation from nestin-expressing neural progenitors in mice. Neoplasia. 2003;5(3):198–204. doi: 10.1016/S1476-5586(03)80052-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H, Hui C, Nakafuku M, Kondoh H. A binding site for Gli proteins is essential for HNF-3beta floor plate enhancer activity in transgenics and can respond to Shh in vitro. Development. 1997;124(7):1313–22. doi: 10.1242/dev.124.7.1313. [DOI] [PubMed] [Google Scholar]
- Schauwecker PE. Modulation of cell death by mouse genotype: differential vulnerability to excitatory amino acid-induced lesions. Exp Neurol. 2002;178(2):219–35. doi: 10.1006/exnr.2002.8038. [DOI] [PubMed] [Google Scholar]
- Serrano-Perez MC, Martin ED, Vaquero CF, Azcoitia I, Calvo S, Cano E, Tranque P. Response of transcription factor NFATc3 to excitotoxic and traumatic brain insults: identification of a subpopulation of reactive astrocytes. Glia. 2011;59(1):94–107. doi: 10.1002/glia.21079. [DOI] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5(2):146–56. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Sims JR, Lee SW, Topalkara K, Qiu J, Xu J, Zhou Z, Moskowitz MA. Sonic hedgehog regulates ischemia/hypoxia-induced neural progenitor proliferation. Stroke. 2009;40(11):3618–26. doi: 10.1161/STROKEAHA.109.561951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RR, Kunkalla K, Qu C, Schlette E, Neelapu SS, Samaniego F, Vega F. ABCG2 is a direct transcriptional target of hedgehog signaling and involved in stroma-induced drug tolerance in diffuse large B-cell lymphoma. Oncogene. 2011;30(49):4874–86. doi: 10.1038/onc.2011.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirko S, Behrendt G, Johansson PA, Tripathi P, Costa M, Bek S, Heinrich C, Tiedt S, Colak D, Dichgans M, et al. Reactive glia in the injured brain acquire stem cell properties in response to sonic hedgehog glia. Cell Stem Cell. 2013a;12(4):426–39. doi: 10.1016/j.stem.2013.01.019. [DOI] [PubMed] [Google Scholar]
- Sirko S, Behrendt G, Johansson PA, Tripathi P, Costa M, Bek S, Heinrich C, Tiedt S, Colak D, Dichgans M, et al. Reactive glia in the injured brain acquire stem cell properties in response to sonic hedgehog. [corrected] Cell Stem Cell. 2013b;12(4):426–39. doi: 10.1016/j.stem.2013.01.019. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32(12):638–47. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone DM, Hynes M, Armanini M, Swanson TA, Gu Q, Johnson RL, Scott MP, Pennica D, Goddard A, Phillips H, et al. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature. 1996;384(6605):129–34. doi: 10.1038/384129a0. [DOI] [PubMed] [Google Scholar]
- Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, Scott MP, Beachy PA. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406(6799):1005–9. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- Vogt DL, Thomas D, Galvan V, Bredesen DE, Lamb BT, Pimplikar SW. Abnormal neuronal networks and seizure susceptibility in mice overexpressing the APP intracellular domain. Neurobiol Aging. 2011;32(9):1725–9. doi: 10.1016/j.neurobiolaging.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Yu S, Simonyi A, Sun GY, Sun AY. Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Mol Neurobiol. 2005;31(1–3):3–16. doi: 10.1385/MN:31:1-3:003. [DOI] [PubMed] [Google Scholar]
- Wang Y, Imitola J, Rasmussen S, O’Connor KC, Khoury SJ. Paradoxical dysregulation of the neural stem cell pathway sonic hedgehog-Gli1 in autoimmune encephalomyelitis and multiple sclerosis. Ann Neurol. 2008;64(4):417–27. doi: 10.1002/ana.21457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A, Piaton G, Lubetzki C. Astrocytes--friends or foes in multiple sclerosis? Glia. 2007;55(13):1300–12. doi: 10.1002/glia.20546. [DOI] [PubMed] [Google Scholar]
- Zhang XM, Zhu J. Kainic Acid-induced neurotoxicity: targeting glial responses and glia-derived cytokines. Curr Neuropharmacol. 2011;9(2):388–98. doi: 10.2174/157015911795596540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Laterra J, Pomper MG. Hedgehog pathway inhibitor HhAntag691 is a potent inhibitor of ABCG2/BCRP and ABCB1/Pgp. Neoplasia. 2009;11(1):96–101. doi: 10.1593/neo.81264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng XY, Zhang HL, Luo Q, Zhu J. Kainic acid-induced neurodegenerative model: potentials and limitations. J Biomed Biotechnol. 2011;2011:457079. doi: 10.1155/2011/457079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Zheng H, Shao X, Wang W, Yao Q, Li Z. Excitotoxicity of TNFalpha derived from KA activated microglia on hippocampal neurons in vitro and in vivo. J Neurochem. 2010;114(2):386–96. doi: 10.1111/j.1471-4159.2010.06763.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supp FigureS1-S2
Supplemental Figure 1. Reactive gliosis reached maximum levels at 7 days post KA-injection. Whole mount GFAP IHC images of KA-treated mice at days 0, 3, 5 and 7 demonstrate maximum activation at day 7.
Supplemental Figure 2. Cyclopamine pre-treatment and subsequent treatment had no significant impact on mouse survival or Gli-light. A, Cyclopamine pre-treatment did not show a statistically significant impact on mouse survival after KA injection but showed some tendency towards worsening symptoms. n=11 for Vehicle+KA and n=12 for Cyclopamine+KA groups. B, There was no significant difference in Gli-light determined by BLI in Vehicle+KA compared to Cyclopamine+KA.