Phospho.ELM: A database of experimentally verified phosphorylation sites in eukaryotic proteins (original) (raw)
Abstract
Background
Post-translational phosphorylation is one of the most common protein modifications. Phosphoserine, threonine and tyrosine residues play critical roles in the regulation of many cellular processes. The fast growing number of research reports on protein phosphorylation points to a general need for an accurate database dedicated to phosphorylation to provide easily retrievable information on phosphoproteins.
Description
Phospho.ELM http://phospho.elm.eu.org is a new resource containing experimentally verified phosphorylation sites manually curated from the literature and is developed as part of the ELM (Eukaryotic Linear Motif) resource. Phospho.ELM constitutes the largest searchable collection of phosphorylation sites available to the research community. The Phospho.ELM entries store information about substrate proteins with the exact positions of residues known to be phosphorylated by cellular kinases. Additional annotation includes literature references, subcellular compartment, tissue distribution, and information about the signaling pathways involved as well as links to the molecular interaction database MINT. Phospho.ELM version 2.0 contains 1703 phosphorylation site instances for 556 phosphorylated proteins.
Conclusion
Phospho.ELM will be a valuable tool both for molecular biologists working on protein phosphorylation sites and for bioinformaticians developing computational predictions on the specificity of phosphorylation reactions.
Keywords: post-transcriptional modification, protein kinase, bioinformatics
Background
The reversible phosphorylation of serine, threonine and tyrosine residues by enzymes of the kinase and phosphatase superfamilies is the most abundant post translational modification in intracellular proteins [1,2] and is an important mechanism for modulating (regulating) many cellular processes such as proliferation, differentiation and apoptosis. Eukaryotic protein kinases form one of the largest multigene families, and the full sequencing of the human genome has allowed the identification of almost all human protein kinases, representing about 1.7% of all human genes [3]. The role of an individual protein kinase in a particular cellular process, however, will be fully explained only when the basis for kinase substrate specificity will be better understood. Determining the substrate specificity of protein kinases is still one of the major challenges in molecular biology.
Phosphorylation site predictors such as the CBS predictor NetPhos [4] based on artificial neural networks [5,6], or Scansite [7] based on peptide library derived position-specific scoring matrices (PSSM) [8] have gone some way to allowing molecular biologists to identify potential kinase substrate sites in query proteins, but suffer to a degree from over-prediction. The ELM resource attempts to reduce such problems using contextual filtering of motifs based on structure, cell compartment, taxonomic limits, and other properties of proteins [9].
Due to the biological importance of protein kinases in cell signaling and the steadily growing volume of reports identifying phosphorylation sites [10] it has become impractical for experimental molecular biologists to keep track of all the phosphorylation modifications of proteins within their area of research. Furthermore, large-scale proteomic and system biology approaches to cell regulation cannot succeed without full access to phosphorylation data. There is therefore a need to create and maintain a comprehensive database of known, experimentally verified phosphorylation sites within proteins.
We describe here Phospho.ELM [11], a server interfaced to a manually curated database of phosphorylation sites (instances) that provides easy access to information from the primary scientific literature concerning experimentally verified serine, threonine and tyrosine phosphorylation sites in eukaryotic proteins.
Construction and content
Phospho.ELM is developed and deployed with open source software. The database management system used is PostgreSQL [12]. The software was developed in Python 2.2 including some modules from the BioPython.org project for retrieval of information from SWISS-PROT and the PyGreSQL module for PostgreSQL interfacing. The web interface software uses the CGI model framework [13].
The Phospho.ELM 1.0 database contained a dataset of 289 proteins. The current release (Phospho.ELM 2.0) has integrated data from PhosphoBase to give a total of 556 proteins (299 human, 52 mouse, 54 rat, and 151 from other species). The Phospho.ELM dataset represents the largest collection of experimentally verified phosphorylation sites: the annotated proteins contain 556 tyrosine, 913 serine and 234 threonine phosphorylation sites (instances) that are verified substrates for 119 different protein kinases (Table 1).
Table 1.
Selected protein kinases, their class, the number of known protein substrates and the instances recorded in Phospho.ELM.
| Kinase | Type | Substrates | Instances |
|---|---|---|---|
| CK2 | Ser/Thr kinase | 54 | 138 |
| PKA | Ser/Thr kinase | 88 | 170 |
| PDKI | Ser/Thr kinase | 12 | 17 |
| Src | non-receptor Tyr Kinase | 40 | 69 |
| Abl | non-receptor Tyr Kinase | 14 | 21 |
| FAK | non-receptor Tyr Kinase | 7 | 11 |
| IR | receptor Tyr Kinase | 11 | 36 |
| EGFR | receptor Tyr Kinase | 19 | 43 |
In the Phospho.ELM database information is presented in two classes, instance and phosphoprotein. The key information consists of the phosphorylated site (instance) and its flanking sequence within a protein, for which experimental evidence has been found in the literature. Moreover, annotations to each instance include (where known) the kinase(s) that phosphorylate(s) the given site, the domain(s) that bind to a phosphorylated motif (this is particularly relevant for tyrosine phosphorylation, e.g. SH2), and a link to the ELM server to retrieve further information about the kinase and the regular expression used for prediction of kinase substrates (see Fig. 1). Where available, hyperlinks are provided to protein structures containing phosphorylated residues [14]. Furthermore, additional information for each protein kinase substrate includes the subcellular compartment (annotated with Gene Ontology terms [15,16]), tissue distribution, a list of interaction partners derived from the MINT database [17], and a diagram of a signaling pathway in which the protein is involved. When one is available we provide a link to the BioCarta-Charting Pathways of Life [18]. Controlled vocabularies to describe experimental evidence [19] will soon be included in the database.
Figure 1.
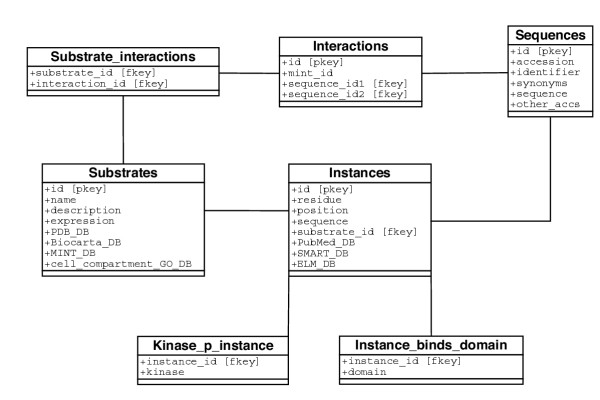
The simplified Phospho.ELM database scheme. The key data objects are Substrates (phosphoprotein) and Instances for which relevant information is stored, as well as links to external databases. pkey and fkey stand for "primary key" and "foreign key", respectively.
The database can be searched by protein name (for the substrate), kinase name to get a list of known substrates, or by phosphopeptide-binding domain to retrieve all instances interacting with the given domain. An example of a search output is given in Fig. 2.
Figure 2.
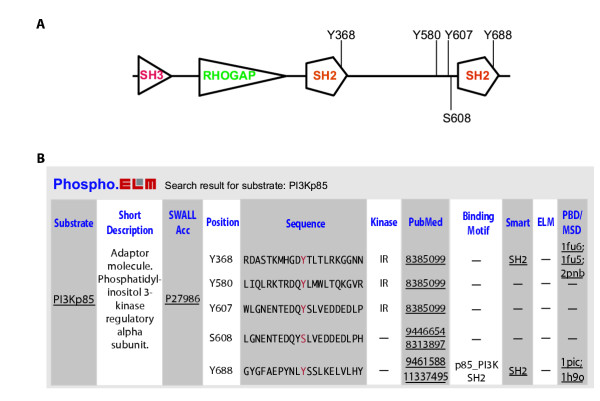
A) Scheme for the PI3Kp85 protein with domains and phosphorylation sites. B) Output example of keyword search using PI3Kp85. Information about the phosphorylated sites includes the flanking sequence, the PubMed reference, the kinase responsible for the phosphorylation and links to additional information for the substrate and other relevant databases.
Utility and discussion
The phospho.ELM server will allow both 'wet-lab' biologists and bioinformaticians to easily retrieve extensive information about phosphoproteins. Indeed, further advance in the field of kinase-specific phosphorylation site prediction requires the combination of advanced algorithms together with high quality annotation of phosphorylation data. As such, Phospho.ELM is a valuable source of reliable data for the development of new predictors. Currently, sufficient data for training a machine learning method (e.g. circa 25 instances are needed for a neural network) are available only for the most well characterized kinases, however this number is expected to increase rapidly as a result of high-throughput proteomics initiatives. A method for kinase-specific substrate prediction of six S/T-kinases has recently been developed at the Center for Biological Sequence Analysis (N. Blom, personal communication).
Conclusions
Currently the set of known protein modification sites that are used to regulate the cell are poorly integrated into bioinformatics resources. This is hampering the research of systems biologists and research groups large and small. With Phospho.ELM we are working towards improving the catalogue for phosphorylation sites. Users are encouraged to help us to keep the database up-to-date by submitting additional information and their datasets of phosphorylation sites for integration into Phospho.ELM. Those interested in becoming data submission partner can send an email to phospho@elm.eu.org.
Availability and requirements
Phospho.ELM can be accessed on the public Apache2 powered website at http://phospho.elm.eu.org.
Author's contributions
FD and SC were responsible for the annotation process and the Web design. Design of the database structure and implementation of the server software is credited to CG. RL contributed to the analysis of the data. AV is involved in linking structural databases. TSP implemented the PhosphoBase database. BK, NB and TJG were responsible for the overall project coordination. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
We wish to thank the EU (grant QLRI-CT-2000-00127) and Cellzome for funding the ELM project. Many thanks to Arnaud Ceol and Ivica Letunic for technical support. We are grateful to Bill Hunter, Sophie Chabanis-Davidson, Aidan Budd and Lars Juhl-Jensen for their insightful comments and suggestions.
Contributor Information
Francesca Diella, Email: diella@embl.de.
Scott Cameron, Email: s.cameron@dundee.ac.uk.
Christine Gemünd, Email: gemuend@embl.de.
Rune Linding, Email: linding@embl.de.
Allegra Via, Email: allegra@cbm.bio.uniroma2.it.
Bernhard Kuster, Email: Bernhard.Kuester@cellzome.com.
Thomas Sicheritz-Pontén, Email: thomas@cbs.dtu.dk.
Nikolaj Blom, Email: nikob@cbs.dtu.dk.
Toby J Gibson, Email: toby.gibson@embl.de.
References
- Hunter T. Signaling-2000 and beyond. Cell. 2000;100:113–127. doi: 10.1016/S0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- Cohen P. The origins of protein phosphorylation. Nat Cell Biol. 2002;4:E127–E130. doi: 10.1038/ncb0502-e127. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- NetPhos http://www.cbs.dtu.dk/services/NetPhos/
- Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294:1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- Kreegipuu A, Blom N, Brunak S. PhosphoBase, a database of phosphorylation sites: release 2.0. Nucleic Acids Res. 1999;27:237–239. doi: 10.1093/nar/27.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scansite http://scansite.mit.edu
- Yaffe MB, Leparc GG, Lai J, Obata T, Volinia S, Cantley LC. A motif-based profile scanning approach for genome-wide prediction of signaling pathways. Nat Biotechnol. 2001;19:348–353. doi: 10.1038/86737. [DOI] [PubMed] [Google Scholar]
- Puntervoll P, Linding R, Gemund C, Chabanis-Davidson S, Mattingsdal M, Cameron S, Martin DM, Ausiello G, Brannetti B, Costantini A, Ferre F, Maselli V, Via A, Cesareni G, Diella F, Superti-Furga G, Wyrwicz L, Ramu C, McGuigan C, Gudavalli R, Letunic I, Bork P, Rychlewski L, Kuster B, Helmer-Citterich M, Hunter WN, Aasland R, Gibson TJ. ELM server: A new resource for investigating short functional sites in modular eukaryotic proteins. Nucleic Acids Res. 2003;31:3625–3630. doi: 10.1093/nar/gkg545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight ZA, Schilling B, Row RH, Kenski DM, Gibson BW, Shokat KM. Phosphospecific proteolysis for mapping sites of protein phosphorylation. Nat Biotechnol. 2003;21:1047–1054. doi: 10.1038/nbt863. [DOI] [PubMed] [Google Scholar]
- Phospho.ELM http://phospho.elm.eu.org
- PostgreSQL http://www.postgresql.org
- Ramu C, Gemund C. CGImodel:CGI programming made easy with python. Linux J. 2000;75:142–149. [Google Scholar]
- Boutselakis H, Dimitropoulos D, Fillon J, Golovin A, Henrick K, Hussain A, lonides J, John M, Keller PA, Krissinel E, McNeil P, Naim A, Newman R, Oldfield T, Pineda J, Rachedi A, Copeland J, Sitnov A, Sobhany S, Suarez-Uruena A, Swaminathan J, Tagari M, Tate J, Tromm S, Velankar S, Vranken W. E-MSD: the European Bioinformatics Institute Macromolecular Structure Database. Nucleic Acids Res. 2003;31:458–462. doi: 10.1093/nar/gkg065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, Richter J, Rubin GM, Blake JA, Bult C, Dolan M, Drabkin H, Eppig JT, Hill DP, Ni L, Ringwald M, Balakrishnan R, Cherry JM, Christie KR, Costanzo MC, Dwight SS, Engel S, Fisk DG, Hirschman JE, Hong EL, Nash RS, Sethuraman A, Theesfeld CL, Botstein D, Dolinski K, Feierbach B, Berardini T, Mundodi S, Rhee SY, Apweiler R, Barrell D, Camon E, Dimmer E, Lee V, Chisholm R, Gaudet P, Kibbe W, Kishore R, Schwarz EM, Sternberg P, Gwinn M, Hannick L, Wortman J, Berriman M, Wood V, de la Cruz N, Tonellato P, Jaiswal P, Seigfried T, White R. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32 Database issue:D258–261. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene Ontology http://www.geneontology.org
- Zanzoni A, Montecchi-Palazzi L, Quondam M, Ausiello G, Helmer-Citterich M, Cesareni G. MINT: a Molecular INTeraction database. FEBS Lett. 2002;513:135–140. doi: 10.1016/S0014-5793(01)03293-8. [DOI] [PubMed] [Google Scholar]
- BioCarta-Charting Pathways of Life http://www.biocarta.com/
- Hermjakob H, Montecchi-Palazzi L, Bader G, Wojcik J, Salwinski L, Ceol A, Moore S, Orchard S, Sarkans U, von Mering C, Roechert B, Poux S, Jung E, Mersch H, Kersey P, Lappe M, Li Y, Zeng R, Rana D, Nikolski M, Husi H, Brun C, Shanker K, Grant SG, Sander C, Bork P, Zhu W, Pandey A, Brazma A, Jacq B, Vidal M, Sherman D, Legrain The HUPO PSI's molecular interaction format – a community standard for the representation of protein interaction data. Nat Biotechnol. 2004;22:177–183. doi: 10.1038/nbt926. [DOI] [PubMed] [Google Scholar]