Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease (original) (raw)
Abstract
Objective:
Diverse autolysosomal proteins were quantified in neurally derived blood exosomes from patients with Alzheimer disease (AD) and controls to investigate disordered neuronal autophagy.
Methods:
Blood exosomes obtained once from patients with AD (n = 26) or frontotemporal dementia (n = 16), other patients with AD (n = 20) both when cognitively normal and 1 to 10 years later when diagnosed, and case controls were enriched for neural sources by anti-human L1CAM antibody immunoabsorption. Extracted exosomal proteins were quantified by ELISAs and normalized with the CD81 exosomal marker.
Results:
Mean exosomal levels of cathepsin D, lysosome-associated membrane protein 1 (LAMP-1), and ubiquitinylated proteins were significantly higher and of heat-shock protein 70 significantly lower for AD than controls in cross-sectional studies (p ≤ 0.0005). Levels of cathepsin D, LAMP-1, and ubiquitinylated protein also were significantly higher for patients with AD than for patients with frontotemporal dementia (p ≤ 0.006). Step-wise discriminant modeling of the protein levels correctly classified 100% of patients with AD. Exosomal levels of all proteins were similarly significantly different from those of matched controls in 20 patients 1 to 10 years before and at diagnosis of AD (p ≤ 0.0003).
Conclusions:
Levels of autolysosomal proteins in neurally derived blood exosomes distinguish patients with AD from case controls and appear to reflect the pathology of AD up to 10 years before clinical onset. These preliminary results confirm in living patients with AD the early appearance of neuronal lysosomal dysfunction and suggest that these proteins may be useful biomarkers in large prospective studies.
There is an urgent need for biomarkers that accurately detect pathogenic components of Alzheimer disease (AD) before appearance of neurologic signs. Early treatments directed to such targets could limit or reverse neuronal damage and prevent development of overt AD. Recent analyses of neurally derived plasma exosomal proteins have shown significantly higher levels of the pathogenic proteins P-T181-tau, P-S396-tau, and β-amyloid (Aβ)1-42 in AD than in case controls.1 Discriminant modeling of these exosomal protein levels correctly classified more than 96% of patients with AD. Neurally derived plasma exosomal levels of P-T181-tau, P-S396-tau, and Aβ1-42 also were significantly higher in preclinical AD than for controls up to 10 years before appearance of neurologic signs.1
Altered levels of phosphorylated forms of the insulin receptor proximal signaling protein, termed insulin receptor substrate (IRS), in neurally derived plasma exosomes supported the possibility of brain insulin resistance in AD.2 Neurally derived exosomal levels of insulin signal-diminishing P-S312-IRS-1 and insulin signal-enhancing P-panY-IRS-1, and the ratio of P-S312-IRS-1 to P-panY-IRS-1 (R, an index of insulin resistance) were significantly different in AD and type 2 diabetes mellitus than for case controls. R levels also were significantly higher in AD than in type 2 diabetes mellitus. In 22 patients with AD studied longitudinally, neurally derived exosomal levels of P-S312-IRS-1, P-panY-IRS-1, and R were significantly different than for controls both 1 to 10 years before and at diagnosis of AD.2
Dystrophic neurons showing segmental distensions with increased numbers of lysosomes and autophagic vacuoles are prominent in AD, but protein biodegradation in resultant autolysosomes fails progressively.3 Upregulation and then failure of neuronal autophagic-lysosomal systems in AD causes lysosomal components to leak into neural extracellular fluid around plaques and into the CSF.4,5 Although fusion of exosome-containing multivesicular bodies with lysosomes has been considered a mechanism for elimination of unnecessary exosomal proteins, autolysosomal dysfunction of neurons in AD may result in addition of lysosomal proteins to exosomal cargo for facilitation of their removal from neurons.3,6
We now examine extracts of neurally derived plasma exosomes from patients with AD, patients with frontotemporal dementia (FTD), and cognitively normal matched controls for differences in quantities of distinct types of lysosome-associated proteins. Results support there being major abnormalities of neural cell autophagic-lysosomal pathways in AD.
METHODS
Experimental design and patient evaluation.
For cross-sectional studies, we retrospectively identified 26 patients with amnestic mild cognitive impairment (aMCI, n = 18) or mild to moderate dementia (n = 8) from AD, who had donated blood once in the Clinical Research Unit of the National Institute on Aging (NIA), Baltimore, MD, or at the Jewish Home of San Francisco (JHSF), CA (table 1). For longitudinal studies, 20 additional patients with AD were identified retrospectively based on their provision of blood twice at the Mayo Clinic or the University of Kentucky, first when cognitively intact and later at diagnosis of AD (table 1). Intervals between the 2 blood samples (number of patients) were 1 to 5 years (7) and 6 to 10 years (13). Each patient in both cross-sectional and longitudinal groups had mental status testing and an MRI, and some had measurements of CSF Aβ1-42, total tau, and P-T181-tau. Cognitively normal control participants (AC group) were from the NIA or JHSF and were matched to characteristics of the patients with AD in cross-sectional studies or preclinical (AP) patients in longitudinal studies. Samples from patients in the longitudinal study were analyzed without knowledge of the clinical data.
Table 1.
Characteristics of patients and control participants
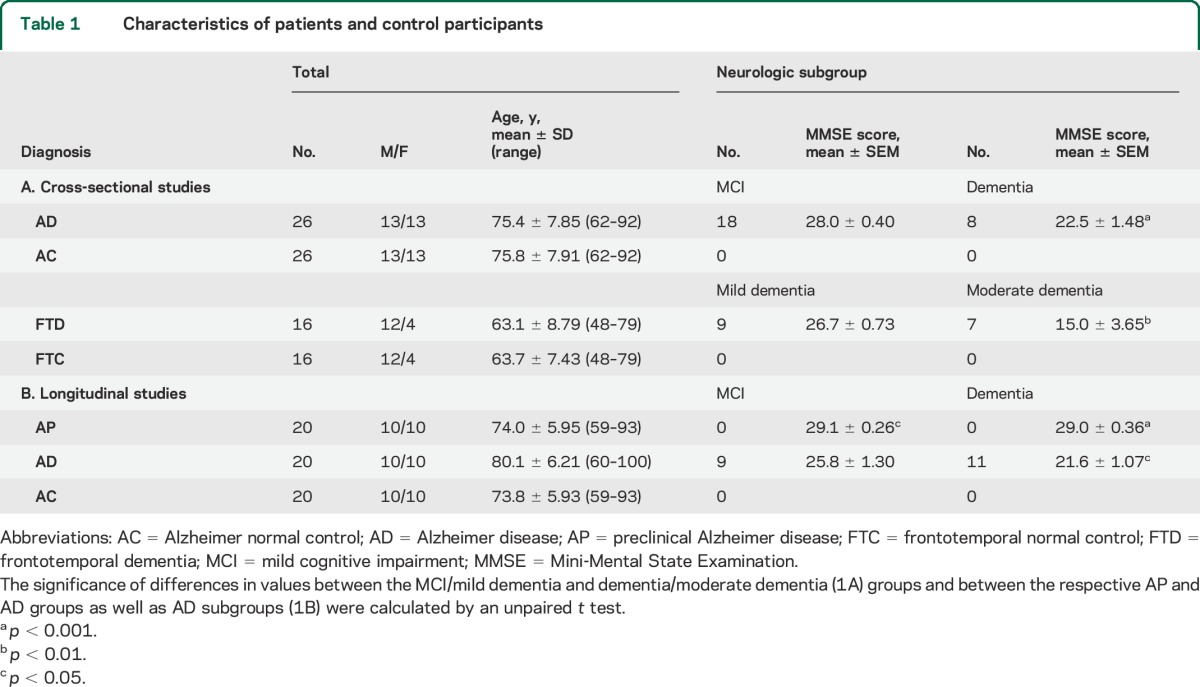
Patients were classified as having aMCI according to Petersen criteria and had Clinical Dementia Rating global scores of 0.5.7,8 Those with probable AD and mild to moderate dementia were diagnosed by Dubois criteria or for University of Kentucky patients by NINCDS-ADRDA (National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Association) criteria and had Clinical Dementia Rating global scores of at least 1.0.9,10 A CSF level of Aβ1-42 <192 pg/mL supported a diagnosis of AD.11 Mini-Mental State Examination (MMSE) and the Alzheimer's Disease Assessment Scale–cognitive subscale were conducted as described.12,13 Twenty-four of the 26 cross-sectional patients with AD were taking an acetyl-cholinesterase inhibitor and/or memantine, and 5 were on antidepressant medications.
Sixteen patients with the behavioral variant of FTD were evaluated at the Memory and Aging Center of the Department of Neurology of the University of California, San Francisco. Their diagnosis and assignment to mild or moderate dementia groups (table 1) were based on standard clinical and mental status criteria, including discriminant analyses of neuropsychiatric and other elements that distinguish FTD from AD.14,15 Seven of the patients with FTD were receiving an antidepressant, 2 were taking an acetyl-cholinesterase inhibitor, and one was on memantine. Cognitively normal control participants (FTC) were matched to patients with FTD as for AC controls.
Blood and CSF sampling of patients and control participants.
Each participant studied and some patient designates signed a consent form approved with the protocol at each institution. Thirty milliliters of venous blood was drawn into 1 mL of saline with ethylenediaminetetraacetic acid or heparin, incubated for 10 minutes at room temperature, and centrifuged for 15 minutes at 2,500_g_.1 Plasma was stored in 0.5-mL aliquots at −80°C. CSF levels of total tau, P-T181-tau, and Aβ1-42 were quantified by Luminex xMAP technology using Innogenetics INNO-BIA Alz Bio3 kits (Innogenetics, Ghent, Belgium).
Isolation of exosomes from plasma for extraction and ELISA quantification of exosome proteins.
One-half milliliter of plasma was incubated with thromboplastin-D (Fisher Scientific, Inc., Hanover Park, IL) followed by addition of calcium- and magnesium-free Dulbecco balanced salt solution (DBS−2) with protease inhibitor cocktail (Roche Applied Sciences, Inc., Indianapolis, IN) and phosphatase inhibitor cocktail (Pierce Halt, Thermo Scientific, Inc., Rockford, IL).1,2 After centrifugation, supernates were incubated with ExoQuick exosome precipitation solution (EXOQ; System Biosciences, Inc., Mountain View, CA) and resultant suspensions centrifuged at 1,500_g_ for 30 minutes at 4°C. Each pellet was resuspended in 250 μL of distilled water with inhibitor cocktails for immunochemical enrichment of exosomes from neural sources.1
Exosome suspensions were incubated with 2 µg of mouse anti-human CD171 (L1CAM neural adhesion protein) biotinylated antibody (clone 5G3; eBioscience, San Diego, CA) in 50 µL of 3% bovine serum albumin (BSA) for 60 minutes at 20°C followed by addition of 10 μL of Streptavidin-Plus UltraLink resin (Pierce-Thermo Scientific, Inc.) in 40 µL of 3% BSA.1,2 After centrifugation, pellets were resuspended in 50 µL of 0.05 M glycine-HCl (pH 3.0), incubated at 4°C for 10 minutes, mixed with 50 µL of 3% BSA, and recentrifuged. Each supernate in a new Eppendorf tube received 5 µL of 1 M Tris-HCl (pH 8.0) followed by 0.40 mL of M-PER mammalian protein extraction reagent (Thermo Scientific, Inc.) containing protease and phosphatase inhibitors, mixed and stored at −80°C.
Exosome proteins were quantified by human-specific ELISAs for total ubiquitin (FIVEphoton Biochemicals, San Diego, CA), lysosome-associated membrane protein 1 (LAMP-1) (USBiological Life Sciences, Salem, MA), heat-shock protein 70 (HSP70; high sensitivity) (Enzo Life Sciences, Farmingdale, NY), cathepsin D (EMD Millipore Corp., Billerica, MA), neuron-specific enolase (R&D Systems, Inc., Minneapolis, MN), neurofilament light chain (American Research Products, Waltham, MA; Cusabio), type 1 neural cell adhesion molecule (NCAM-1) (RayBiotech, Norcross, GA), and tetraspanin exosome marker human CD81 (American Research Products, Cusabio) with verification of the CD81 antigen standard curve using human purified recombinant CD81 antigen (OriGene Technologies, Inc., Rockville, MD), according to suppliers' directions. The mean value for all determinations of CD81 in each assay group was set at 1.00 and the relative values for each sample used to normalize their recovery. Evidence for enrichment of exosomes from neural sources comes from comparison of the levels of known neural markers relative to the CD81 exosome marker after initial precipitation and after immuno-isolation by anti-L1CAM antibody absorption (table e-1 on the _Neurology_® Web site at Neurology.org). Although there are differences in exosomal levels of most neural markers between patients with AD and controls, immune-specific enrichment increased the levels of all neural markers in both groups by 8- to 13-fold.
Statistical analyses.
The statistical significance of differences between means for cross-sectional patient groups and between each patient group and their respective control group was determined with an unpaired t test including a Bonferroni correction (GraphPad Prism 6, La Jolla, CA). Discriminant classifier analyses were conducted by the Wilks Λ method to assess the performance of each exosomal protein and the combined set in patient classification, as described.1 Receiver operating characteristic (ROC) analyses were conducted under the nonparametric distribution assumption for standard error of area to determine the performance of classifier models (SPSS version 21.0; IBM Corp., Armonk, NY).1 For longitudinal analyses, the significance of differences between serial values for patients with AD taken before and after onset of aMCI or dementia was calculated with a paired t test (GraphPad).
RESULTS
Patient characteristics.
More of the patients with AD in cross-sectional studies had aMCI than dementia and the latter group had significantly lower MMSE scores than the former (p < 0.001) (table 1A). The MMSE scores for patients with AD who had dementia in the longitudinal group also were significantly lower than those with aMCI (table 1B). MMSE scores for both neurologic subgroups of patients with AD in the longitudinal study were significantly higher in their preclinical phase (AP) than after development of AD (table 1B). The 16 patients with FTD were a younger group than those with AD and had more with mild dementia than moderate dementia, with the latter group having significantly lower MMSE scores (p < 0.01) (table 1A).
Exosomal protein levels in cross-sectional studies and relationship to severity of cognitive loss.
The aspartyl endoproteinase cathepsin D normally is localized in lysosomes and endosomes in several types of neural cells, where it cleaves diverse proteins.16–18 Neuronal lysosomal proteolytic dysfunction in AD results in massive accumulation of cathepsin D–rich autolysosomes and elevated secretion of cathepsin D, which we hypothesized was exosome-associated.6,19,20 Exosomal levels of cathepsin D for the patients with AD in cross-sectional studies were 17.7 ± 0.80 ng/mL (mean ± SEM), which were significantly higher than the level of 8.35 ± 0.27 ng/mL for the control AC participants (p < 0.0001) (figure 1). Cathepsin D levels for patients with FTD of 12.8 ± 0.75 ng/mL were significantly lower than those for patients with AD (p = 0.0001), but were significantly higher than those of 6.23 ± 0.15 ng/mL for their FTC control group (p < 0.0001).
Figure 1. Levels of plasma exosomal proteins in patients with AD, FTD, and cognitively normal case controls.

The horizontal line in each cluster here and in figure 2 depicts the mean for that set. AC = Alzheimer normal control; AD = Alzheimer disease; FTC = frontotemporal normal control; FTD = frontotemporal dementia; HSP70 = heat-shock protein 70; LAMP-1 = lysosome-associated membrane protein 1.
The lysosomal outer membrane proteins, termed types 1 and 2 lysosome-associated membrane proteins (LAMP-1 and LAMP-2), associate with and stabilize polypeptide translocational systems, such as transporter associated with antigen processing like (TAPL).21 If the autophagic-lysosomal dysfunction of neurons in AD results in exosomal uptake of lysosomal membrane proteins as well as lysosomal granule cathepsin D, we hypothesized that LAMP-1 levels are elevated in neurally derived plasma exosomes from patients with AD as contrasted with those from AC controls. Exosomal levels of LAMP-1 for the patients with AD in cross-sectional studies were 1,808 ± 204 pg/mL (mean ± SEM), which were significantly higher than the level of 946 ± 119 pg/mL for the control AC participants (p = 0.00051) (figure 1). The LAMP-1 levels for the patients with FTD of 1,071 ± 62.7 pg/mL were significantly lower than those of the patients with AD (p = 0.0058) and were not significantly different than those of 1,147 ± 88.9 pg/mL for their FTC control group (p = 0.4892).
Ubiquitinylation and elimination from brain of some neuropathic proteins by proteosomal and autophagic-lysosomal systems are abnormal in AD.3 Neural localization of proteins with critical functions, such as synaptic transmission, and efficient sorting of proteins into exosome-containing multivesicular bodies both require precise ubiquitin labeling, which is abnormal in AD.22–24 To examine exosomal consequences of neural protein ubiquitinylation defects in AD, total ubiquitin-labeled proteins were quantified in neural-derived plasma exosomes. Exosomal levels of ubiquitinylated proteins for the patients with AD in cross-sectional studies were 477 ± 25.4 pg/mL (mean ± SEM), which were significantly higher than the level of 225 ± 10.1 pg/mL for control AC participants (p < 0.0001) (figure 1). The levels of ubiquitinylated proteins for the patients with FTD of 255 ± 11.5 pg/mL were significantly lower than those of the patients with AD (p < 0.0001), but were not significantly different than those of 228 ± 8.53 pg/mL for their FTC control group (p = 0.0726).
The HSP70 family of protein chaperones exhibits diverse effects on configuration, oligomerization, transmembrane transport, and survival of many proteins, and promotes removal of damaged proteins.25,26 HSP70 proteins also prevent abnormal alterations in lysosomal permeability and inhibit apoptosis.27,28 HSP70 release from some cells is largely dependent on exosome secretion, rather than the common secretory or lipid raft-mediated pathways, and the level of HSP70 in exosomal cargo is increased by cellular stress.29 Levels of HSP70 in neural-derived plasma exosomes were quantified to identify deficiencies that might diminish autolysosomal clearance of neuropathic proteins in AD.
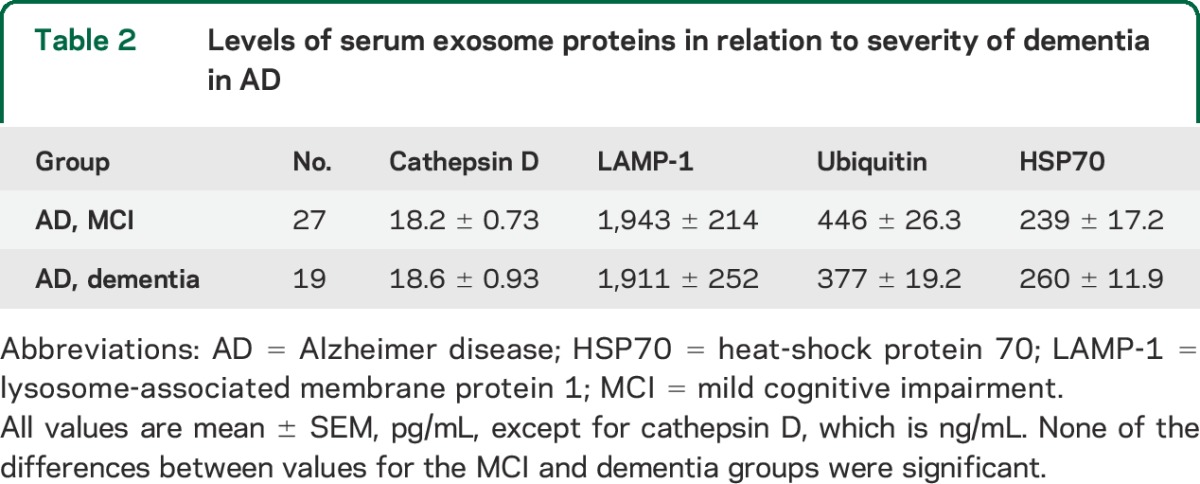
Exosomal levels of HSP70 for the patients with AD in cross-sectional studies were 246 ± 18.0 pg/mL (mean ± SEM), which were significantly lower than the level of 394 ± 15.2 pg/mL for the control AC participants (p < 0.0001) (figure 1). The levels of HSP70 for the patients with FTD of 165 ± 4.39 pg/mL were significantly lower than those of the patients with AD (p = 0.0012) and those of 429 ± 15.6 pg/mL for their FTC control group (p < 0.0001). Exosomal levels of cathepsin D, LAMP-1, ubiquitinylated proteins, and HSP70 for patients with AD in the cross-sectional studies were the same for the mild cognitive impairment (MCI) and dementia neurologic subgroups (table 2). Furthermore, there were no differences in exosomal levels of any of the autolysosomal proteins in cross-sectional or longitudinal studies between patients with AD who had APOE 4/4 genotype (5) or x/4 genotype (9) and those without a type 4 APOE allele.
Table 2.
Levels of serum exosome proteins in relation to severity of dementia in AD
Stepwise discriminant classification of AD vs AC incorporated cathepsin D, then ubiquitinylated proteins, and finally HSP70, but not LAMP-1 (figure e-1). ROC curves of AD vs AC showed an area under the curve of 1.0 for both cathepsin D and composite scores from the final model with correct classification of 100% of the patients with AD. Similar ROC analyses correctly classified 100% of patients with FTD vs FTC controls and 95.8% of patients with AD vs patients with FTD.
Longitudinal studies of exosomal protein levels in conversions from preclinical to overt AD.
The exosomal levels of cathepsin D, LAMP-1, ubiquitinylated proteins, and HSP70 for the longitudinal AC control group were indistinguishable from those of the cross-sectional AC control group at 8.50 ± 0.36 ng/mL (mean ± SEM), 1,035 ± 119 pg/mL, 206 ± 7.46 pg/mL, and 392 ± 14.2 pg/mL, respectively (figure 2). The exosomal levels of cathepsin D, LAMP-1, ubiquitinylated proteins, and HSP70 for the AD group at time of diagnosis of MCI or dementia were 19.0 ± 0.70 ng/mL, 2,080 ± 257 pg/mL, 347 ± 13.9 pg/mL, and 250 ± 11.8 pg/mL, respectively, which were significantly higher or for HSP70 lower than corresponding values for the AC group (p < 0.0001 for all, except LAMP-1 where p = 0.0003). For the preclinical AP group, 1 to 10 years before diagnosis of AD, exosomal levels of the same proteins were 18.4 ± 0.68 ng/mL, 2,638 ± 354 pg/mL, 364 ± 13.9 pg/mL, and 244 ± 16.4 pg/mL, respectively, which were significantly different from those of the AC control group (p < 0.0001 for all) (figure 2). There were no significant differences between any of the exosomal protein levels of the preclinical AP group and those of the AD group at neurologic conversion. Finally, there were no distinctions between AP or AD exosomal protein levels for those patients who converted to MCI as contrasted with dementia or for those who converted neurologically in 1 to 5 years as contrasted with 6 to 10 years.
Figure 2. Sequential levels of plasma exosomal proteins in patients with AD measured first at a time of normal cognition (preclinical, AP) and later at the time of development of aMCI or dementia (AD), as contrasted with cognitively normal control participants (AC) matched to individuals in their preclinical phase.

AC = Alzheimer normal control; AD = Alzheimer disease; aMCI = amnestic mild cognitive impairment; AP = preclinical Alzheimer disease; FTC = frontotemporal normal control; FTD = frontotemporal dementia; HSP70 = heat-shock protein 70; LAMP-1 = lysosome-associated membrane protein 1.
DISCUSSION
The results of analyses of neural-derived plasma exosome proteins derived from lysosomes and related cellular organelles increase our understanding of lysosomal dysfunction in AD and may provide useful biomarkers for recognizing preclinical AD. The levels of cathepsin D, LAMP-1, and ubiquitinylated proteins all were significantly higher and those of HSP70 significantly lower in neural-derived plasma exosomes from AD plasmas than from control plasmas and there was no overlap in values for cathepsin D (figure 1). Thus, autophagocytic-lysosomal dysfunction in AD results in increased cargo levels per exosome of the intrinsic lysosomal components cathepsin D and LAMP-1, as well as ubiquitinylated proteins destined for elimination in autolysosomes, but lower cargo levels of cytosolic HSP70 that normally is secreted predominantly in exosomes. AD was statistically distinguished from FTD by levels of all these proteins, but differences between FTD and FTC were significant only for cathepsin D and HSP70. Furthermore, none of the lysosome-related neural-derived plasma exosome proteins distinguished AD from FTD as well as P-S396-tau, where there was no overlap.1
Longitudinal studies of lysosome-related neural-derived plasma exosomal proteins showed significant differences in levels between those of controls and of patients with AD both at their preclinical phase 1 to 10 years before diagnosis (AP) and when manifesting clinically apparent signs of AD (figure 2). None of the exosomal levels of lysosome-related proteins was significantly higher in AD than in the preclinical phase (figure 2), as for neural-derived plasma exosome levels of proteins implicated in pathogenesis of AD, except Aβ1-42, and of abnormally phosphorylated forms of IRS.1,2
The assumption that levels of lysosome-related neurally derived plasma exosomal proteins reflect their relative concentrations in neural cells is based on several findings. Support for their neural source comes from observing >95% cross-binding of neurally enriched exosomes by anti-NCAM-1 and anti-L1CAM antibodies, and 8- to 13-fold mean increases by neural enrichment in content of multiple neural markers relative to total precipitated plasma exosomes (table e-1). The number and size distribution of neural-derived exosomes isolated from a given volume of plasma have been the same for patients with AD and AC controls in our limited series.1 Thus, differences between patients with AD and AC controls in neurally derived plasma exosomal levels of proteins implicated in AD pathogenesis presumably reflect principally those of cargo per exosome.1,2 However, other variables that affect exosome biogenesis, secretion, and cargo content must be examined in much larger numbers of patients.
Exosome-containing multivesicular bodies normally either fuse with the plasma membrane for exosome secretion or dock with lysosomes for exosome biodegradation, but in AD appear to import cargo from dysfunctional autolysosomes for transport to the extracellular space.1,6 Neural cellular stress at some stages of AD thereby increase amounts of proteins per exosome, as for stressed leukocyte-derived exosome content of HSP70.29 Furthermore, lysosomal dysfunction induced by some neurotoxic proteins may increase the exosomal cargo of proteins, as we have found (table e-1).30 These diverse mechanisms of autophagic-lysosomal dysfunction in AD provide new targets for potentially therapeutic agents and the proteins are useful biomarkers for large prospective studies.
Numerous problems inherent in retrospective studies of small numbers of carefully selected patients suggest that conclusions of the preliminary investigations here reported may be modified by results of larger prospective analyses. All of our control groups excluded subjects with a family history of AD. The patients in the longitudinal set were selected based on availability of a paired earlier plasma sample for analysis, which restricts assessment of predictive accuracy. Many aspects of assay method reproducibility have met accepted criteria, but full validation series are still in progress. Nonetheless, quantification of 3 other clusters of neural derived exosome proteins has shown the same capacity as that reported here for distinguishing neurodegenerative diseases from matched controls cross-sectionally and longitudinally.1,2,31
Supplementary Material
Data Supplement
ACKNOWLEDGMENT
The authors are grateful to Lynn Kane (JHSF), Anna Karydas (UCSF MAC), Dana Swenson-Dravis and Matthew Miller (Mayo Clinic), Sonya Anderson (University of Kentucky), and Melissa Swaby (NIA) for organizing and distributing clinical materials and data. The authors thank Judith H. Goetzl for expert preparation of graphic illustrations.
GLOSSARY
Aβ
β-amyloid
AD
Alzheimer disease
aMCI
amnestic mild cognitive impairment
AP
preclinical Alzheimer disease
BSA
bovine serum albumin
FTD
frontotemporal dementia
HSP70
heat-shock protein 70
IRS
insulin receptor substrate
JHSF
Jewish Home of San Francisco
LAMP-1
lysosome-associated membrane protein 1
MCI
mild cognitive impairment
MMSE
Mini-Mental State Examination
NCAM-1
type 1 neural cell adhesion molecule
NIA
National Institute on Aging
ROC
receiver operating characteristic
Footnotes
AUTHOR CONTRIBUTIONS
E.J.G. developed analytical methodology, performed laboratory bench work, analyzed data, wrote and edited manuscript. A.B. evaluated patients, analyzed data, edited manuscript. J.B.S. analyzed data, wrote and edited manuscript. E.L.A. evaluated patients, analyzed data, edited manuscript. R.C.P. evaluated patients, analyzed data, edited manuscript. B.L.M. evaluated patients, analyzed data, edited manuscript. D.K. evaluated patients, analyzed data, performed statistical analyses, edited manuscript.
STUDY FUNDING
Intramural Research Program of the National Institute on Aging (NIA; D.K.), UK ADC P30, AG028383 (E.L.A.), and an unrestricted grant for method development from NanoSomiX, Inc. (E.J.G.).
DISCLOSURE
E. Goetzl has filed a provisional application with the US Patent Office for the platform and methodologies described in this report; he was a founder of Nanosomix, but now only receives from them support for purchasing laboratory supplies. A. Boxer declares that outside the submitted studies he has grants from NIH/NIA, grants from Tau Research Consortium, grants from Corticobasal Degeneration Solutions, grants, personal fees, and nonfinancial support from Archer Biosciences, grants from Allon Therapeutics, personal fees from Acetylon, personal fees from iPierian, grants from Genentech, grants from Bristol-Myers Squibb, grants from TauRx, grants from Alzheimer's Association, grants from Bluefield Project to Cure FTD, grants from Association for Frontotemporal Degeneration, grants from Alzheimer's Drug Discovery Foundation, grants from EnVivo, grants from C2N Diagnostics, grants from Pfizer, and grants from Eli Lilly. J. Schwartz and E. Abner report no disclosures relevant to the manuscript. R. Petersen is chair of the Data Monitoring Committee, Pfizer, Inc., and Janssen Alzheimer Immunotherapy, and a consultant for Merck, Inc., Roche, Inc., and Genentech, Inc. B. Miller and D. Kapogiannis report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Fiandaca MS, Kapogiannis D, Mapstone M, et al. Identification of preclinical Alzheimer's disease by a profile of pathogenic proteins in neurally derived blood exosomes: a case-control study. Alzheimers Dement Epub 2014 Aug 14. [DOI] [PMC free article] [PubMed]
- 2.Kapogiannis D, Boxer A, Schwartz JB, et al. Dysfunctionally phosphorylated IRS-1 in neural derived blood exosomes of preclinical Alzheimer's disease. FASEB J 2015;29:589–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ihara Y, Morishima-Kawashima M, Nixon R. The ubiquitin-proteasome system and the autophagic-lysosomal system in Alzheimer disease. Cold Spring Harb Perspect Med 2012;2:a006361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwagerl AL, Mohan PS, Cataldo AM, Vonsattel JP, Kowall NW, Nixon RA. Elevated levels of the endosomal-lysosomal proteinase cathepsin D in cerebrospinal fluid in Alzheimer disease. J Neurochem 1995;64:443–446. [DOI] [PubMed] [Google Scholar]
- 5.Nixon RA, Cataldo AM. Lysosomal system pathways: genes to neurodegeneration in Alzheimer's disease. J Alzheimers Dis 2006;9:277–289. [DOI] [PubMed] [Google Scholar]
- 6.Urbanelli L, Magini A, Buratta S, et al. Signaling pathways in exosomes biogenesis, secretion and fate. Genes 2013;4:152–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med 2004;256:183–194. [DOI] [PubMed] [Google Scholar]
- 8.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging–Alzheimer's Association Workgroups on Diagnostic Guidelines for Alzheimer's Disease. Alzheimers Dement 2011;7:280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dubois B, Feldman HH, Jacova C, et al. Research criteria for the diagnosis of Alzheimer's disease: revising the NINCDS-ADRDA criteria. Lancet Neurol 2007;6:734–746. [DOI] [PubMed] [Google Scholar]
- 10.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 11.Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol 2009;65:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Irizarry MC, Webb DJ, Bains C, et al. Predictors of placebo group decline in the Alzheimer's Disease Assessment Scale–cognitive subscale (ADAS-Cog) in 24 week clinical trials of Alzheimer's disease. J Alzheimers Dis 2008;14:301–311. [DOI] [PubMed] [Google Scholar]
- 13.Cano SJ, Posner HB, Moline ML, et al. The ADAS-cog in Alzheimer's disease clinical trials: psychometric evaluation of the sum and its parts. J Neurol Neurosurg Psychiatry 2010;81:1363–1368. [DOI] [PubMed] [Google Scholar]
- 14.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011;134:2456–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology 2011;76:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Faust PL, Kornfeld S, Chirgwin JM. Cloning and sequence analysis of cDNA for human cathepsin D. Proc Natl Acad Sci USA 1985;82:4910–4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benes P, Vetvicka V, Fusek M. Cathepsin D: many functions of one aspartic protease. Crit Rev Oncol Hematol 2008;68:12–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masson O, Bach AS, Derocq D, et al. Pathophysiological functions of cathepsin D: targeting its catalytic activity versus its protein binding activity? Biochimie 2010;92:1635–1643. [DOI] [PubMed] [Google Scholar]
- 19.Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett 2010;584:1393–1398. [DOI] [PubMed] [Google Scholar]
- 20.Kim S, Ock J, Kim AK, et al. Neurotoxicity of microglial cathepsin D revealed by secretome analysis. J Neurochem 2007;103:2640–2650. [DOI] [PubMed] [Google Scholar]
- 21.Demirel O, Jan I, Wolters D, et al. The lysosomal polypeptide transporter TAPL is stabilized by interaction with LAMP-1 and LAMP-2. J Cell Sci 2012;125:4230–4240. [DOI] [PubMed] [Google Scholar]
- 22.Bingol B, Sheng M. Deconstruction for reconstruction: the role of proteolysis in neural plasticity and disease. Neuron 2011;69:22–32. [DOI] [PubMed] [Google Scholar]
- 23.Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol 2001;2:211–216. [DOI] [PubMed] [Google Scholar]
- 24.Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem 2011;80:125–156. [DOI] [PubMed] [Google Scholar]
- 25.Wegele H, Muller L, Buchner J. Hsp70 and Hsp90: a relay team for protein folding. Rev Physiol Biochem Pharmacol 2004;151:1–44. [DOI] [PubMed] [Google Scholar]
- 26.Jinwal UK, Akoury E, Abisambra JF, et al. Imbalance of Hsp70 family variants fosters tau accumulation. FASEB J 2013;27:1450–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nylandsted J, Gyrd-Hansen M, Danielewicz A, et al. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med 2004;200:425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beere HM, Wolf BB, Cain K, et al. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol 2000;2:469–475. [DOI] [PubMed] [Google Scholar]
- 29.Lancaster GI, Febbraio MA. Exosome-dependent trafficking of HSP70: a novel secretory pathway for cellular stress proteins. J Biol Chem 2005;280:23349–23355. [DOI] [PubMed] [Google Scholar]
- 30.Alvarez-Erviti L, Seow Y, Schapira AH, et al. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol Dis 2011;42:360–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goetzl EJ, Boxer A, Schwartz JB, et al. Low neural exosomal levels of cellular survival factors in Alzheimer's disease. Ann Clin Transl Neurol 2015. doi:10.1002/acn3.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Supplement