Antiangiogenic therapy: impact on invasion, disease progression, and metastasis (original) (raw)
. Author manuscript; available in PMC: 2015 Aug 18.
Published in final edited form as: Nat Rev Clin Oncol. 2011 Mar 1;8(4):210–221. doi: 10.1038/nrclinonc.2011.21
Abstract
Antiangiogenic drugs targeting the VEGF pathway have slowed metastatic disease progression in some patients, leading to progression-free survival (PFS) and overall survival benefits compared with controls. However, the results are more modest than predicted by most preclinical testing and benefits in PFS are frequently not accompanied by overall survival improvements. Questions have emerged about the basis of drug resistance and the limitations of predictive preclinical models, and also about whether the nature of disease progression following antiangiogenic therapy is different to classic cytotoxic therapies—in particular whether therapy may lead to more invasive or metastatic behavior. In addition, because of recent clinical trial failures of antiangiogenic therapy in patients with early-stage disease, and the fact that there are hundreds of trials underway in perioperative neoadjuvant and adjuvant settings, there is now greater awareness about the lack of appropriate preclinical testing that preceded these studies. Improved preclinical assessment of all stages of metastatic disease should be a priority for future antiangiogenic drug discovery and development.
Introduction
Antiangiogenic therapy is based on the theory that blocking new blood vessel formation in tumors will stop or slow their growth. Currently, four molecular-targeted drugs are approved by the FDA for six tumor indications; all act to disrupt the VEGF pathway.1 Thus, nearly four decades after the antiangiogenesis concept was introduced by Judah Folkman,2 antiangiogenic therapy is considered a major anticancer treatment modality.3 However, with hundreds of clinical trials currently underway in multiple cancer indications and pathological stages, and dozens of other VEGF and other angiogenic-pathway-targeted agents now in experimental or clinical testing, an urgent issue is understanding why the majority of patients stop responding—or do not respond at all—to such drugs and how such limitations can be overcome. Numerous mechanisms of resistance to antiangiogenic therapy have been proposed4 highlighting that over two decades of positive preclinical studies have yielded only modest incremental changes in the clinic. While this is an unfortunate and common occurrence among cancer treatments, the question remains: are the challenges facing antiangiogenic drugs unique?
In theory, targeting the host ‘tumor-supporting’ angiogenic processes has many benefits but it might also have limitations. Antiangiogenic therapies might initiate an array of stromal and microenvironmental defense mechanisms4 that contribute to eventual drug inefficacy and, more provocatively, may lead to a more aggressive and invasive tumor phenotype—one with an increased ability to metastasize. Though perhaps surprising, this latter property is not distinct from other anticancer treatment modalities—surgery, radiation and chemotherapy can also produce similar unwanted ‘prometastatic’ effects in certain isolated experimental settings (Box 1). However, the possibility that VEGF-pathway inhibitors, and perhaps other ‘host-targeted’ drugs as well, could augment invasive or metastatic potential (despite controlling primary tumor growth or initially slowing the growth of metastasis) could be significant and has become a topic of considerable controversy. The debate has been fuelled by modest clinical benefits, high drug cost, and adverse side effects, in addition to converging findings published in the past 2 years, which relate to limited drug efficacy in early-stage disease. The first finding comes from two preclinical studies showing that the benefits from VEGF-pathway-inhibitor monotherapy can depend on disease stage and treatment circumstances and can, in certain settings, be offset by increased aggressive invasiveness and augmented metastatic potential.5,6 The second finding comes from two large phase III clinical trials involving bevacizumab, a monoclonal antibody to VEGF, used in combination with chemotherapy and administered as adjuvant therapy to patients with early-stage colorectal carcinoma; the treatment combination showed no benefit in the primary end point of progression-free survival (PFS) compared with the chemotherapy-alone arm.7,8 These studies have raised questions about the expectations for antiangiogenic agents in blocking different stages of tumor progression and, in particular, the benefits of these drugs in micrometastatic disease settings.
Box 1. Therapy-accelerated tumor growth and metastasis—not a new phenomenon.
Nearly all anticancer treatments have been shown in some preclinical settings to enhance or facilitate metastatic disease growth and distribution (Supplementary Table 1 online). For example, antitumor effects of radiation can be offset by effects on adjacent ‘bystander’ tissues (the radiation-induced ‘tumor bed effect’) that, in turn, allow for a more hospitable site for tumor extravasation and metastatic growth.130,131 However, preclinical studies involving therapy-induced metastasis must be put into context. This phenomenon only occurs under certain conditions, and can be directly contrasted with positive preclinical examples of beneficial effects in cancer where treatment is sustained. Moreover, decades of clinical use of chemotherapy and radiation clearly demonstrate that antitumor effects outweigh any potential prometastatic effects. Nevertheless, no anticancer therapy has been consistently curative for patients, and prometastatic effects could counteract, or limit, the beneficial antitumor effects of any treatment strategy. Molecular host-targeted drugs such as antiangiogenics could warrant more careful consideration—particularly in micrometastatic disease settings. Chemotherapy and radiation mainly act by direct tumor cytotoxicity and are administered for defined periods (usually brief), whereas antiangiogenic agents are typically cytostatic inhibitors and meant to be administered for longer periods because of their reduced toxic effects.
We summarize evidence that suggests antiangiogenic drugs might alter the natural history of disease progression, depending on the disease stage and tumor type, and focus on limitations that antiangiogenic drugs might have to overcome to bring about greatly improved clinical benefits. It is possible that antiangiogenic therapy may induce a different disease progression pattern than cytotoxics and lead to worse outcomes in terms of progression, invasion, and metastasis. However, this result might never materialize outside of certain limited preclinical scenarios. It remains theoretically possible that such ‘evasive resistance’ mechanisms have a role in the clinical limitations of successful antiangiogenic drugs and, perhaps most importantly, might provide a clue as to how they can be made more effective. There is no compelling clinical evidence that antiangiogenesis treatment will make disease worse or decrease survival;9 however, neither is there a large pool of supporting preclinical evidence that such therapy will be beneficial in blocking early-stage disease, particularly in potentially curative and preventive settings where detailed analysis is rarely performed. With thousands of patients projected to be enrolled to clinical trials over the next 5 years to assess neoadjuvant or long-term adjuvant use of VEGF-pathway-targeted drugs,10 a rigorous assessment of actual and predicted outcomes for antiangiogenic therapy should be conducted using improved preclinical models to better understand when and to what extent these new drugs are likely to work.
Successful therapy—but challenges remain
Bevacizumab was the first molecular-targeted antiangiogenic therapy approved by the FDA and is used as first-line therapy in colorectal cancer (CRC), metastatic breast cancer (MBC), non-small-cell lung cancer (NSCLC), and metastatic renal cell carcinoma (mRCC), and as second-line therapy in CRC and glioblastoma multiforme (GBM).11 With the exception of GBM, bevacizumab is only approved when combined with chemotherapy or cytokine therapy, as monotherapy failed to show robust activity in most instances of advanced-stage disease.12 A second class of approved inhibitors (sunitinib, sorafenib and pazopanib) include oral small-molecule tyrosine kinase inhibitors (TKIs) that target VEGFRs, platelet-derived growth factor (PDGF) receptors, and other kinases including KIT, Ret, BRAF and Flt-3.13 All three of these VEGFR TKIs have been approved as monotherapies for the treatment of mRCC; sunitinib is approved to treat imatinib-refractory gastrointestional stromal tumors (GIST), and sorafenib is approved for hepatocellular carcinoma (HCC). But, these clinical successes have been accompanied by questions that have emerged in the phase III trial setting, which represent potential challenges that must be addressed in order to overcome the limited efficacy of VEGF-pathway inhibitors (Tables 1 and 2).
Table 1.
Successful completed phase III trials with anti-VEGF pathway agents
| Combined with | Tumor (setting) | ↑ PFS? | ↑OS? | Trial identifier |
|---|---|---|---|---|
| Bevacizumab | ||||
| FOLFIRI | CRC (1st) | Yes | Yes* | AVF210796 |
| FOLFOX or XELOX | CRC (1st) | Yes* | Yes | NO1696615 |
| FOLFOX | CRC (2nd) | Yes | Yes* | E320097 |
| Paclitaxol | MBC (1st) | Yes* | No | E210098 |
| Docetaxol | MBC (1st) | Yes* | NA | AVADO99 |
| Capecitabine and taxane or anthracycline | MBC (1st) | Yes* | No | Ribbon1100 |
| Chemotherapy‡ | MBC (2nd) | Yes* | NA | Ribbon2101 |
| Carboplatin and paclitaxel | NSCLC (1st) | Yes | Yes* | E4599102 |
| Cisplatin and gemcitabine | NSCLC (1st) | Yes* | No | AVAiL103 |
| Erlotinib | NSCLC (2nd) | Yes* | NA | ATLAS104 |
| Interferon-2α | RCC (1st) | Yes | No* | AVOREN105 |
| Interferon-2α | RCC (1st) | Yes | No* | CALGB90206106 |
| Carboplatin and paclitaxel | OC (1st) | Yes* | NA | GOG 021837 |
| Monotherapy | GBM (2nd)§ | Yes‖ | Yes‖ | AVF3708107 |
| Sunitinib | ||||
| Monotherapy | RCC (1st) | Yes* | Yes | NCT00083889108 |
| Monotherapy | GIST (2nd) | Yes¶ | NA | SUN 1112109 |
| Monotherapy | PIC (2nd) | Yes* | Yes | NCT00428597110 |
| Sorafenib | ||||
| Monotherapy | RCC (1st) | Yes | No*# | TARGET111 |
| Monotherapy | HCC (1st) | No | Yes* | SHARP112 |
| Pazopanib | ||||
| Monotherapy | RCC (1st and 2nd) | Yes* | NA | VEG105192113 |
| Vandetanib | ||||
| Docetaxol | NSCLC (2nd) | Yes* | NA | ZODIAC114 |
Table 2.
Unsuccessful or terminated phase III trials with anti-VEGF pathway agents
| Combined with | Tumor (setting) | ↑ PFS? | ↑ OS? | Identifier |
|---|---|---|---|---|
| Bevacizumab | ||||
| XELOX and cetuximab | CRC (1st) | No*‡ | NA | PACCE115 |
| Oxaliplatin or irinotecan and panitumumab | CRC (1st) | No*‡ | NA | CAIRO2116 |
| FOLFOX | CRC (adjuvant) | No§ | NA | NSABP-C-0889 |
| Capecitabine | MBC (2nd) | No* | No | AVF2119117 |
| Erlotinib | NSCLC (2nd) | Yes | No* | BeTa118 |
| Capecitabine or 5-FU and cisplatin | AGC (1st) | Yes | No* | AVAGAST119 |
| Gemcitabine | PC (1st) | No | No* | CALGB80303120 |
| Gemcitabine and erlotinib | PC (1st) | Yes | No* | AviTA121 |
| Docetaxol and prednisone | PC (1st) | Yes | No* | CALGB90401122 |
| FOLFOX or XELOX | CRC (adjuvant) | No§ | NA | AVANT24 |
| Aflibercept | ||||
| Gemcitabine | PC (1st) | NA | No* | VANILLA‖ |
| Sunitinib | ||||
| Paclitaxol | MBC (1st) | No* | NA | SUN 1094‖ |
| Capcitabine | MBC (2nd) | No* | No | SUN 1099123 |
| Docetaxol | MBC (1st) | No* | No | SUN 1064‖ |
| FOLFIRI | CRC (1st) | No* | NA | SUN 1122‖ |
| Erlotinib | NSCLC (2nd) | Yes | No* | SUN 1087‖ |
| Monotherapy | MBC (2nd) | No* | No | SUN 1107124 |
| Monotherapy | HCC (2nd) | NA | No | SUN 1107‖ |
| Hormone therapy and prednisone | PR (2nd) | NA | No* | SUN 1120‖ |
| Sorafenib | ||||
| Carboplatin and paclitaxol | MM (2nd) | No | No* | PRISM‖ |
| Carboplatin and paclitaxol | NSCLC (1st) | No | No* | ESCAPE125 |
| PTK787 | ||||
| FOLFOX | CRC (2nd) | Yes | No* | CONFIRM 2126 |
| FOLFOX | CRC (1st) | No | No* | CONFIRM 1‖ |
| Semaxanib | ||||
| FOLFIRI | CRC (1st) | NA | No* | NCT00021281‖ |
| Leucovorin and 5-FU | CRC (1st) | NA | No* | NCT00004252‖ |
| Axitinib | ||||
| Gemcitabine | PC (1st) | NA | No* | A4061028‖ |
| Vandetanib | ||||
| Monotherapy | NSCLC (2nd) | No* | No | ZEST127 |
| Pemetrexed | NSCLC (2nd) | No* | No | ZEAL128 |
| Cediranib | ||||
| FOLFOX | CRC (1st) | Yes | No* | HORIZON III‖ |
| Monotherapy or lomustine | GBM (2nd) | No* | No | REGAL129 |
PFS gains and overall survival
In terms of objective benefits, such as disease stabilization and PFS or overall survival, VEGF-pathway-targeted therapy has largely yielded only modest gains. Despite the presence of VEGF and VEGFR2, tumors either do not respond or eventually become unresponsive with PFS or overall survival benefits in patients receiving antiangiogenic therapy being, in most cases, measured in months.14 In some instances, trials in indications that initially yielded significant improvements in overall survival when bevacizumab was combined with chemotherapy have sometimes not shown similar benefits when compared with more-effective chemotherapies in follow-up studies.15
Perhaps more concerning, however, is the emerging trend where patient response rate and PFS does not translate into significantly increased overall survival in phase III trials (Tables 1 and 2 and Supplementary Box 1 online). Currently, overall survival remains the gold standard for determining therapeutic benefit but the potential use of PFS as an ‘overall survival surrogate’ has been introduced because of a typically strong correlation between the hazard ratios for overall survival and PFS.16 However, there remains a lack of consensus on the use of PFS in this manner and results with antiangiogenic drugs suggest an example where PFS benefits are often not translated into overall survival benefits. It remains a major question as to why such robust gains in PFS seen in the majority of completed phase III trials with bevacizumab and chemotherapy, or VEGFR TKIs as monotherapy, have not frequently corresponded to robust gains in overall survival.
The paradox of chemotherapy combination
Failed trials with VEGF-pathway inhibitors have uncovered a disparity between the efficacy of different treatment modalities with and without chemotherapy. With only two exceptions to date (Supplementary Box 2 online), bevacizumab monotherapy has proven ineffective and VEGFR TKIs have failed to improve results obtained with chemotherapy when given in combination in randomized phase III trials. Nevertheless, inhibition of the VEGF pathway can have striking effects (Table 1) but the molecular basis of why this effect is dependent on the drug strategy employed is unknown (Supplementary Box 3 online). It is also not clear why clinical limitations of bevacizumab monotherapy contrast with preclinical data that indicated efficacy, or why the effects of VEGFR TKIs are not at least additive with chemotherapy despite efficacy in some indications, such as mRCC or HCC, when used as monotherapies (Table 1).
Is disease bound to ‘rebound’?
The potential that sustained suppression of the VEGF pathway, once discontinued, may lead to a ‘rebound’ in tumor growth is important—raising the possibility that initial positive effects of treatment, such as rapid reduction in tumor vascularity and inhibition of tumor growth (which could lead to improved PFS), may be negated or reversed (which could influence overall survival). In the case of VEGFR TKIs, such rebounds have been reported during ‘drug holiday’ periods in the 6-week sunitinib cycle and when treatment is stopped in patients with RCC.17,18 Enhanced tumor regrowth rates after therapy cessation was noted with liver metastases in patients with CRC when bevacizumab was combined with chemotherapy,19 and in patients with RCC treated with bevacizumab alone.20 There is also preclinical evidence for rapid revascularization21 and rebound tumor growth22,23 in studies using imaging and immunohistochemical techniques in mice. Though similar rebounds have not been observed in all instances,24 further study of this concept is critical because drug discontinuation or dose reduction can occur with high frequency, as shown in RCC where 30–50% of patients halted therapy either because of inefficacy or toxic effects.25–27 Moreover, drug discontinuation rates are reported to be higher outside of clinical trials28 and in patients with a genetic background that makes them susceptible to toxic effects.29 Disease stage and treatment combinations could be important in the observation of rebound; Miles et al.9 demonstrated using data from phase III trials in metastatic diseases that halting bevacizumab (and chemotherapy) did not alter mortality rates (Box 2).9
Box 2. Therapy-induced metastasis—preclinical anomaly or clinical reality?
It remains a controversial issue whether mechanisms of resistance to antiangiogenic therapy might involve increased invasive behavior with enhanced metastatic potential and there is debate about how to make the proper assessments. In terms of tumor rebound when VEGF-targeted therapy is stopped, there is no consensus in preclinical studies. Revasularization and regrowth has been observed when treatment with VEGFR tyrosine kinase inhibitors (TKIs) is stopped,21–23 but similar rebounds were not observed in localized tumors when treated with different TKIs24 or with anti-VEGF antibodies.132 Perhaps the critical distinction is that the latter studies did not monitor micrometastatic disease progression. Increases in invasive characteristics have been confirmed after treatment with VEGFR TKIs;79 however, acceleration of metastasis has not been observed in similar circumstances,133,134 including with antibody treatment.82 Crucially, overall survival improvement in mouse models of clinically relevant metastasis is not regularly tested or observed (Supplementary Table 2 online).
In a meta-analysis of phase III trial data from over 4,000 patients with colorectal (NO16966 and AVF2107g), breast (AVADO), renal (AVOREN), and pancreas (AViTA) cancer treated with bevacizumab, disease progression was not accelerated when therapy was stopped.9 Unfortunately, there are caveats. First, the trials incorporate chemotherapy or immunotherapy whereas preclinical studies tested antiangiogenic drugs as monotherapy, using anti-VEGFR2 antibodies or VEGFR TKIs. Second, the patients included have established metastatic (often refractory) disease, and there are no preclinical equivalents that mirror such clinical trials. Thus the question of whether VEGF-pathway inhibition could negatively influence micrometastatic disease remains outstanding135,136 and further testing is required.
A case for treatment beyond progression?
Also of potential importance is that rebounds (if real) may be reversed; in the case of VEGFR TKI-treated patients that have been taken off therapy or fail to respond, benefits have been observed from treatment resumption after a break period,30 or from switching drugs (for example sunitinib to sorafenib, or vice versa),31–33 suggesting resistance may be transient in some cases.34 An observational study of nearly 2,000 patients with CRC (BRiTE Registry) suggests that continuation of bevacizumab treatment while discontinuing and/or switching to additional chemotherapy may substantially increase survival times, indicating that treatment beyond progression may have value.35 This finding was recently confirmed in another study (ARIES observational cohort study).36 Indeed, even without progression, bevacizumab as a maintenance therapy significantly improved PFS in two phase III trials (GOG0218 and ICON7) in the primary treatment of advanced ovarian cancers when chemotherapy (carboplatin or paclitaxel) was halted but bevacizumab treatment continued—raising the question of whether administration of an anti-VEGF therapy should continue for longer periods.37,38 Thus, continued dosing and/or alternative antiangiogenic drug ‘switching’ might reduce any rebound effects, giving insight into resistance mechanisms and providing a clue as to how such rebounds (if any) may be minimized.
VEGF-pathway inhibition—disease progression
Effect on local tumor invasion
There is a small but growing supportive body of literature that indicates initial tumor response, and even tumor shrinkage, during or after antiangiogenic therapy can sometimes be followed not only by eventual relapse and regrowth, but also an enhanced invasive or infiltrative phenotype.39 Supportive evidence is largely anecdotal and limited to small studies or case reports; therefore the concept remains speculative (Box 2). GBM is the most notable example, as 30–50% of patients treated with bevacizumab develop progressive disease accompanied by a high rate of diffuse infiltrative lesions.40,41 Although GBM is already a highly infiltrative, invasive tumor, this finding has been noted in several studies42–50 and suggests an adaptive response to anti-angiogenic therapy that leads to more invasive behavior. In preclinical mouse models of GBM where VEGF or hypoxia inducible factor 1α is genetically or therapeutically blocked, initial tumor stabilization and/or shrinkage can be followed by recurrent or existing tumor regrowth, as well as increases in new microsatellite lesions in adjacent sites with infiltrative behavior and wide fronts of invasion (Supplementary Box 4 online).6,51–59 The caveat is that such findings are not uniformly observed60 and could manifest primarily from the initial success of therapy rather than from a direct negative effect. If patients with GBM survive longer because of bevacizumab treatment, then this could create more time for tumors to become invasive. Thus, a PFS benefit might have uncovered progression patterns of a rapidly progressing tumor type that had not been observed as frequently and that may shorten the period between relapse and death and compromise overall survival benefits (Figure 1).
Figure 1.
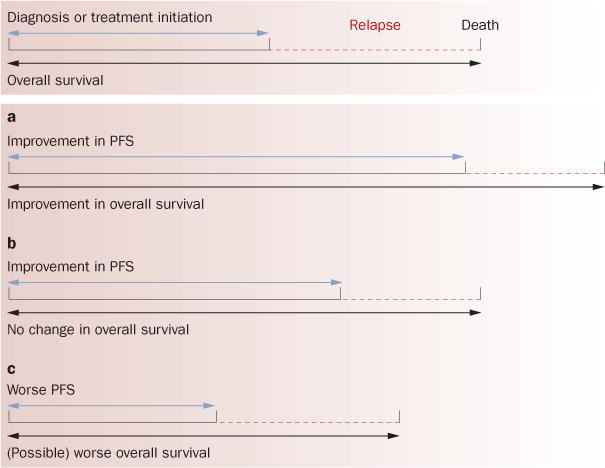
Clinical results of combinations of PFS and overall survival. There are several different combinations of PFS and overall survival, including no change in either (not shown here). a | Improvement in PFS translates into improved overall survival. In completed phase III trials with anti-VEGF-pathway therapy (Tables 1 and 2), two additional scenarios have occurred: b | PFS benefit does not translate into improved overall survival, and c | reduced PFS (Table 2). Worse overall survival has not been shown in a phase III trial though a recent interim analysis of the AVANT trial indicated that this trend is possible.24 It is possible that response to anti-VEGF therapy (even if leading to improved PFS) can change the natural history of disease progression to include a more aggressive phenotype—possibly explaining lack of changes in overall survival. This figure is based on conceptual ideas outlined by David Reardon. Abbreviation: PFS, progression-free survival.
Effect on tumor dissemination and metastasis
Paez-Ribes et al.6 observed increased numbers of meta-stases in distant organs after VEGF-pathway inhibition. It is critical to note that—as for previous preclinical studies—these results were observed only after objective tumor growth inhibition in localized disease that led to prolonged overall survival.6 Therefore, despite an initial and overall benefit in survival after treatment, tumor-response mechanisms to therapy may eventually facilitate induction of invasive and metastatic tumor outgrowths. This, in turn, might limit the overall benefits in survival times. If these findings suggest a tumor-dependent response to therapy, then it is also possible that host-dependent responses to VEGF-pathway inhibition could facilitate metastasis. Similar potent antitumor properties were observed using short-term and sustained VEGFR TKI monotherapy treatment in orthotopically implanted tumors, but when mice were treated before intravenous inoculation (experimental metastasis) or immediately following primary tumor removal (spontaneous metastasis) an increase in metastatic disease was observed that translated into shortened survival times for mice receiving therapy.5 Thus, short-term treatment could influence early-stage micrometastatic disease initiation, independent of direct effects of drug on tumor cells, suggesting that systemic reactions to VEGF-pathway disruption could facilitate tumor dissemination. These preclinical studies demonstrate that early-stage micrometastatic growth, under certain conditions, can be elicited rather than inhibited by VEGF-pathway inhibition, and might involve both adaptive tumor-dependent and tumor-independent (host-mediated) mechanisms.5,6
The clinical relevance of these findings is unclear; however, clinical results that seem consistent with these preclinical findings have emerged. For VEGFR TKIs, similar instances where treatment cessation and rebound regrowth has been accompanied by increases in local foci and/or distant metastasis in retrospective analyses of patients with RCC who discontinued either sunitinib or sorafenib,30 and in isolated case reports.61 In one study, the anatomical sites of disease progression were similar in patients who eventually failed to respond to either interferon or VEGF-pathway inhibitors, however, in the latter group there was an increase in metastases in previously uninvolved anatomical sites, suggesting that efficacy of therapy in sites of established metastases is superior to the prevention or inhibition of microscopic tumor growth in new ones.62
Mechanisms of evasive resistance
Modes of resistance to VEGF-pathway inhibition have been discussed,4 and themes have emerged that could be related to disease progression changes in response to therapy, including tumor and host responses (Box 3 and Supplementary Box 5 online).39,63 For cytotoxic therapies, drug-resistance mechanisms involve a multitude of tumor-dependent changes, including multidrug-resistance gene or protein upregulation; clonal selection or repopulation; and resistance of cancer stem cells.4 The microenvironment can also be affected by cytotoxic therapies; however, for anti-angiogenic agents—where the microenvironment is the primary target—it is clearly possible that microenvironment effects are of greater influence (Box 3). Disruption of the VEGF pathway could affect these functions with eventual tumor progression and disease relapse.
Box 3. Possible mechanisms influencing invasion and metastasis after therapy.
Tumor-dependent mechanisms
- ▪
Increased expression of prometastatic proteins: c-met,69,70 interleukin (IL)-6,137 IL-8,138 and urokinase-type plasminogen activator receptor139 - ▪
Suppression of antimetastatic mediators: myoglobulin140 - ▪
Altered adhesion: upregulation or activation and secretion of exomal proteolytic enzymes, such as matrix metalloproteinases141,142 - ▪
Bone-marrow-derived dendritic cell (BMDC) mobilization creates ‘premetastatic niches’143 - ▪
Acute hypoxic stress144–146 - ▪
Instigation of tumor epithelial–mesenchymal transition147 - ▪
Increased vascular co-optive behavior133 - ▪
Activation of alternative angiogenic pathways: FGF and ephrin148 - ▪
Induction of stromal autophagy149 - ▪
Vascular mimicry or cancer stem cells150,151
Tumor-independent—host-mediated—mechanisms
- ▪
Compensatory upregulation of proangiogenic or prometastatic factors contribute to ‘rebound’21 and/or increased extravasive potential: VEGF, PlGF, G-CSF, osteopontin, Bv8 (prokineticin), G-CSF, angiopoieten2, PDGFA and SDF1α14,152–158 - ▪
BMDC mobilization recruits VEGFR1-positive bone marrow cells to distant sites to facilitate ‘premetastatic niches’;159,160 this has not been confirmed in all cases92 - ▪
BMDC mobilization of Gr1+CD11b+ myeloid suppressortype cells, TIE2 expressing monocytes, and tumorassociated macrophages to home to the tumor microenvironment and produce compensatory proangiogenic factors14,155–158 - ▪
Pericyte dysfunction increases vessel leakiness and allows for increased extravasive and metastatic tumor potential4,74,161,162 - ▪
Increased prothrombotic events caused by vessel damage as a result of therapy allows for increased tumor cell ‘seeding’ and growth in distant organs163 - ▪
Altered endothelial cell adhesion molecule function may enhance VEGF-driven angiogenesis and tumor growth164 - ▪
Inflammatory pathway activation alters the endothelial microenvironment increasing intravasive and extravasive potential of tumor cells72
A change in the seed, the soil, or both?
Although acquired drug resistance is an accepted reality for antiangiogenic therapy, how would resistance lead to a tumor phenotype of increased invasion or metastasis? When a locally growing primary tumor progresses to form distant metastases, several steps are involved including loss of cellular adhesion; enhanced motility and invasion capabilities; intravasation into the bloodstream; homing and survival; extravasation and seeding of micrometastases; and colonization and growth in a distant site.64 Critically, as Stephen Paget theorized as the ‘seed and soil hypo thesis’,65 both the tumor (seed) and host organ environment (soil) must allow for dissemination of disease. There are many preclinical studies showing that anticancer treatments can facilitate the dissemination of tumor cells and metastases (Box 1), and there are mechanisms that could account for antiangiogenic-therapy-induced invasion or metastasis (Box 2), some driven by the host and others by the tumor, though it is likely that both have a role.
Perhaps the most important compensatory mechanism a tumor can acquire in response to VEGF-pathway inhibition is an elevation in tumor hypoxia, which could select for tumor populations able to grow in low oxygen environments66,67 and/or provide alternate compensatory proangiogenic pathways to allow persistent neovascularization.68 Though the connection between antiangiogenic therapy and an increase in invasive and metastatic phenotypes needs further validation, the evidence linking hypoxia to a more aggressive metastatic phenotype is established. Both acute and systemic oxygen deprivation facilitate tumor metastasis and studies have demonstrated that hypoxia-induced mechanisms, such as c-met upregulation (among others), can force tumors to branch and disseminate despite therapy-induced hypoxia being a key initial controller of tumor growth.69–71
A second important potential mediator of increased metastatic potential after therapy could include inflammatory mechanisms of the host, perhaps as a result of alteration (or injury) to the endothelial microenvironment, which assist in both the intravasive and extravasive potential of tumors.72,73 It is possible (though unproven), that such favorable conditions (or ‘premetastatic niches’) could differ significantly depending on the therapy. Chemotherapy and radiation, for example, could primarily act in this manner to promote metastasis (Box 1 and Supplementary Table 1 online), and it is possible that this effect could differ between VEGF-pathway inhibitors. For example, the less-specific multitargeted small-molecule TKIs could cause an increased metastatic potential, whereas antibodies or other large-molecule inhibitors, which may not evoke a systemic inflammatory response, could lack or have attenuated ‘prometastatic’ capacity. In addition, perivascular pericytes might act as a barrier to limit tumor cell intravasation and extravasation and targeting these cells using VEGFR TKIs (which block PDGFRs) could promote aspects of the metastatic process.74 Future investigations could illuminate the differences between how the tumor and micro environment react to therapy, whether positively or negatively, with respect to tumor growth and metastatic dissemination.
Early-stage disease
There are several important ramifications for the field of antiangiogenesis therapy if one or more of the theoretical mechanisms of resistance and/or preclinical findings manifest into altered disease progression in the clinical setting. The most obvious question is how can such data be reconciled with the numerous preclinical and clinical data indicating that antiangiogenic therapy inhibits, not promotes, disease progression in localized and metastatic settings? Indeed, experimental conditions (such as animal model, tumors, drugs, doses, treatment duration, or combinations with chemotherapy) may explain some differences in outcomes; however, antiangiogenic therapies may have different efficacies in established localized primary tumors and micrometastatic and macrometastatic disease.
The gap between bedside and bench
Perhaps foremost among the challenges in predicting disease-progression patterns and mechanisms of drug resistance to antiangiogenic therapy is a general disconnect between how VEGF-pathway inhibitors (and all anticancer therapies for that matter) are tested in experimental versus clinical settings. In preclinical evaluations, the majority of analyses have been conducted either in genetically engineered mouse models (GEMMS) or, more frequently, in locally grown primary ectopic (or orthotopic) tumors using human (xenograft) or mouse (syngenic) models.75 Conversely, most cancer patients receiving VEGF-pathway inhibitors have late-stage (sometimes refractory) disease involving established metastases in more than one site.71 In the preclinical setting, only a negligible fraction of studies have tested VEGF-pathway inhibitors in similar late-stage models and even fewer have compared directly antitumor efficacy in such circumstances to locally grown primary tumors (Figure 2). Also, preclinical metastasis models are often quantitatively assessed (for example visual nodule counts, immunohistochemistry, and imaging76) and disease is measured at a defined end point—usually when a primary or localized tumor has reached an institutional ethical limit. This means studies are stopped short of overt systemic metastatic disease, and therefore the majority of preclinical studies involving VEGF-pathway inhibitors and metastasis have included non-survival-based analyses. These limitations have resulted in relatively few studies that are designed to investigate the impact of VEGF-pathway inhibitors on established metastasis when compared with the hundreds of publications dedicated to localized or primary disease. In addition, there are even fewer studies that include clinically relevant, survival-based evaluations of therapy in models of metastasis, and there is disparity in the tumor models employed and modes of metastasis quantification used (Figure 2 and Supplementary Table 2 online). These preclinical studies have shown that VEGF-pathway-targeted therapy leads to the inhibition of metastasis when quantified empirically, either after short treatment periods or when studies are terminated because of primary tumor growth. However, considering that the vast majority of patients receiving similar drugs in the clinic have established metastases, more relevant preclinical analyses should be conducted to determine the consequences of this on overall survival. In such rare preclinical studies, the results have been mixed, with some reporting treatment benefit77 and others noting more moderate or negligible effects78,79 (Supplementary Table 2 online). The limitations of these studies are of particular relevance because metastasis is generally the cause of patient mortality,80 and antiangiogenic agents are now being evaluated in earlier stages of disease, such as the adjuvant setting, which may involve treating early-stage occult micrometastatic disease. Moreover, as the studies by Paez-Ribes et al.6 and Ebos et al.5 show, positive effects in primary tumor models do not always translate into beneficial effects in blocking hematogenous micrometastatic-disease progression (the outcomes may even be worse), and comparisons of drug effects in the primary tumor and micrometastasis treatment settings can be very different. Results from similar studies have varied,79,81,82 and thus it is critical when interpreting potential conflicting data sets from preclinical studies using inhibitors of the VEGF pathway to consider variables such as disease stage, the types of drug employed (antibodies versus TKIs), and the models of metastasis that are used.
Figure 2.
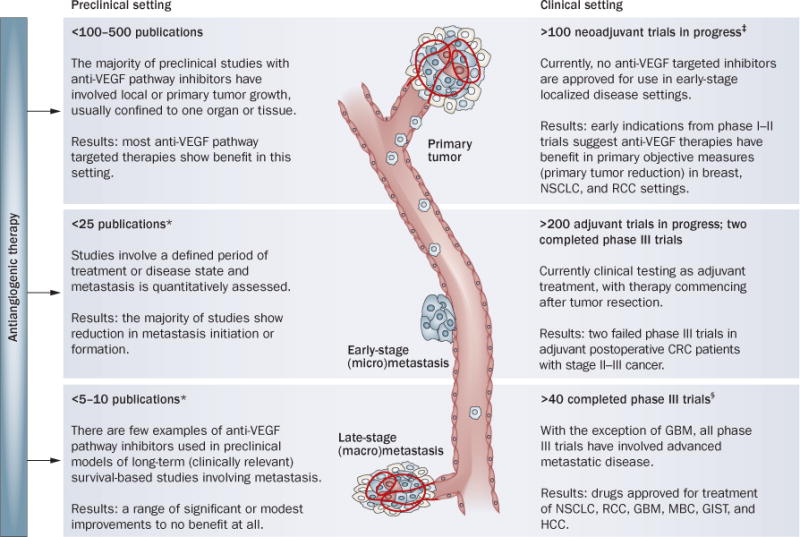
Variable efficacy of VEGF pathway-targeted therapies: exposing the gap between preclinical and clinical testing. The number of studies that have been completed in the clinic in each setting are inversely correlated with the number of preclinical publications that model each setting. *See Supplementary Table 2 online. ‡See Table 3. §See Table 1. Abbreviations: CRC, colorectal cancer; GBM, glioblastoma muliforme; GIST, gastrointestional stromal tumors; HCC, hepatocellular carcinoma; MBC, metastatic breast cancer; NSCLC, non-small-cell lung cancer; RCC, renal cell carcinoma.
The need for optimal mouse models to study metastasis has taken on a greater urgency, particularly in the setting of micrometastatic disease. A recent Review covered this topic in detail and listed models that could be employed10—an example is a model of NSCLC where sunitinib prolonged survival but longer treatments (initiated earlier) did not translate into greater benefit.83,84 This situation emphasizes the importance of developing models that can clearly distinguish between macroscopic and microscopic disease. In addition, use of models that employ clinically relevant end points such as PFS are promising for improving their predictions of clinical potential.85
The perioperative setting
Perhaps the area where disease progression after therapy presents the biggest challenge (and the potential to show benefit) is in the perioperative setting, when treatments are administered either before (neoadjuvant) or after (adjuvant) surgery to remove the tumor. With studies underway in patients with CRC, RCC, NSCLC, breast and central nervous system cancers, it will be critical to determine safety parameters for wound healing and therapy toxicity to optimize guidelines,86 and to determine the efficacy of VEGF-pathway inhibition in these settings.
Adjuvant therapy
Currently, there are over 200 adjuvant clinical trials planned or underway assessing antiangiogenesis drugs either alone or in combination with chemotherapy in cancer types including breast, RCC, prostate, head and neck, NSCLC, and ovarian.87 The rationale for therapeutic intervention with VEGF-pathway inhibitors in the postoperative setting was summarized by Bagri et al.10 who highlighted the advantages of antiangiogenic blockade in preventing occult micrometastatic growth in distant sites. Most obvious is that because of the integral role of the vasculature in the step-wise process of metastasis, antiangiogenic therapy could compromise some of these steps in primary tumors such as preventing or delaying intravasation (for example via the destruction of the immature vasculature) and the ‘angiogenic switch’ in avascular metastases at distant sites.10 Recently, two phase III postoperative adjuvant trials (C-08 and AVANT) that assessed bevacizumab in patients with stage II–III CRC were completed. Patients in both trials received either bevacizumab for 1 year (in combination with chemotherapy for the first 6 months) or 6 months of chemotherapy alone. The chemotherapy regimen FOLFOX (5-fluorouracil, leucovorin and oxaliplatin) was compared with bevacizumab in C-08, and FOLFOX or XELOX (oxaliplatin and capecitabine) was compared with bevacizumab in AVANT. The primary end point of a benefit in PFS after 3 years was not met in either trial, although in both the C-08 and AVANT trials indications of PFS improvement was observed following the 6-month bevacizumab maintenance period at the 1-year interim analysis, and at subsequent interim analyses (in C-08 only)—but the extent of the benefit diminished over time in both trials.88,89 The basis of this ‘fading’ effect is unknown, and questions remain as to whether long-term bevacizumab maintenance should be tested in follow-up studies to potentially prolong the observed PFS benefits (as was seen in the GOG0218 and ICON7 ovarian cancer trials37). However, it is important to question if PFS benefits translate into overall survival benefits and, if not, do they justify the associated costs and toxicity of using a drug such as bevacizumab? As well, and perhaps over shadowing such questions, the AVANT trial results demonstrated that patients receiving bevacizumab with chemotherapy had numerical increases in disease relapse and death compared with chemotherapy alone.24 Though firm conclusions cannot be made based on early reporting of trial results, and it is possible that patient crossover in the control group may have had a role in these observations (these patients later received bevacizumab), it remains an open question whether bevacizumab was a detriment in this trial—a point raised by the trial organizers.8 Regardless, in both trials, the fact that PFS changed rapidly after bevacizumab was halted requires further study and highlights the importance of undertaking appropriate preclinical studies to examine the mechanisms by which antiangiogenic treatments lose their activity and/or alter tumor progression and metastasis over time, especially in the adjuvant setting (Figure 2).
Neoadjuvant therapy
In neoadjuvant therapy, the theoretical advantages of antiangiogenesis treatment are twofold. First, to elicit an objective reduction in tumor size, usually to downstage an unresectable tumor or improve the impact of surgery of resectable tumors and second, to prevent micrometastatic outgrowth, increasing the potential for PFS and overall survival benefits.90,91 There are over 100 ongoing neoadjuvant trials using VEGF-pathway inhibitors, either alone or in combination with chemotherapy, radiation, or other therapies (Table 3 and Supplementary Box 6 online).87
Table 3.
VEGF pathway-targeted drugs currently in neoadjuvant clinical trials
| Search criteria* | Cancer | Number of trials (drugs used) | |||||
|---|---|---|---|---|---|---|---|
| Total | Monotherapy ‡ | With MTT | With hormone therapy | With chemotherapy§ | With radiation | ||
| Neoadjuvant and sunitinib | Breast, renal, bladder, soft-tissue sarcoma, GIST, prostate | 17 | 10 | – | 2 (exemestane, LHRH agonist) | 4 (TCARB, gemcitabine, cisplatin, docetaxel) | 2 |
| Neoadjuvant and sorafenib | Breast, renal, soft-tissue sarcoma, prostate, rectal, SCCHN | 12 | 5 | – | 1 (letrozole) | 6 (cisplatin, capecitabine, IE, ifosfamide, ixabepilone, LDM CTX) | 5 |
| Neoadjuvant and pazopanib | Breast, NSCLC | 3 | 3 | – | – | 1 (docetaxel) | – |
| Neoadjuvant and zactima | Breast, NSCLC, esophageal | 3 | 2 | – | 1 (anastrozole) | 1 (docetaxel and carboplatin combination) | 1 |
| Neoadjuvant and cediranib | Breast | 1 | – | – | – | 1 (docetaxel, doxorubicin and cyclophosphamide combination) | – |
| Neoadjuvant and semaxinib | Soft-tissue sarcoma | 1 | – | – | – | 1 (doxorubicin, ifosfamide and dacarbazine combination) | 1 |
| Neoadjuvant and bevacizumab | Breast, renal, bladder, soft-tissue sarcoma, prostate, esophageal, cervical, colorectal, urothelial, rectal, NSCLC, glioblastoma, pancreatic, ovarian, uveal melanoma, gastric or adrenal | 63 | 2 | 3 (trastuzumab, cetuximab) | 4 (letrozole, AI) | 65 (docetaxel, carboplatin, capecitabine, gemcitabine, irinotecan, cisplatin, 5-FU, XELOX, FOLFOX, FOLFOXIRI, TC, M-VAC, TAC, TCARB, DC, AC, FEC, IC, DG, OCFL-BC, DECNC, ECX) | 10 |
To date, there are few, if any, preclinical studies that have been conducted with antiangiogenic drugs (or any other drug types) that analyze neoadjuvant or presurgical treatments,81,92 and virtually none that compare treatment effects on primary tumors to metastatic-disease progression (or prevention) after resection. Similar to post-operative adjuvant studies, preclinical neoadjuvant studies could be useful to distinguish between relative efficacy effects of a drug in both the primary and metastatic settings and to evaluate the usefulness of antiangiogenic therapies in this treatment setting (Supplementary Box 7 online).
Opportunities and challenges
With the success of VEGF-pathway inhibitors in the clinic, and the discovery of limitations to their use, what should be the priority for researchers and clinicians? First, clearly there is a need to evaluate new therapies in the most appropriate cancer models possible, at various stages of disease and metastatic spread, even if this means observed benefits may not be as significant as has traditionally been the case in preclinical testing. Less impressive gains in more challenging disease models may equate to more clinical relevance.93 Second, in using such models it might be possible to uncover therapeutic agents that have differential activity in different settings, for example, efficacy in localized tumor growth but not in slowing (or preventing) micrometastatic or macrometastatic disease. Of course, uncovering drugs or therapies that have the opposite properties could be extremely important as well, that is, no effect in established tumors but with antimetastatic activity.94 Several examples of inhibitors or treatment strategies with such effects have been noted, including those targeting the TGF-β, integrin, and c-met/HGF pathways; nuclear and cellular protein inhibitors (for example, agents targeting NF-κB, Grb-2, and RhoC); and other compounds, such as propranolol and cyclopamine.80 Such antimetastasic (but not ‘antiprimary’) properties have also been noted with chemotherapy regimens administered continuously at low doses (termed ‘metronomic chemotherapy’) in preclinical models of MBC (cyclophosphomide and UFT, a 5-fluorouracil prodrug), and melanoma (cyclophosphomide and vinblastine).75 These findings raise the possibility of more rational combination studies; for example, could the limitations of antiangiogenic agents be overcome (or delayed) by combination with an antimetastatic agent, which itself may not have potent antitumor properties? Third, it is critical to perform studies in preclinical models that assess drug treatments or combinations that closely mimic phase III clinical trials, ideally using similar (or equivalent) quantitative assessments to PFS and overall survival. An example was performed by Singh et al.85 who used two different GEMMs involving KRAS mutations (modeling NSCLC and pancreatic cancer) to compare ‘standard of care’ chemotherapy regimens with inhibitors of EGFR (erlotinib) and VEGF, using a bevacizumab-equivalent mouse-specific antibody. Such large-scale preclinical studies, despite a high expense, could be used as surrogates for clinical trials as retrospective study tools to understand failure (as in the Singh et al.85 study) or prospectively to assess and predict results for ongoing or planned phase III trials.95 Finally, with respect to determining whether VEGF-pathway inhibitors can lead to more invasive and metastatic phenotypes (despite initial positive effects in terms of tumor shrinkage, PFS or overall survival benefits), it will be critical to properly assess (and compare) different modes of inhibition (antibodies versus TKIs) in different disease stages (localized verses micrometastasis and macrometastasis) to understand disease progression on (or off) therapy. This will be essential for understanding the basis of any future rationale for extended treatments in patients, such as the initial benefits seen in certain settings where treatment extends beyond disease progression. In addition, the differences between PFS and overall survival benefits in clinical trials investigating anti-angiogenic drugs could have important implications for the future use of VEGF-pathway-targeted agents. For example, in 2010, the FDA rejected full approval of bevacizumab with chemotherapy in patients with MBC based on toxicities and the lack of overall survival benefits and diminishing PFS benefits in the AVADO and RIBBON-1 trials compared to the earlier E2100 trial (Tables 1 and 2 and Box 3).
Conclusions
While efficacies of VEGF-pathway-targeted therapies in certain cancer settings represent a conceptual and practical medical success, the lack of substantial benefits for the vast majority of patients in terms of increased long-term overall survival times remains an ongoing challenge. Understanding the basis of these treatment limitations will likely be key to devising improved strategies and to overcome the possible difficulties facing further development of antiangiogenic therapies used at all stages of tumor progression.
Supplementary Material
supplemental material
Key points.
- ▪
Successful clinical trials with various VEGF-pathway inhibitors have been accompanied by numerous phase III failures - ▪
Trial failures in adjuvant disease, and ongoing trials in early-stage settings, could highlight differences in antiangiogenic drug efficacy depending on disease stage - ▪
There is a gap between how antiangiogenics are usually tested in the clinic (late-stage metastatic) and in preclinical mouse models (localized primary tumors) - ▪
There is debate whether anti-VEGF therapy may lead to ‘rebound growth’ when halted or if it may fuel more invasive and metastatic disease phenotypes - ▪
Future testing of antiangiogenic therapies should be conducted in clinically relevant animal models of all disease stages
Acknowledgments
The authors thank David Reardon for editorial comments and permission to use the concept in Figure 1, and William Cruz-Munoz and Christina R. Lee for critically reading the manuscript.
Footnotes
Author contributions
J. M. L. Ebos researched the data for the article and wrote the manuscript. Both authors had a substantial contribution to discussion and editing of content.
Supplementary information is linked to the online version of the paper at www.nature.com/nrclinonc
Competing interests
R. S. Kerbel declares associations with the following companies: GlaxoSmithKline, MetronomX, MolMed, Pfizer, Taiho Pharmaceutical, YM Biosciences. See the article online for full details of the relationships. J. M. L. Ebos declares no competing interests.
References
- 1.US Department of Health and Human Services. FDA US Food and Drug Administration [online] 2011 www.fda.gov.
- 2.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 3.Abdollahi A, Folkman J. Evading tumor evasion: current concepts and perspectives of anti-angiogenic cancer therapy. Drug Resist Updat. 2010;13:16–28. doi: 10.1016/j.drup.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 4.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ebos JM, et al. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232–239. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pàez-Ribes M, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allegra CJ, et al. Phase III trial assessing bevacizumab in stages II and III carcinoma of the colon: results of NSABP protocol C-08. J Clin Oncol. 2011;29:11–16. doi: 10.1200/JCO.2010.30.0855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Gramont A, et al. AVANT: Results from a randomized, three-arm multinational phase III study to investigate bevacizumab with either XELOX or FOLFOX4 versus FOLFOX4 alone as adjuvant treatment for colon cancer [abstract] J Clin Oncol. 2011;29(Suppl. 4):a362. [Google Scholar]
- 9.Miles D, et al. Disease course patterns after discontinuation of bevacizumab: pooled analysis of randomized phase III trials. J Clin Oncol. 2011;29:83–88. doi: 10.1200/JCO.2010.30.2794. [DOI] [PubMed] [Google Scholar]
- 10.Bagri A, Kouros-Mehr H, Leong KG, Plowman GD. Use of anti-VEGF adjuvant therapy in cancer: challenges and rationale. Trends Mol Med. 2010;16:122–132. doi: 10.1016/j.molmed.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 11.Heath VL, Bicknell R. Anticancer strategies involving the vasculature. Nat Rev Clin Oncol. 2009;6:395–404. doi: 10.1038/nrclinonc.2009.52. [DOI] [PubMed] [Google Scholar]
- 12.Boere IA, Hamberg P, Sleijfer S. It takes two to tango: combinations of conventional cytotoxics with compounds targeting the vascular endothelial growth factor-vascular endothelial growth factor receptor pathway in patients with solid malignancies. Cancer Sci. 2010;101:7–15. doi: 10.1111/j.1349-7006.2009.01369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ivy SP, Wick JY, Kaufman BM. An overview of small-molecule inhibitors of VEGFR signaling. Nat Rev Clin Oncol. 2009;6:569–579. doi: 10.1038/nrclinonc.2009.130. [DOI] [PubMed] [Google Scholar]
- 14.Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358:2039–2049. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saltz LB, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol. 2008;26:2013–2019. doi: 10.1200/JCO.2007.14.9930. [DOI] [PubMed] [Google Scholar]
- 16.Wilkerson J, Fojo T. Progression-free survival is simply a measure of a drug’s effect while administered and is not a surrogate for overall survival. Cancer J. 2009;15:379–385. doi: 10.1097/PPO.0b013e3181bef8cd. [DOI] [PubMed] [Google Scholar]
- 17.Wolter P, Beuselinck B, Pans S, Schöffski P. Flare-up: an often unreported phenomenon nevertheless familiar to oncologists prescribing tyrosine kinase inhibitors. Acta Oncol. 2009;48:621–624. doi: 10.1080/02841860802609574. [DOI] [PubMed] [Google Scholar]
- 18.Desar IM, et al. The reverse side of the victory: flare up of symptoms after discontinuation of sunitinib or sorafenib in renal cell cancer patients. A report of three cases. Acta Oncol. 2009;48:927–931. doi: 10.1080/02841860902974167. [DOI] [PubMed] [Google Scholar]
- 19.Cacheux W, et al. Reversible tumor growth acceleration following bevacizumab interruption in metastatic colorectal cancer patients scheduled for surgery. Ann Oncol. 2008;19:1659–1661. doi: 10.1093/annonc/mdn540. [DOI] [PubMed] [Google Scholar]
- 20.Stein WD, Yang J, Bates SE, Fojo T. Bevacizumab reduces the growth rate constants of renal carcinomas: a novel algorithm suggests early discontinuation of bevacizumab resulted in a lack of survival advantage. Oncologist. 2008;13:1055–1062. doi: 10.1634/theoncologist.2008-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mancuso MR, et al. Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J Clin Invest. 2006;116:2610–2621. doi: 10.1172/JCI24612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levashova Z, et al. Molecular imaging of changes in the prevalence of vascular endothelial growth factor receptor in sunitinib-treated murine mammary tumors. J Nucl Med. 2010;51:959–966. doi: 10.2967/jnumed.109.072199. [DOI] [PubMed] [Google Scholar]
- 23.Nagengast WB, et al. VEGF-PET imaging is a noninvasive biomarker showing differential changes in the tumor during sunitinib treatment. Cancer Res. 2011;71:143–153. doi: 10.1158/0008-5472.CAN-10-1088. [DOI] [PubMed] [Google Scholar]
- 24.di Tomaso E, et al. Glioblastoma recurrence after cediranib therapy in patients: lack of “rebound” revascularization as mode of escape. Cancer Res. 2011;71:19–28. doi: 10.1158/0008-5472.CAN-10-2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.La Vine DB, Coleman TA, Davis CH, Carbonell CE, Davis WB. Frequent dose interruptions are required for patients receiving oral kinase inhibitor therapy for advanced renal cell carcinoma. Am J Clin Oncol. 2010;33:217–220. doi: 10.1097/COC.0b013e3181a650a6. [DOI] [PubMed] [Google Scholar]
- 26.Gore ME, et al. Safety and efficacy of sunitinib for metastatic renal-cell carcinoma: an expanded-access trial. Lancet Oncol. 2009;10:757–763. doi: 10.1016/S1470-2045(09)70162-7. [DOI] [PubMed] [Google Scholar]
- 27.van der Veldt AA, et al. Predictive factors for severe toxicity of sunitinib in unselected patients with advanced renal cell cancer. Br J Cancer. 2008;99:259–265. doi: 10.1038/sj.bjc.6604456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Riechelmann RP, et al. Sorafenib for metastatic renal cancer: the Princess Margaret experience. Am J Clin Oncol. 2008;31:182–187. doi: 10.1097/COC.0b013e3181574084. [DOI] [PubMed] [Google Scholar]
- 29.Yoo C, et al. The efficacy and safety of sunitinib in Korean patients with advanced renal cell carcinoma: high incidence of toxicity leads to frequent dose reduction. Jpn J Clin Oncol. 2010;40:980–985. doi: 10.1093/jjco/hyq073. [DOI] [PubMed] [Google Scholar]
- 30.Johannsen M, et al. Can tyrosine kinase inhibitors be discontinued in patients with metastatic renal cell carcinoma and a complete response to treatment? A multicentre, retrospective analysis. Eur Urol. 2009;55:1430–1438. doi: 10.1016/j.eururo.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 31.Sablin MP, et al. Sequential sorafenib and sunitinib for renal cell carcinoma. J Urol. 2009;182:29–34. doi: 10.1016/j.juro.2009.02.119. [DOI] [PubMed] [Google Scholar]
- 32.Dudek AZ, Zolnierek J, Dham A, Lindgren BR, Szczylik C. Sequential therapy with sorafenib and sunitinib in renal cell carcinoma. Cancer. 2009;115:61–67. doi: 10.1002/cncr.24009. [DOI] [PubMed] [Google Scholar]
- 33.Tamaskar I, et al. Antitumor effects of sunitinib or sorafenib in patients with metastatic renal cell carcinoma who received prior antiangiogenic therapy. J Urol. 2008;179:81–86. doi: 10.1016/j.juro.2007.08.127. [DOI] [PubMed] [Google Scholar]
- 34.Hammers HJ, et al. Reversible epithelial to mesenchymal transition and acquired resistance to sunitinib in patients with renal cell carcinoma: evidence from a xenograft study. Mol Cancer Ther. 2010;9:1525–1535. doi: 10.1158/1535-7163.MCT-09-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ellis LM, Haller DG. Bevacizumab beyond progression: does this make sense? J Clin Oncol. 2008;26:5313–5315. doi: 10.1200/JCO.2008.17.4540. [DOI] [PubMed] [Google Scholar]
- 36.Cohn AL, et al. Clinical outcomes in bevacizumab (BV)-treated patients (pts) with metastatic colorectal cancer (mCRC): Results from ARIES observational cohort study (OCS) and confirmation of BRiTE data on BV beyond progression (BBP) [abstract] J Clin Oncol. 2010;28(Suppl):a3596. [Google Scholar]
- 37.Burger RA, et al. Phase III trial of bevacizumab (BEV) in the primary treatment of advanced epithelial ovarian cancer (EOC), primary peritoneal cancer (PPC), or fallopian tube cancer (FTC): A Gynecologic Oncology Group study [abstract] J Clin Oncol. 2010;28(Suppl):aLBA1. [Google Scholar]
- 38.Perren T, et al. ICON7: A phase III Gynaecologic Cancer InterGroup (GCIG) trial of adding bevacizumab to standard chemotherapy in women with newly diagnosed epithelial ovarian, primary peritoneal or fallopian tube cancer [abstract] Ann Oncol. 2010;21(Suppl. 8):aLBA3. [Google Scholar]
- 39.Ebos JM, Lee CR, Kerbel RS. Tumor and host-mediated pathways of resistance and disease progression in response to antiangiogenic therapy. Clin Cancer Res. 2009;15:5020–5025. doi: 10.1158/1078-0432.CCR-09-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Norden AD, et al. Bevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrence. Neurology. 2008;70:779–787. doi: 10.1212/01.wnl.0000304121.57857.38. [DOI] [PubMed] [Google Scholar]
- 41.Narayana A, et al. Antiangiogenic therapy using bevacizumab in recurrent high-grade glioma: impact on local control and patient survival. J Neurosurg. 2009;110:173–180. doi: 10.3171/2008.4.17492. [DOI] [PubMed] [Google Scholar]
- 42.Fischer I, et al. High-grade glioma before and after treatment with radiation and Avastin: initial observations. Neuro Oncol. 2008;10:700–708. doi: 10.1215/15228517-2008-042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ellis LM, Reardon DA. Cancer: The nuances of therapy. Nature. 2009;458:290–292. doi: 10.1038/458290a. [DOI] [PubMed] [Google Scholar]
- 44.Mathews MS, Linskey ME, Hasso AN, Fruehauf JP. The effect of bevacizumab (Avastin) on neuroimaging of brain metastases. Surg Neurol. 2008;70:649–652. doi: 10.1016/j.surneu.2007.06.029. [DOI] [PubMed] [Google Scholar]
- 45.Zuniga RM, et al. Efficacy, safety and patterns of response and recurrence in patients with recurrent high-grade gliomas treated with bevacizumab plus irinotecan. J Neurooncol. 2009;91:329–336. doi: 10.1007/s11060-008-9718-y. [DOI] [PubMed] [Google Scholar]
- 46.Iwamoto FM, et al. Patterns of relapse and prognosis after bevacizumab failure in recurrent glioblastoma. Neurology. 2009;73:1200–1206. doi: 10.1212/WNL.0b013e3181bc0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zuniga RM, et al. Rebound tumour progression after the cessation of bevacizumab therapy in patients with recurrent high-grade glioma. J Neurooncol. 2010;99:237–242. doi: 10.1007/s11060-010-0121-0. [DOI] [PubMed] [Google Scholar]
- 48.Verhoeff JJ, et al. Concerns about anti-angiogenic treatment in patients with glioblastoma multiforme. BMC Cancer. 2009;9:444. doi: 10.1186/1471-2407-9-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Narayana A, et al. Bevacizumab in recurrent high-grade pediatric gliomas. Neuro Oncol. 2010;12:985–990. doi: 10.1093/neuonc/noq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Narayana A, et al. Change in pattern of relapse after antiangiogenic therapy in high-grade glioma. Int J Radiat Oncol Biol Phys. doi: 10.1016/j.ijrobp.2010.10.038. [DOI] [PubMed] [Google Scholar]
- 51.Rubenstein JL, et al. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia. 2000;2:306–314. doi: 10.1038/sj.neo.7900102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kunkel P, et al. Inhibition of glioma angiogenesis and growth in vivo by systemic treatment with a monoclonal antibody against vascular endothelial growth factor receptor-2. Cancer Res. 2001;61:6624–6628. [PubMed] [Google Scholar]
- 53.Blouw B, et al. The hypoxic response of tumors is dependent on their microenvironment. Cancer Cell. 2003;4:133–146. doi: 10.1016/s1535-6108(03)00194-6. [DOI] [PubMed] [Google Scholar]
- 54.Verhoeff JJ, et al. Tumour control by whole brain irradiation of anti-VEGF-treated mice bearing intracerebral glioma. Eur J Cancer. 2009;45:3074–3080. doi: 10.1016/j.ejca.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 55.Lucio-Eterovic AK, Piao Y, de Groot JF. Mediators of glioblastoma resistance and invasion during antivascular endothelial growth factor therapy. Clin Cancer Res. 2009;15:4589–4599. doi: 10.1158/1078-0432.CCR-09-0575. [DOI] [PubMed] [Google Scholar]
- 56.Gomez-Manzano C, et al. VEGF Trap induces antiglioma effect at different stages of disease. Neuro Oncol. 2008;10:940–945. doi: 10.1215/15228517-2008-061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Groot JF, et al. Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro Oncol. 2010;12:233–242. doi: 10.1093/neuonc/nop027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lamszus K, et al. Inhibition of glioblastoma angiogenesis and invasion by combined treatments directed against vascular endothelial growth factor receptor-2, epidermal growth factor receptor, and vascular endothelial-cadherin. Clin Cancer Res. 2005;11:4934–4940. doi: 10.1158/1078-0432.CCR-04-2270. [DOI] [PubMed] [Google Scholar]
- 59.Leenders WP, et al. Antiangiogenic therapy of cerebral melanoma metastases results in sustained tumor progression via vessel co-option. Clin Cancer Res. 2004;10:6222–6230. doi: 10.1158/1078-0432.CCR-04-0823. [DOI] [PubMed] [Google Scholar]
- 60.Chamberlain MC. Radiographic patterns of relapse in glioblastoma. J Neurooncol. 2011;101:319–323. doi: 10.1007/s11060-010-0251-4. [DOI] [PubMed] [Google Scholar]
- 61.Petrelli F, Cabiddu M, Carpo M, Ghilardi M, Barni S. Progression of intramedullary metastasis during perioperative cessation of sunitinib. Nat Rev Urol. 2010;7:634–637. doi: 10.1038/nrurol.2010.161. [DOI] [PubMed] [Google Scholar]
- 62.Plimack ER, Tannir N, Lin E, Bekele BN, Jonasch E. Patterns of disease progression in metastatic renal cell carcinoma patients treated with antivascular agents and interferon: impact of therapy on recurrence patterns and outcome measures. Cancer. 2009;115:1859–1866. doi: 10.1002/cncr.24211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Crawford Y, Ferrara N. Tumor and stromal pathways mediating refractoriness/resistance to anti-angiogenic therapies. Trends Pharmacol Sci. 2009;30:624–630. doi: 10.1016/j.tips.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 64.Nguyen DX, Bos PD, Massagué J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 65.Paget S. The distribution of secondary growths in cancer of the breast. Lancet. 1889;133:571–573. [PubMed] [Google Scholar]
- 66.Hirota K, Semenza GL. Regulation of angiogenesis by hypoxia-inducible factor 1. Crit Rev Oncol Hematol. 2006;59:15–26. doi: 10.1016/j.critrevonc.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 67.Yu JL, Rak JW, Coomber BL, Hicklin DJ, Kerbel RS. Effect of p53 status on tumor response to antiangiogenic therapy. Science. 2002;295:1526–1528. doi: 10.1126/science.1068327. [DOI] [PubMed] [Google Scholar]
- 68.Rapisarda A, Melillo G. Role of the hypoxic tumor microenvironment in the resistance to anti-angiogenic therapies. Drug Resist Updat. 2009;12:74–80. doi: 10.1016/j.drup.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pennacchietti S, et al. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003;3:347–361. doi: 10.1016/s1535-6108(03)00085-0. [DOI] [PubMed] [Google Scholar]
- 70.Steeg PS. Angiogenesis inhibitors: motivators of metastasis? Nat Med. 2003;9:822–823. doi: 10.1038/nm0703-822. [DOI] [PubMed] [Google Scholar]
- 71.Blagosklonny MV. Antiangiogenic therapy and tumor progression. Cancer Cell. 2004;5:13–17. doi: 10.1016/s1535-6108(03)00336-2. [DOI] [PubMed] [Google Scholar]
- 72.Bidard FC, Pierga JY, Vincent-Salomon A, Poupon MF. A “class action” against the microenvironment: do cancer cells cooperate in metastasis? Cancer Metastasis Rev. 2008;27:5–10. doi: 10.1007/s10555-007-9103-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Solinas G, Marchesi F, Garlanda C, Mantovani A, Allavena P. Inflammation-mediated promotion of invasion and metastasis. Cancer Metastasis Rev. 2010;29:243–248. doi: 10.1007/s10555-010-9227-2. [DOI] [PubMed] [Google Scholar]
- 74.Xian X, et al. Pericytes limit tumor cell metastasis. J Clin Invest. 2006;116:642–651. doi: 10.1172/JCI25705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Francia G, Cruz-Munoz W, Man S, Xu P, Kerbel RS. Mouse models of advanced spontaneous metastasis for experimental therapeutics. Nat Rev Cancer. 2011;11:135–141. doi: 10.1038/nrc3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bos PD, Nguyen DX, Massagué J. Modeling metastasis in the mouse. Curr Opin Pharmacol. 2010;10:571–577. doi: 10.1016/j.coph.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hu-Lowe DD, et al. Nonclinical antiangiogenesis and antitumor activities of axitinib (AG-013736), an oral, potent, and selective inhibitor of vascular endothelial growth factor receptor tyrosine kinases 1, 2, 3. Clin Cancer Res. 2008;14:7272–7283. doi: 10.1158/1078-0432.CCR-08-0652. [DOI] [PubMed] [Google Scholar]
- 78.Schomber T, et al. Differential effects of the vascular endothelial growth factor receptor inhibitor PTK787/ZK222584 on tumor angiogenesis and tumor lymphangiogenesis. Mol Cancer Ther. 2009;8:55–63. doi: 10.1158/1535-7163.MCT-08-0679. [DOI] [PubMed] [Google Scholar]
- 79.Yin JJ, Zhang L, Munasinghe J, Linnoila RI, Kelly K. Cediranib/AZD2171 inhibits bone and brain metastasis in a preclinical model of advanced prostate cancer. Cancer Res. 2010;70:8662–8673. doi: 10.1158/0008-5472.CAN-10-1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Steeg PS, Theodorescu D. Metastasis: a therapeutic target for cancer. Nat Clin Pract Oncol. 2008;5:206–219. doi: 10.1038/ncponc1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Padera TP, et al. Differential response of primary tumor versus lymphatic metastasis to VEGFR-2 and VEGFR-3 kinase inhibitors cediranib and vandetanib. Mol Cancer Ther. 2008;7:2272–2279. doi: 10.1158/1535-7163.MCT-08-0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee YJ, et al. Differential effects of VEGFR-1 and VEGFR-2 inhibition on tumor metastases based on host organ environment. Cancer Res. 2010;70:8357–8367. doi: 10.1158/0008-5472.CAN-10-1138. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 83.Gandhi L, et al. Sunitinib prolongs survival in genetically engineered mouse models of multistep lung carcinogenesis. Cancer Prev Res (Phila) 2009;2:330–337. doi: 10.1158/1940-6207.CAPR-08-0213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Grandis JR, Argiris A. Targeting angiogenesis from premalignancy to metastases. Cancer Prev Res (Phila) 2009;2:291–294. doi: 10.1158/1940-6207.CAPR-09-0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Singh M, et al. Assessing therapeutic responses in Kras mutant cancers using genetically engineered mouse models. Nat Biotechnol. 2010;28:585–593. doi: 10.1038/nbt.1640. [DOI] [PubMed] [Google Scholar]
- 86.Bose D, et al. Vascular endothelial growth factor targeted therapy in the perioperative setting: implications for patient care. Lancet Oncol. 2010;11:373–382. doi: 10.1016/S1470-2045(09)70341-9. [DOI] [PubMed] [Google Scholar]
- 87.US National Institutes of Health. Clinical Trials [online] 2011 www.clinicaltrials.gov.
- 88.Allegra CJ, et al. Initial safety report of NSABP C-08: A randomized phase III study of modified FOLFOX6 with or without bevacizumab for the adjuvant treatment of patients with stage II or III colon cancer. J Clin Oncol. 2009;27:3385–3390. doi: 10.1200/JCO.2009.21.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wolmark N, et al. A phase III trial comparing mFOLFOX6 to mFOLFOX6 plus bevacizumab in stage II or III carcinoma of the colon: Results of NSABP Protocol C-08 [abstract] J Clin Oncol. 2009;27(Suppl):aLBA4. [Google Scholar]
- 90.Kinoshita T, et al. Preoperative induction with sorafenib pathologically downstaged advanced renal cell carcinoma: a case report. Int J Urol. 2010;17:286–288. doi: 10.1111/j.1442-2042.2009.02444.x. [DOI] [PubMed] [Google Scholar]
- 91.Altorki N, et al. Phase II proof-of-concept study of pazopanib monotherapy in treatment-naive patients with stage I/II resectable non-small-cell lung cancer. J Clin Oncol. 2010;28:3131–3137. doi: 10.1200/JCO.2009.23.9749. [DOI] [PubMed] [Google Scholar]
- 92.Dawson MR, Duda DG, Chae SS, Fukumura D, Jain RK. VEGFR1 activity modulates myeloid cell infiltration in growing lung metastases but is not required for spontaneous metastasis formation. PLoS ONE. 2009;4:e6525. doi: 10.1371/journal.pone.0006525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Steeg PS, et al. Preclinical drug development must consider the impact on metastasis. Clin Cancer Res. 2009;15:4529–4530. doi: 10.1158/1078-0432.CCR-09-1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mack GS, Marshall A. Lost in migration. Nat Biotechnol. 2010;28:214–229. doi: 10.1038/nbt0310-214. [DOI] [PubMed] [Google Scholar]
- 95.Francia G, Kerbel RS. Raising the bar for cancer therapy models. Nat Biotechnol. 2010;28:561–562. doi: 10.1038/nbt0610-561. [DOI] [PubMed] [Google Scholar]
- 96.Hurwitz H, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 97.Giantonio BJ, et al. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern Cooperative Oncology Group Study E3200. J Clin Oncol. 2007;25:1539–1544. doi: 10.1200/JCO.2006.09.6305. [DOI] [PubMed] [Google Scholar]
- 98.Miller K, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357:2666–2676. doi: 10.1056/NEJMoa072113. [DOI] [PubMed] [Google Scholar]
- 99.Miles DW, et al. Phase III study of bevacizumab plus docetaxel compared with placebo plus docetaxel for the first-line treatment of human epidermal growth factor receptor 2-negative metastatic breast cancer. J Clin Oncol. 2010;28:3239–3247. doi: 10.1200/JCO.2008.21.6457. [DOI] [PubMed] [Google Scholar]
- 100.Robert NJ, et al. RIBBON-1: Randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab (B) for first-line treatment of HER2-negative locally recurrent or metastatic breast cancer (MBC) [abstract] J Clin Oncol. 2009;27(Suppl):a1005. doi: 10.1200/JCO.2010.28.0982. [DOI] [PubMed] [Google Scholar]
- 101.Brufsky A, et al. Progression-free survival (PFS) in patient subgroups in RIBBON-2, a phase III trial of chemotherapy (chemo) plus or minus bevacizumab (BV) for second-line treatment of HER2-negative, locally recurrent or metastatic breast cancer (MBC) [abstract] J Clin Oncol. 2010;28(Suppl):a1021. [Google Scholar]
- 102.Sandler A, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–2550. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 103.Reck M, et al. Overall survival with cisplatin-gemcitabine and bevacizumab or placebo as first-line therapy for nonsquamous non-small-cell lung cancer: results from a randomised phase III trial (AVAiL) Ann Oncol. 2010;21:1804–1809. doi: 10.1093/annonc/mdq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kabbinavar FF, Miller VA, Johnson BE, O’Connor PG, Soh C. Overall survival (OS) in ATLAS, a phase IIIb trial comparing bevacizumab (B) therapy with or without erlotinib (E) after completion of chemotherapy (chemo) with B for first-line treatment of locally advanced, recurrent, or metastatic non-small cell lung cancer (NSCLC) [abstract] J Clin Oncol. 2010;28(Suppl):a7526. [Google Scholar]
- 105.Escudier B, et al. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol. 2010;28:2144–2150. doi: 10.1200/JCO.2009.26.7849. [DOI] [PubMed] [Google Scholar]
- 106.Rini BI, et al. Phase III trial of bevacizumab plus interferon alfa versus interferon alfa monotherapy in patients with metastatic renal cell carcinoma: final results of CALGB 90206. J Clin Oncol. 2010;28:2137–2143. doi: 10.1200/JCO.2009.26.5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cohen MH, Shen YL, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist. 2009;14:1131–1138. doi: 10.1634/theoncologist.2009-0121. [DOI] [PubMed] [Google Scholar]
- 108.Motzer RJ, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009;27:3584–3590. doi: 10.1200/JCO.2008.20.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Goodman VL, et al. Approval summary: sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clin Cancer Res. 2007;13:1367–1373. doi: 10.1158/1078-0432.CCR-06-2328. [DOI] [PubMed] [Google Scholar]
- 110.Raymond E, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:501–513. doi: 10.1056/NEJMoa1003825. [DOI] [PubMed] [Google Scholar]
- 111.Escudier B, et al. Sorafenib for treatment of renal cell carcinoma: final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J Clin Oncol. 2009;27:3312–3218. doi: 10.1200/JCO.2008.19.5511. [DOI] [PubMed] [Google Scholar]
- 112.Llovet JM, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 113.Sternberg CN, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061–1068. doi: 10.1200/JCO.2009.23.9764. [DOI] [PubMed] [Google Scholar]
- 114.Herbst RS, et al. Vandetanib plus docetaxel versus docetaxel as second-line treatment for patients with advanced non-small-cell lung cancer (ZODIAC): a double-blind, randomised, phase 3 trial. Lancet Oncol. 2010;11:619–626. doi: 10.1016/S1470-2045(10)70132-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hecht JR, et al. A randomized phase IIIB trial of chemotherapy, bevacizumab, and panitumumab compared with chemotherapy and bevacizumab alone for metastatic colorectal cancer. J Clin Oncol. 2009;27:672–680. doi: 10.1200/JCO.2008.19.8135. [DOI] [PubMed] [Google Scholar]
- 116.Tol J, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med. 2009;360:563–572. doi: 10.1056/NEJMoa0808268. [DOI] [PubMed] [Google Scholar]
- 117.Miller KD, et al. Randomized phase III trial of capecitabine compared with bevacizumab plus capecitabine in patients with previously treated metastatic breast cancer. J Clin Oncol. 2005;23:792–799. doi: 10.1200/JCO.2005.05.098. [DOI] [PubMed] [Google Scholar]
- 118.Jones D. Avastin-Tarceva combination fails in lung cancer. Nat Biotechnol. 2009;27:108–109. doi: 10.1038/nbt0209-108. [DOI] [PubMed] [Google Scholar]
- 119.Kang Y, et al. AVAGAST: A randomized double-blind, placebo-controlled, phase III study of first-line capecitabine and cisplatin plus bevacizumab or placebo in patients with advanced gastric cancer (AGC) [abstract] J Clin Oncol. 2010;28(Suppl):aLBA4007. [Google Scholar]
- 120.Kindler HL, et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the cancer and Leukemia Group B (CALGB 80303) J Clin Oncol. 2010;28:3617–3622. doi: 10.1200/JCO.2010.28.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Van Cutsem E, et al. Phase III trial of bevacizumab in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. J Clin Oncol. 2009;27:2231–2237. doi: 10.1200/JCO.2008.20.0238. [DOI] [PubMed] [Google Scholar]
- 122.Kelly WK, et al. A randomized, double-blind, placebo-controlled phase III trial comparing docetaxol, prednisone, and placebo with docetaxel, prednisone, and bevacizumab in men with metastatic castration-resistant prostate cancer (mCRPC): Survival results of CALB 90401 [abstract] J Clin Oncol. 2010;28(Suppl):aLBA4511. doi: 10.1200/JCO.2011.39.4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Crown J, et al. Phase III trial of sunitinib (SU) in combination with capecitabine (C) versus C in previously treated advanced breast cancer (ABC) [abstract] J Clin Oncol. 2010;28(Suppl):aLBA1011. [Google Scholar]
- 124.Barrios CH, et al. Phase III randomized trial of sunitinib versus capecitabine in patients with previously treated HER2-negative advanced breast cancer. Breast Cancer Res Treat. 2010;121:121–131. doi: 10.1007/s10549-010-0788-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Scagliotti G, et al. Phase III study of carboplatin and paclitaxel alone or with sorafenib in advanced non-small-cell lung cancer. J Clin Oncol. 2010;28:1835–1842. doi: 10.1200/JCO.2009.26.1321. [DOI] [PubMed] [Google Scholar]
- 126.Kohne C, et al. Final results of CONFIRM 2: A multinational, randomized, double-blind, phase III study in 2nd line patients (pts) with metastatic colorectal cancer (mCRC) receiving FOLFOX4 and PTK787/ZK 222584 (PTK/ZK) or placebo [abstract] J Clin Oncol. 2007;25(Suppl):a4033. [Google Scholar]
- 127.Natale RB, et al. Phase III trial of vandetanib compared with erlotinib in patients with previously treated advanced non-small-cell lung cancer. J Clin Oncol. doi: 10.1200/JCO.2010.28.5981. [DOI] [PubMed] [Google Scholar]
- 128.de Boer RH, et al. Vandetanib plus pemetrexed for the second-line treatment of advanced non-small-cell lung cancer: a randomized, double-blind phase III trial. J Clin Oncol. doi: 10.1200/JCO.2010.29.5717. [DOI] [PubMed] [Google Scholar]
- 129.Batchelor TT, et al. A phase III randomized study comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, with lomustine alone in recuttrent glioblastoma patients [abstract] Ann Oncol. 2010;21(Suppl. 8):aLBA7. doi: 10.1200/JCO.2012.47.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.von Essen CF. Radiation enhancement of metastasis: a review. Clin Exp Metastasis. 1991;9:77–104. doi: 10.1007/BF01756381. [DOI] [PubMed] [Google Scholar]
- 131.Kargiotis O, Geka A, Rao JS, Kyritsis AP. Effects of irradiation on tumor cell survival, invasion and angiogenesis. J Neurooncol. 2010;100:323–338. doi: 10.1007/s11060-010-0199-4. [DOI] [PubMed] [Google Scholar]
- 132.Bagri A, et al. Effects of anti-VEGF treatment duration on tumor growth, tumor regrowth, and treatment efficacy. Clin Cancer Res. 2010;16:3887–3900. doi: 10.1158/1078-0432.CCR-09-3100. [DOI] [PubMed] [Google Scholar]
- 133.Kienast Y, et al. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med. 2010;16:116–122. doi: 10.1038/nm.2072. [DOI] [PubMed] [Google Scholar]
- 134.Lee SL, et al. Hypoxia-induced pathological angiogenesis mediates tumor cell dissemination, invasion, and metastasis in a zebrafish tumor model. Proc Natl Acad Sci USA. 2009;106:19485–19490. doi: 10.1073/pnas.0909228106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ellis LM, Reardon DA. Is there really a yin and yang to VEGF-targeted therapies? Lancet Oncol. 2010;11:809–811. doi: 10.1016/S1470-2045(10)70161-3. [DOI] [PubMed] [Google Scholar]
- 136.Van Cutsem E, Lambrechts D, Prenen H, Jain RK, Carmeliet P. Lessons from the adjuvant bevacizumab trial on colon cancer: what next? J Clin Oncol. 2011;29:1–4. doi: 10.1200/JCO.2010.32.2701. [DOI] [PubMed] [Google Scholar]
- 137.Saidi A, et al. Combined targeting of interleukin-6 and vascular endothelial growth factor potently inhibits glioma growth and invasiveness. Int J Cancer. 2009;125:1054–1064. doi: 10.1002/ijc.24380. [DOI] [PubMed] [Google Scholar]
- 138.Rofstad EK, Halsør EF. Hypoxia-associated spontaneous pulmonary metastasis in human melanoma xenografts: involvement of microvascular hot spots induced in hypoxic foci by interleukin 8. Br J Cancer. 2002;86:301–308. doi: 10.1038/sj.bjc.6600052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rofstad EK, et al. Hypoxia promotes lymph node metastasis in human melanoma xenografts by up-regulating the urokinase-type plasminogen activator receptor. Cancer Res. 2002;62:1847–1853. [PubMed] [Google Scholar]
- 140.Galluzzo M, Pennacchietti S, Rosano S, Comoglio PM, Michieli P. Prevention of hypoxia by myoglobin expression in human tumor cells promotes differentiation and inhibits metastasis. J Clin Invest. 2009;119:865–875. doi: 10.1172/JCI36579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cairns RA, Khokha R, Hill RP. Molecular mechanisms of tumor invasion and metastasis: an integrated view. Curr Mol Med. 2003;3:659–671. doi: 10.2174/1566524033479447. [DOI] [PubMed] [Google Scholar]
- 142.Park JE, et al. Hypoxia tumor cell modulates its microenvironment to enhance angiogenic and metastastic potential by secretion of proteins and exosomes. Mol Cell Proteomics. 2010;9:1085–1099. doi: 10.1074/mcp.M900381-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Du R, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cairns RA, Hill RP. Acute hypoxia enhances spontaneous lymph node metastasis in an orthotopic murine model of human cervical carcinoma. Cancer Res. 2004;64:2054–2061. doi: 10.1158/0008-5472.can-03-3196. [DOI] [PubMed] [Google Scholar]
- 145.Cairns RA, Kalliomaki T, Hill RP. Acute (cyclic) hypoxia enhances spontaneous metastasis of KHT murine tumors. Cancer Res. 2001;61:8903–8908. [PubMed] [Google Scholar]
- 146.Rofstad EK, Gaustad JV, Egeland TA, Mathiesen B, Galappathi K. Tumors exposed to acute cyclic hypoxic stress show enhanced angiogenesis, perfusion and metastatic dissemination. Int J Cancer. 2010;127:1535–1546. doi: 10.1002/ijc.25176. [DOI] [PubMed] [Google Scholar]
- 147.Higgins DF, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117:3810–3820. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;8:299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 149.Martinez-Outschoorn UE, et al. The autophagic tumor stroma model of cancer or “battery operated tumor growth”: A simple solution to the autophagy paradox. Cell Cycle. 2010;9:4297–4306. doi: 10.4161/cc.9.21.13817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ricci-Vitiani L, et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;468:824–828. doi: 10.1038/nature09557. [DOI] [PubMed] [Google Scholar]
- 151.Wang R, et al. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. 2010;468:829–833. doi: 10.1038/nature09624. [DOI] [PubMed] [Google Scholar]
- 152.Crawford Y, et al. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell. 2009;15:21–34. doi: 10.1016/j.ccr.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 153.Ebos JM, Lee CR, Christensen JG, Mutsaers AJ, Kerbel RS. Multiple circulating proangiogenic factors induced by sunitinib malate are tumor-independent and correlate with antitumor efficacy. Proc Natl Acad Sci USA. 2007;104:17069–17074. doi: 10.1073/pnas.0708148104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Batchelor TT, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11:83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lewis CE, De Palma M, Naldini L. Tie2-expressing monocytes and tumor angiogenesis: regulation by hypoxia and angiopoietin-2. Cancer Res. 2007;67:8429–8432. doi: 10.1158/0008-5472.CAN-07-1684. [DOI] [PubMed] [Google Scholar]
- 156.Shojaei F, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25:911–920. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- 157.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Shojaei F, et al. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci USA. 2009;106:6742–6747. doi: 10.1073/pnas.0902280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kaplan RN, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Steeg PS. Cancer biology: emissaries set up new sites. Nature. 2005;438:750–751. doi: 10.1038/438750b. [DOI] [PubMed] [Google Scholar]
- 161.Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003;111:1287–1295. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hirschi KK, D’Amore PA. Control of angiogenesis by the pericyte: molecular mechanisms and significance. EXS. 1997;79:419–428. doi: 10.1007/978-3-0348-9006-9_18. [DOI] [PubMed] [Google Scholar]
- 163.Elice F, Rodeghiero F, Falanga A, Rickles FR. Thrombosis associated with angiogenesis inhibitors. Best Pract Res Clin Haematol. 2009;22:115–128. doi: 10.1016/j.beha.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 164.Reynolds AR, et al. Stimulation of tumor growth and angiogenesis by low concentrations of RGD-mimetic integrin inhibitors. Nat Med. 2009;15:392–400. doi: 10.1038/nm.1941. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplemental material