Pre-Columbian mycobacterial genomes reveal seals as a source of New World human tuberculosis (original) (raw)
. Author manuscript; available in PMC: 2015 Aug 27.
Published in final edited form as: Nature. 2014 Aug 20;514(7523):494–497. doi: 10.1038/nature13591
Abstract
Modern strains of Mycobacterium tuberculosis from the Americas are closely related to those from Europe, supporting the assumption that human tuberculosis was introduced post-contact1. This notion, however, is incompatible with archaeological evidence of pre-contact tuberculosis in the New World2. Comparative genomics of modern isolates suggests that M. tuberculosis attained its worldwide distribution following human dispersals out of Africa during the Pleistocene epoch3, although this has yet to be confirmed with ancient calibration points. Here we present three 1,000-year-old mycobacterial genomes from Peruvian human skeletons, revealing that a member of the M. tuberculosis complex caused human disease before contact. The ancient strains are distinct from known human-adapted forms and are most closely related to those adapted to seals and sea lions. Two independent dating approaches suggest a most recent common ancestor for the M. tuberculosis complex less than 6,000 years ago, which supports a Holocene dispersal of the disease. Our results implicate sea mammals as having played a role in transmitting the disease to humans across the ocean.
Mycobacterium tuberculosis has had a long history with humans, although consensus has not been reached on when this interaction began1,3,4. Previous models held that the human-adapted pathogen evolved from a zoonotic transfer of Mycobacterium bovis following animal domestication during the Neolithic age5. Comparative genomic analyses, however, suggest that the bovine form and those adapted to other animal hosts are in fact derived from human strains3,6. This supports a rather different disease history where humans may have been the most susceptible host species for early progenitors of strains currently circulating. Today the majority of M. tuberculosis diversity exists in Africa7, implying that the pathogen probably originated from a monoclonal expansion therein and achieved its worldwide distribution via human movements1,3,4. The observations that M. tuberculosis strains tend to be associated with human populations8 and that selection in the bacterium exists at loci associated with host immune responses9 indicate that host and pathogen have had sufficient time to co-evolve. Dating approaches that use human demographic events for calibration generate substitution rates that differ by over an order of magnitude depending on the model3,10, and would thus contribute to vastly different coalescence estimates for all M. tuberculosis lineages, collectively referred to as the M. tuberculosis complex (MTBC).
Given the pathogen’s phylogeography, current models are unable to explain the abundant archaeological evidence for the presence of tuberculosis in the Americas before European contact. Strains currently circulating in the Americas are most closely related to those of European origin, and this has been used to support a European dissemination from either early settlement or trade associations1. This model, however, is incompatible with bioarchaeological data indicating the presence of tuberculosis in the pre-contact New World2 (see Supplementary Information). Molecular investigations using ancient pre-Columbian material have identified short conserved regions of mobile elements considered to be diagnostic for tuberculosis, although these markers offer no information about phylogenetic placement, and are thus difficult to authenticate as ancient11. While a Pleistocene dispersal following human movements out of Africa could explain its presence in the pre-contact New World3,4, the dominance of European-derived lineages in the Americas today makes this difficult to reconcile without data to support a complete strain replacement within the past 500 years.
Genomic reconstructions of ancient pathogens provide robust evidence of DNA authenticity and permit genome-level comparisons12. The success of DNA capture13 and genomic assembly of an historical MTBC strain via metagenomic sequencing14 implies that DNA preservation of this pathogen may be adequate to address outstanding evolutionary questions requiring use of archaeological material. Here we apply these techniques to demonstrate that a previously uncharacterized member of the MTBC caused human infection in the Americas before European contact.
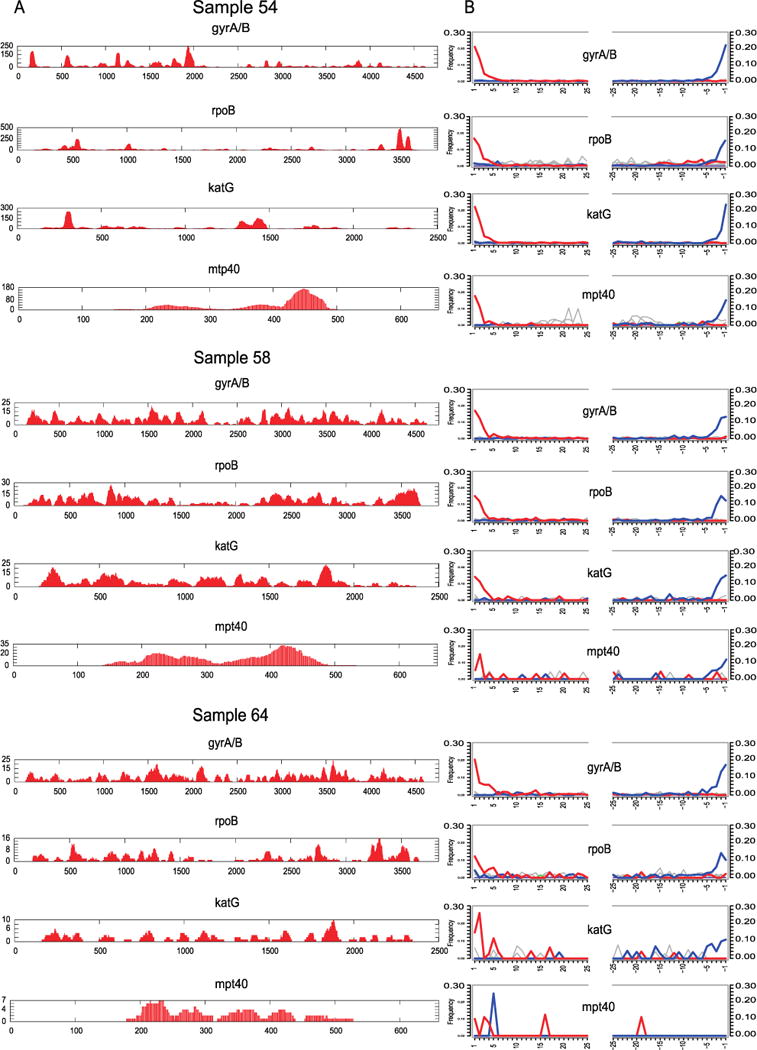
We screened 68 skeletal samples representing New World pre- and post-contact sites (Supplementary Table 1). All individuals showed skeletal indicators associated with tuberculosis infections. Samples were processed via established protocols and were screened for M. tuberculosis DNA by an in-solution capture assay designed for the rpoB, gyrA, gyrB, katG, and mpt40 genes (Supplementary Table 2). Capture products for samples and negative controls were sequenced on an Illumina MiSeq and mapped to the corresponding regions in the M. tuberculosis H37Rv reference genome (NC_000962.2). No tuberculosis-specific fragments were found in our negative controls. Only three of the 68 samples, referred to here as samples 54, 58, and 64, showed convincing preservation of tuberculosis DNA (see Supplementary Information, Extended Data Fig. 1, and Supplementary Table 1): all three samples were recovered from excavations in Peru and derive from Chiribaya cultures associated with the Middle Horizon/Late Intermediate period (AD 750–1350) (Fig. 1). Radiocarbon dates ranging from AD 1028 to AD 1280 (at not less than 98.5% probability) (Supplementary Table 3) confirm that they predate European contact. Spectra of DNA damage displayed a pattern expected of ancient molecules15. For comparison, non-enriched libraries were sequenced on an Illumina MiSeq producing 34,780 to 112,428 reads for each of the three samples, of which 4.6% to 1.6% mapped to the human genome (hg19). In contrast, a maximum of only 1.8% of the reads mapped to the M. tuberculosis reference genomes (Supplementary Table 1), indicating that DNA capture would be necessary for genome retrieval.
Figure 1. Archaeological description of the skeletal samples.
a, Map of Peru showing locations of archaeological sites; CGIAR SRTM 90m Digital Elevation Database version 4.1 (http://srtm.csi.cgiar.org). b, c, Skeletal lesions of active tuberculosis from two individuals positive for M. tuberculosis DNA (b, individual 58; c, individual 64). Arrows show vertebral lesions, collapse, fusion, and kyphosis.
DNA libraries treated with uracil DNA glycosylase were generated to remove and repair damaged nucleotides, and were subsequently used for full genome hybridization capture (Agilent). Array probes were designed to accommodate known genetic diversity in the MTBC, as well as portions of the Mycobacterium avium and Mycobacterium kansasii genomes (Supplementary Table 4). Enriched products were sequenced on one lane of an Illumina HiSeq 2000. For comparison against a larger data set of 259 modern MTBC genomes including the outgroup Mycobacterium canettii3, all ancient reads were mapped against a computationally constructed ancestor for the MTBC9. The recently published genome from an eighteenth century Hungarian mummy14, as well as 14 animal strains from the Mycobacterium caprae, Mycobacterium microti, and Mycobacterium pinnipedii lineages, were added, along with a strain recently isolated from a wild chimpanzee16. Standard mapping resulted in heterozygous positions for the mummy, Peruvian samples 54 and 64, and all modern samples (Extended Data Fig. 2). Increased mapping stringency removed many heterozygous positions for the Peruvian samples, suggesting they derived from non-tuberculosis reads; however, the Hungarian mummy and eight modern samples still displayed heterozygosity consistent with mixed strains14 (Extended Data Fig. 3). Our more stringent mapping reduced overall genomic coverage for all samples. The final data set thus consisted of 262 genomes with a minimum of 75% coverage, four of which were ancient (Supplementary Table 5). A minimum of 20-fold average coverage was obtained for each of the Peruvian genomes (Fig. 2 and Supplementary Table 6), implying a 40-to 120-fold enrichment (Supplementary Table 6).
Figure 2. Coverage plots for three ancient genomes.
Inner ring: purple, AT content; gold, GC content. Coverage rings for samples 64, 58, and 54 shown in red, green, and blue, respectively. Vertical lines indicate locations of unique SNPs. SNPs were identified before exclusion of positions with missing data from the full 262 genome data set.
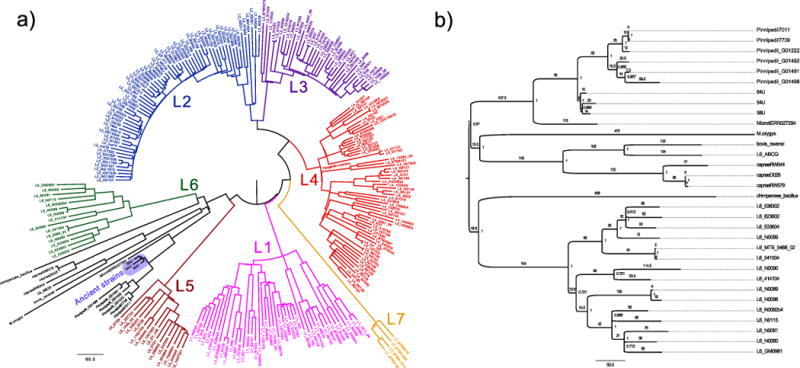
Single nucleotide polymorphism (SNP) analyses were performed by comparing all genomes against the constructed ancestor. This identified 53,177 SNPs for the entire data set, which ranged between 489 and 1,415 per genome (Supplementary Table 8). As input for phylogenetic assessments we used an alignment of 22,480 variable positions after removing all positions with missing data. Tree reconstructions revealed that our Peruvian genomes did not cluster with other human strains, but rather were more closely related to the animal lineage (Extended Data Figs 4–6), sharing 76 SNPs with modern M. pinnipedii strains (Fig. 3). Genomic architecture revealed a region of difference (RD) deletion pattern common to all animal lineages (Supplementary Table 7), as well as absence of the _M. microti_-specific RDmic and presence of the _M. pinnipedii_-specific RDseal. To our knowledge, M. pinnipedii strains have been isolated only from seal species restricted to the Southern Hemisphere17. Here they were harvested from captive and wild animals from South America and Australia. The three ancient strains share five unique SNPs, all of which are non-synonymous (Supplementary Table 9); this indicates that these strains derive from a common progenitor, with subsequent accumulation of 10–23 substitutions along the three strain-specific branches. To investigate possible signals of adaptation, we screened these five shared SNPs for putative functional effects. Our computational analysis predicted a functional impact of the P44L mutation in Rv2258c, encoding a methyltransferase involved in ubiquinone metabolism (Supplementary Table 9). The SNP in the ctpA gene at codon 62 (D62N) was not predicted to have a functional impact; however, we identified two other non-synonymous SNPs (D62G and D62E), also not predicted to have functional impacts, in the same codon of ctpA at different positions, each in a lineage 4 modern strain. The occurrence of homoplasies is uncommon in the MTBC, and therefore potentially indicates positive selective pressure18. A site-wise analysis of positive selection on codon 62 of ctpA confirmed that all three SNPs may be under diversifying selection (Supplementary Table 10). The ctp genes encode efflux ion pumps that are thought to prevent metal accumulation in the bacterium19, hence adaptation may relate to host metal-ion availability. This notion is supported by the existence of homoplasies in other genes of the efflux pump family in modern MTBC strains (Supplementary Table 9).
Figure 3. Phylogenetic analysis.
a, Bayesian maximum clade credibility tree of 261 MTBC genomes (excluding Hungarian mummy), with estimated divergence dates shown in years before present using a model of population expansion. b, Maximum parsimony tree for lineage 6 and animal-adapted MTBC genomes with SNPs that define all branches. Bootstrap values in grey italics. Deletions specific to the animal lineages are shown as triangles.
Bayesian dating analysis used radiocarbon dates as tip calibration (Supplementary Table 3). The Hungarian mummy sample was excluded because of the presence of multiple strains. A clock test rejected the molecular clock for all 258 modern genomes (_P_= 5×10−147) (Extended Data Fig. 7). Dating analysis using a relaxed clock model and a constant population size generated a mutation rate of 4.6×10−8 substitutions per site per year (3×10−8 to 6.2×10−8 95% highest posterior density (HPD) interval). Bayesian skyline plots revealed constant population sizes for the animal strains and clear indications of expansions in the human-adapted lineages (Extended Data Fig. 8b). An expansion model had a negligible influence on the mutation rate, generating 4.9×10−8 substitutions per site per year (3.4×10−8 to 6.4×10−8 95% HPD), which corresponds to 0.20 and 0.21 substitutions per genome per year for a constant and expanding population model, respectively. This rate agrees well with estimates of MTBC evolution in modern epidemiological contexts20, and is more than tenfold faster than those using human dispersals out of Africa as calibration3. Our mutation rates date the most recent common ancestor (MRCA) for the MTBC (excluding M. canettii) at 4,449 years before present (yr BP) (2,990–6,062 yr BP 95% HPD) and 4,064 yr BP (2,951–5,339 yr BP 95% HPD) for constant size and expansion models, respectively (Extended Data Fig. 8a). This dating was corroborated by an independent analysis using the sequences from the Hungarian mummy14 sample as the only ancient calibration point. We separated the individual variants of the two mummy strains by reconstructing them onto the MTBC lineage 4 phylogeny (Extended Data Fig. 9). Lengths from the terminal branches were estimated by using the number of heterozygous variants not present in the modern strains, under the assumption that both isolates were equidistant from the root of the tree. The year of death 1797 and estimated ages for the penultimate nodes were used as priors for Bayesian phylogenetic reconstruction. Using only synonymous variants, a relaxed clock, and constant population size, we estimate the age of the MRCA of the MTBC (excluding M. canettii) to be 5268.5 yr BP (2689.6–8417.7 95% HPD) with a synonymous substitution rate of 7.07×10−8 (3.70× 10−8 to 1.12×10−7 95% HPD) per site per year (Extended Data Fig. 10 and Supplementary Table 11). This higher substitution rate may be due to lower selective pressure on synonymous sites.
Our results provide unequivocal evidence of human infection caused by members of the MTB Cinpre-Columbian South America. Our MRCA, which is at least an order of magnitude younger than previous estimates3,4, presented us with a challenge to explain how a mammalian pathogen could have reached human populations in the Americas about 10,000 years after inundation of the Bering land bridge21. The fact that our ancient genomes share a common ancestor with strains that are restricted to seals and sea lions17 provides a plausible, if unexpected, route of entry into the New World: within the past 2,500 years pinnipeds probably contracted the disease from an African host species, carried the disease across ocean waters, and exploitation of marine mammals among coastal peoples of South America facilitated a zoonotic transfer of the bacterium within the first millennium AD. This parallels similar zoo-noses of marine parasites acquired from seal consumption among archaeological coastal populations22 (Supplementary Information).
Owing to the abundance of publications reporting morphological evidence of pre-Columbian tuberculosis in the region, the coasts of Peru and northern Chile have long been recognized in the archaeological literature as locations where tuberculosis first came into view in the New World2. Some have even suggested marine mammals as a potential source of the infection23. The three individuals considered here show pathological changes consistent with either pulmonary or disseminated tuberculosis, so a non-contagious infection acquired from consumption of contaminated animal products in each case cannot be ruled out. In the absence of these data, however, the five unique derived positions shared by the ancient Peruvian genomes may provide preliminary evidence of host specificity. All three genomes share a common ancestor that predates the radiocarbon age of our skeletal material by more than 100 years, and two SNPs show potential signals of adaptation. These observations could support a single zoonotic transfer from pinnipeds to humans between AD 700 and AD 1000 (Fig. 3). Subsequent host adaptation and dissemination is a compelling prospect for future work. If confirmed, this would constitute the first example of a zoonotic transfer followed by re-adaptation to the human host in the MTBC.
Such a model could explain the abundance of tuberculosis-like lesions in the region that accumulate beginning at approximately AD 700 (refs 24, 25). The later appearance of similar skeletal lesions in North America that first appear at about AD 900 is consistent with either a transcontinental spread of the pathogen via established trade routes26 or a later independent introduction of tuberculosis from a different source. The lack of representation of this or any other American-specific strain in modern groups supports replacement by a European strain after contact that quickly moved through indigenous populations on account of additional adverse factors such as social marginalization, food insecurity, and potentially facilitative co-circulating infections that reached epidemic levels, such as those recorded in northern North America during the decline of the fur trade27. Our data also indicate a subsequent introduction of M. pinnipedii to Australian seal colonies within the past 700 years (Fig. 3); the potential for similar zoonotic transfers, therefore, exists in Oceanian populations, although lesions suggestive of tuberculosis have not been identified in relevant skeletal material2.
M. pinnipedii has caused infection in several mammalian host species, including humans, in the context of zoo outbreaks28. Further sampling of animal-adapted MTBC from both modern and ancient contexts will be of great value in determining its range of potential host species and in clarifying directions of transmission. While a human transfer of the bacterium to marine mammals cannot be ruled out from our data, we consider this extremely unlikely: humans did not herd or farm seals, and close, regular contacts would be required for anthroponotic transmission, as is observed in domestic cattle29.
The above assertion of an introduction of MTBC via pinnipeds followed by human adaptation and subsequent transmission throughout the Americas can only be confirmed by comparison with additional North and South American pre-Columbian MTBC genomes from non-coastal groups, which remain elusive despite the inclusion of suitable material in our screening (Supplementary Table 1). In addition, our dating analyses are based on two independent approaches, although each relies on (effectively) a single calibration point. Mutation rate heterogeneity is documented in other clonal pathogens30, and the rejection of our molecular clock indicates that MTBC evolution is not constant among lineages. Additional calibration points from ancient MTBC lineages around the world will be essential to evaluate the legitimacy of our proposed models. Such caveats are of paramount importance considering the many investigations that report on members of the MTBC identified in skeletal samples that predate our inferred MRCA, or American material from periods that predate our proposed time of MTBC entry. Such claims could only be reconciled with what we propose here if (1) rate heterogeneity or horizontal gene transfer is obscuring our dating analysis, perhaps as a result of human population expansions which increase the availability of susceptible hosts and allow selection to operate more quickly, (2) the pathogens identified in the earlier archaeological material are in fact not members of the MTBC, but rather are ancestral forms that have since undergone replacements, or (3) certain techniques for MTBC identification in archaeological material lack specificity.
Extended Data
Extended Data Figure 1.
Coverage and damage plots for the M. tuberculosis capture regions for samples 54, 58, and 64.
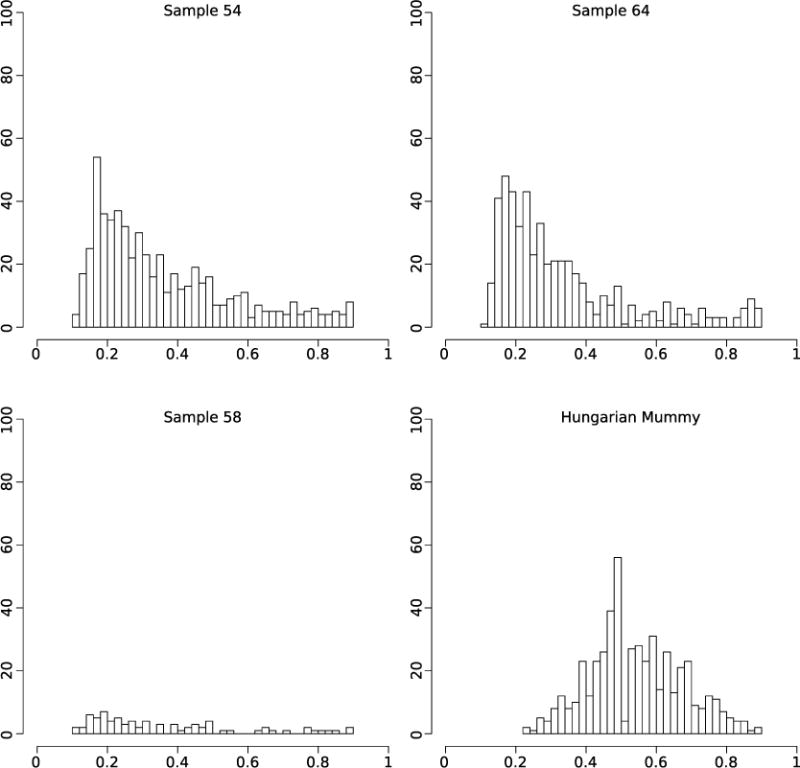
Extended Data Figure 2. Histograms of SNP allele frequency distributions for the ancient samples and the Hungarian mummy sample using standard mapping parameters.
The x axis denotes the frequency of reads covering a SNP position in which the SNP was detected. The y axis denotes the number of observed SNP calls with the respective frequency. All variants with a SNP allele frequency below 90% are shown.
Extended Data Figure 3. Histograms of SNP allele frequency distributions for the ancient samples, the Hungarian mummy sample, and two modern isolates using stricter mapping and filtering parameters.
The x axis denotes the frequency of reads covering a SNP position in which the SNP was detected. The y axis denotes the number of observed SNP calls with the respective frequency. All variants with a SNP allele frequency below 90% are shown.
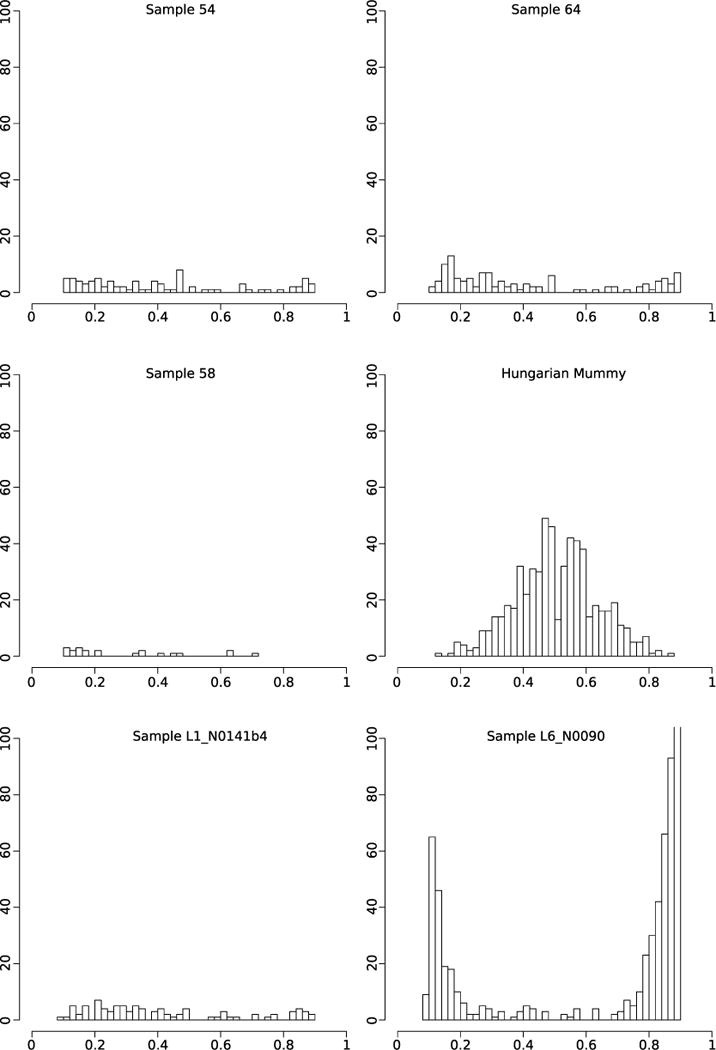
Extended Data Figure 4. Maximum parsimony analysis.
a, Maximum parsimony tree of all 262 samples of the complete data set. Positions with missing data were excluded. b, Subtree of the full maximum parsimony tree showing the lineage 6 and animal strains. Positions with missing data were excluded. Branches are labelled with the absolute number of substitutions. Internal nodes are labelled with bootstrap statistics obtained from 1,000 replicates.
Extended Data Figure 5. Maximum likelihood analysis.
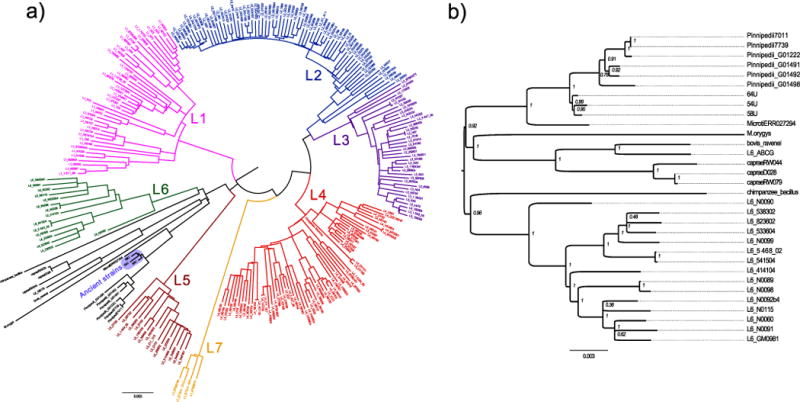
a, Maximum likelihood tree of all 262 samples of the complete data set. Positions with missing data were excluded. b, Maximum likelihood subtree showing the lineage 6 and animal strains. Positions with missing data were excluded. Internal nodes are labelled with bootstrap statistics obtained from 200 replicates.
Extended Data Figure 6. Neighbour joining analysis.
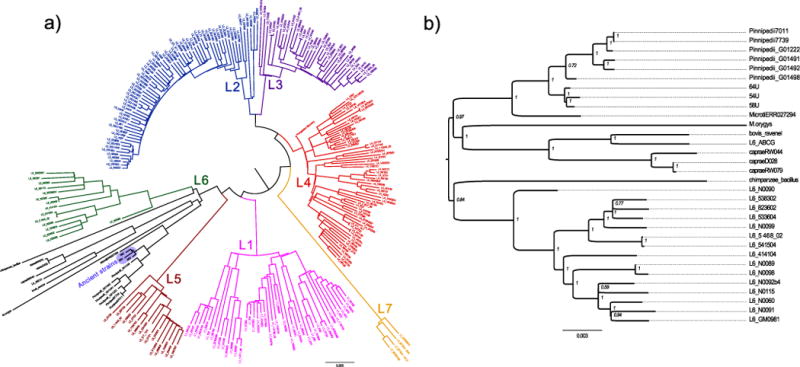
a, Neighbour joining tree of all 262 samples of the complete data set. Positions with missing data were excluded. b, Neighbour joining subtree showing the lineage 6 and animal strains. Positions with missing data were excluded. Internal nodes are labelled with bootstrap statistics obtained from 1,000 replicates.
Extended Data Figure 7. Maximum clade credibility tree of M. tuberculosis.
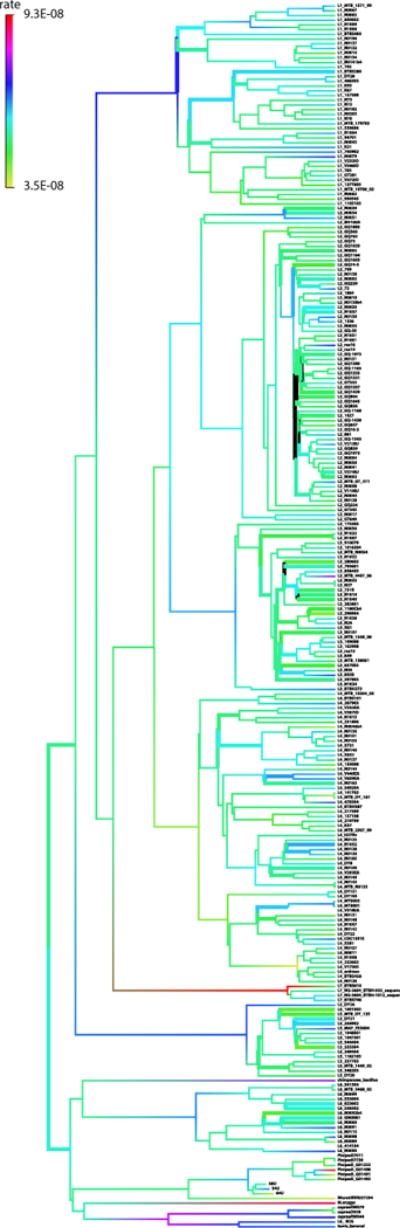
The tree was estimated using the uncorrelated log-normal relaxed clock model in BEAST 1.7.5 (ref. 31). The radiocarbon dates of the ancient Peruvian strains were used as temporal estimates to date the tree. Branch lengths are scaled to years. Branch colours indicate the estimated branch substitution rate on the logarithmic scale shown in the legend at the left.
Extended Data Figure 8.
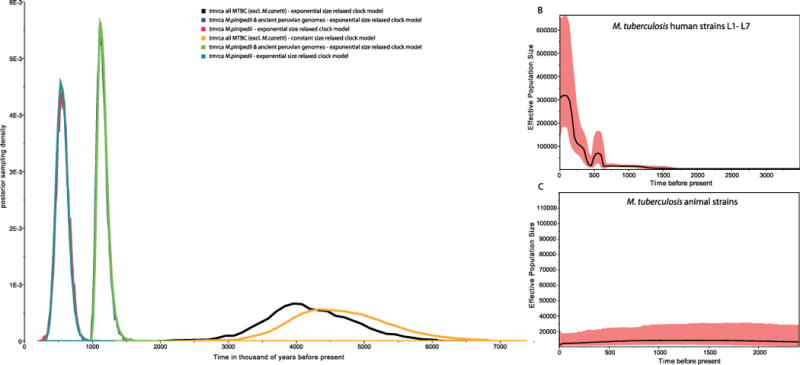
a, Posterior distributions of times to most recent common ancestor (TMRCA) for different MTBC branches, and exponential growth and constant size models. b, Bayesian skyline plot showing estimated effective population sizes for the human lineages. c, Bayesian skyline plot showing estimated effective population sizes for the animal lineages.
Extended Data Figure 9. Maximum likelihood phylogeny of L4 lineage including modern and ancient strains.
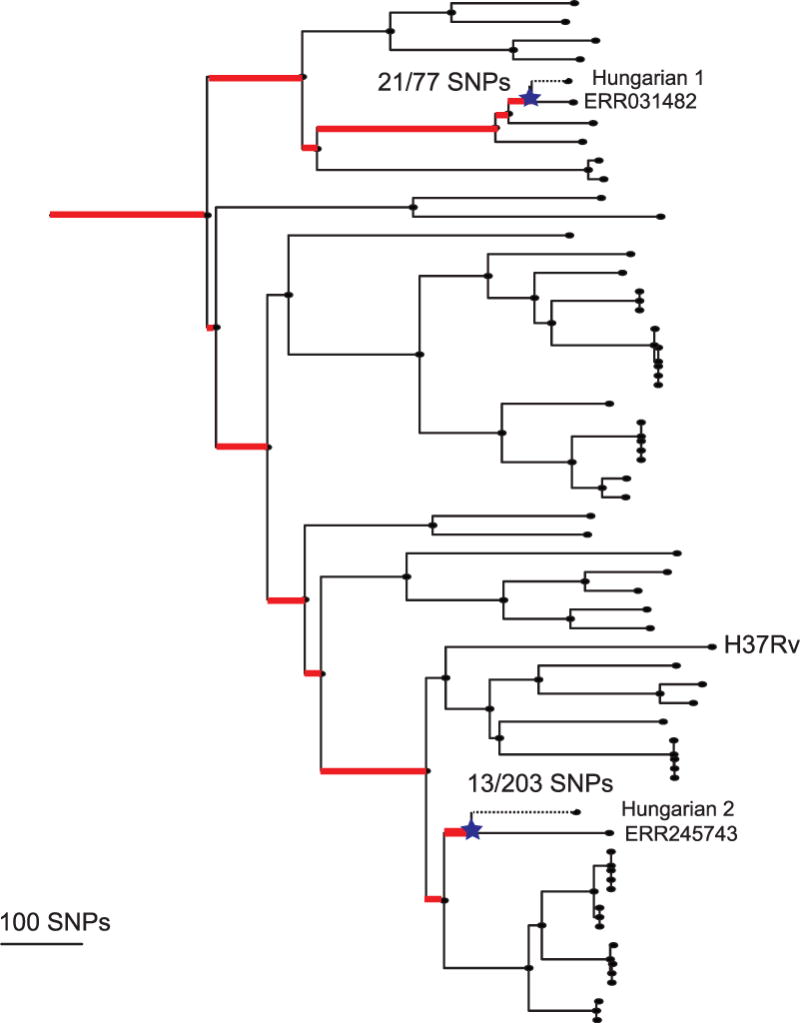
The mixed samples are separated out into Hungarian 1 and 2. SNPs were mapped back onto the phylogeny, and branches marked in red are those defined by variants found to be mixed in the Hungarian sample. This allowed us to determine the ancestral nodes and branches for each of the two strains on the tree. The dotted lines represent the unknown length of the terminal branches, with the stars representing the theoretical penultimate node for which age priors were determined.
Extended Data Figure 10. Maximum clade credibility tree produced using BEAST31.
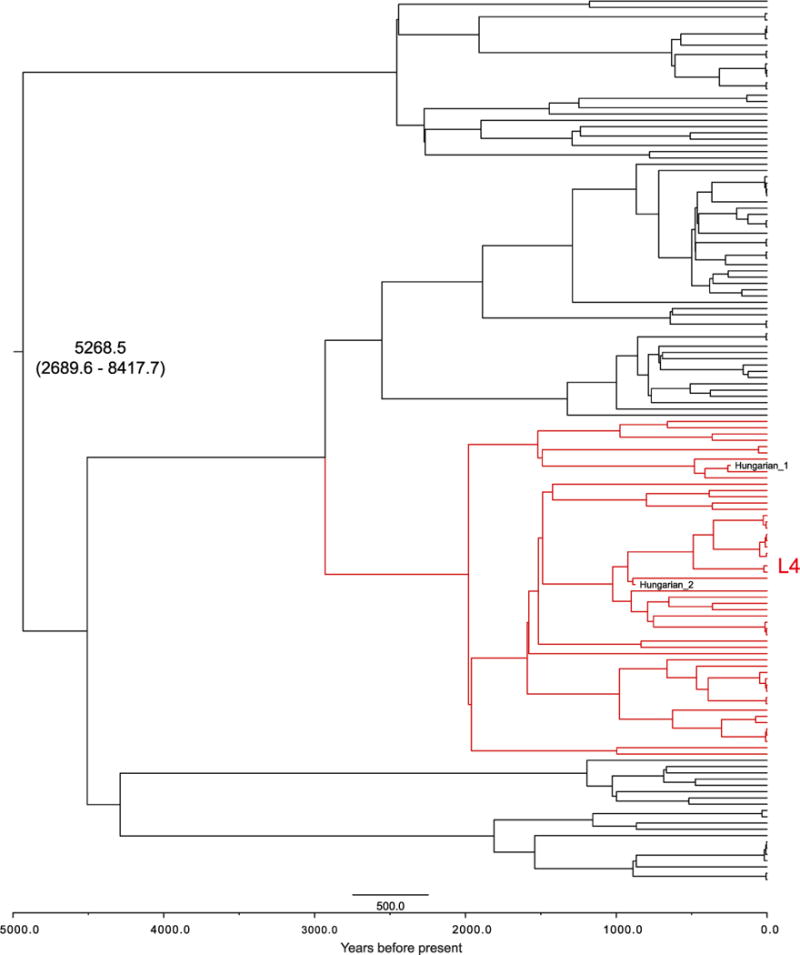
Produced using TreeAnnotator from 9,000 trees. Branch lengths are scaled by age. The mean age (yr BP) of the MRCA plus 95% HPD, and the position of the separated Hungarian ancient strains, are marked on the phylogeny.
Supplementary Material
Supplementary Table 1
Supplementary Table 5
Supplementary Table 6
Supplementary Table 8
Supplementary Table 9
Supplementary Tables 2-4, 7, 10-11
Acknowledgments
We thank the following people and institutions for assistance and/or permission for sampling: Museo Contisuyo, Centro Mallqui, S. Guillen, Instituto Nacional de Cultura, Peru, G. Cock, C. Gaither, M. Murphy, M. C. Lozada, S. Burgess, D. Blom, B. Owen, A. Oquiche Hernani, P. Palacios Filinich, S. Williams, B. Vargas, D. Rice, and H. Klaus, S. Pfieffer, the University of Toronto, the Upper Mississippi Valley Archaeological Research Foundation, L. Conrad, Indiana University, G. Milner, the Pennsylvania State University, the American Museum of Natural History, A. Stodder, B. Brier, I. Tattersall, K. Mowbray, the National Museum of Natural History (Smithsonian Institution), B. Billeck, the Smithsonian Institution, N. Tuross, L.-A. Pfister, Rochester Museum & Science Center, L. P. Saunders, C. Grivas, G. Housman, and M. Nieves-Colon. We thank H. Poinar for discussions about capture regions for M. tuberculosis screening. We thank B. Coombes and B. Krause-Kyora for providing modern tuberculosis for bait manufacture. The Huron Wendat Nation is aware of the sampling of Uxbridge bone and is a recipient of information from this study. We acknowledge the following sources of funding: European Research Council starting grant APGREID (to J.K.), the National Science Foundation (to A.C.S. and J.E.B.) for NSF BCS-1063939, NSF-REU BCS-0612222, and NSF BCS-0612222, the George E. Burch Fellow in Theoretic Medicine and Affiliated Sciences at the Smithsonian Institution (2003–2007, to J.E.B.), Social Sciences and Humanities Research Council of Canada postdoctoral fellowship grant 756-2011-501 (to K.I.B.), National Science Foundation Graduate Research Fellowship DGE-1311230 and Jacob K. Javits Fellowship (to K.M.H.), Ramón y Cajal Spanish research grant RYC-2012–10627 (to I.C.), Swiss National Science Foundation PP0033_119205 (to S.G.), National Institutes of Health AI090928 (to S.G.), European Research Council 309540 (to S.G.), PICT0575 Argentina (to R.A.G.), Wadsworth Fellowship from the Wenner-Gren Foundation (to T.J.C.), Wellcome Trust 098051 (to J.P., J.M.B. and S.R.H.), and funding from the Medical Research Council (to J.M.B.).
Footnotes
Online Content Methods, along with any additional Extended Data display items and Source Data, are available in the online version of the paper; references unique to these sections appear only in the online paper.
Author Contributions A.C.S., J.E.B., J.K., K.I.B., and K.M.H. conceived the investigation. J.K., K.I.B., A.C.S., S.A.F., N.W., and A.K.W. designed experiments. J.P., R.A.G., D.L.W.S., D.C.C., S.N., M.A.B., M.Z., and R.B. provided samples for analysis. K.I.B., K.M.H., V.J.S., T.J.C., and A.K.W. performed laboratory work. A.H., J.K., S.G., M.C., N.W., K.I.B., I.C., D.Y., J.P., J.M.B., S.R.H., D.H., K.N., A.C.S., K.M.H., J.E.B., T.J.C., D.C.C., and D.L.W.S. performed analyses. K.I.B. wrote the manuscript with contributions from all co-authors.
Supplementary Information is available in the online version of the paper.
Author Information Raw sequencing data have been deposited in the National Center for Biotechnology Information Sequence Read Archive under accession numbers SRP041177 for the ancient Peruvian samples and SRP041181 for the M. pinnipedii strains. The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper.
References
- 1.Hershberg R, et al. High functional diversity in Mycobacterium tuberculosis driven by genetic drift and human demography. PLoS Biol. 2008;6:e311. doi: 10.1371/journal.pbio.0060311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts CA, Buikstra JE. The Bioarchaeology of Tuberculosis A Global View on a Reemerging Disease. Univ. Press of Florida; 2003. pp. 187–213. [Google Scholar]
- 3.Comas I, et al. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nature Genet. 2013;45:1176–1182. doi: 10.1038/ng.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wirth T, et al. Origin, spread, and demography of the Mycobacterium tuberculosis complex. PLoS Pathogens. 2008;4:e1000160. doi: 10.1371/journal.ppat.1000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cockburn A. The Evolution and Eradication of Infectious Diseases. Johns Hopkins Press; 1963. [Google Scholar]
- 6.Brosch R, et al. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci USA. 2002;99:3684–3689. doi: 10.1073/pnas.052548299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gagneux S, Small PM. Global phylogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. Lancet Infect Dis. 2007;7:328–337. doi: 10.1016/S1473-3099(07)70108-1. [DOI] [PubMed] [Google Scholar]
- 8.Gagneux S, et al. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2006;103:2869–2873. doi: 10.1073/pnas.0511240103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Comas I, et al. Human T-cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nature Genet. 2010;42:498–503. doi: 10.1038/ng.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pepperell CS, et al. The role of selection in shaping diversity of natural M. tuberculosis populations. PLoS Pathogens. 2013;9:e1003543. doi: 10.1371/journal.ppat.1003543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shapiro B, Gilbert MPT. No proof that typhoid fever caused the Plague of Athens (a reply to Papagrigorakis et al) Int J Infect Dis. 2006;10:334–340. doi: 10.1016/j.ijid.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 12.Bos KI, et al. A draft genome of Yersinia pestis from victims of the Black Death. Nature. 2011;478:506–510. doi: 10.1038/nature10549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bouwman A, et al. Genotype of a historic strain of Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2012;109:18511–18516. doi: 10.1073/pnas.1209444109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan JZM, et al. Metagenomic analysis of tuberculosis in a mummy. N Engl J Med. 2013;369:289–290. doi: 10.1056/NEJMc1302295. [DOI] [PubMed] [Google Scholar]
- 15.Briggs AW, et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc Natl Acad Sci USA. 2007;104:14616–14621. doi: 10.1073/pnas.0704665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coscolla M, et al. Novel Mycobacterium tuberculosis complex from a wild chimpanzee. Emerg Infect Dis. 2013;19:969–976. doi: 10.3201/eid1906.121012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bastida R, et al. Tuberculosis in a wild subantarctic fur seal from Argentina. J Wildl Dis. 1999;35:796–798. doi: 10.7589/0090-3558-35.4.796. [DOI] [PubMed] [Google Scholar]
- 18.Comas I, et al. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nature Genet. 2012;18:106–110. doi: 10.1038/ng.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Botella H, et al. Metallobiology of host-pathogen interactions: an intoxicating new insight. Trends Microbiol. 2012;20:106–112. doi: 10.1016/j.tim.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 20.Bryant JM, et al. Inferring patient to patient transmission of Mycobacterium tuberculosis from whole genome sequencing data. BMC Infect Dis. 2013;13:110. doi: 10.1186/1471-2334-13-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elias SA, Short SK, Nelson CH, Birks HH. Life and times of the Bering land bridge. Nature. 1996;382:60–63. [Google Scholar]
- 22.Patrucco R, et al. Parasitological studies of coprolites of pre-Hispanic Peruvian populations. Curr Anthropol. 1983;24:393–394. [Google Scholar]
- 23.Bastida R, et al. La tuberculosis en grupos de cazadores recolectores de Patagonia y Tierra del Fuego: nuevas alternativas de contagio a través de la fauna silvestre. Rev Arg Antropol Biol. 2011;13:83–95. [Google Scholar]
- 24.Salo W, et al. Identification of Mycobacterium tuberculosis DNA in a pre-Columbian Peruvian mummy. Proc Natl Acad Sci USA. 1994;91:2091–2094. doi: 10.1073/pnas.91.6.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arriaza RT, et al. Pre-Columbian tuberculosis in northern Chile: molecular and skeletal evidence. Am J Phys Anthropol. 1995;98:37–45. doi: 10.1002/ajpa.1330980104. [DOI] [PubMed] [Google Scholar]
- 26.Anawalt PR. Cultural contacts between Ecuador, west Mexico, and the American Southwest: clothing similarities. Lat Am Antiq. 1992;3:114–129. [Google Scholar]
- 27.Herring DA, Sattenspiel L. Social contexts, syndemics, and infectious disease in northern Aboriginal populations. Am J Hum Biol. 2007;19:190–202. doi: 10.1002/ajhb.20618. [DOI] [PubMed] [Google Scholar]
- 28.Jurczynski K, et al. Pinniped tuberculosis in Malayan tapirs (Tapirus indicus) and its transmission to other terrestrial mammals. J Zoo Wildl Med. 2011;42:222–227. doi: 10.1638/2009-0207.1. [DOI] [PubMed] [Google Scholar]
- 29.Berg S, et al. The burden of mycobacterial disease in Ethiopian cattle: implications for public health. PLoS ONE. 2009;4:e5068. doi: 10.1371/journal.pone.0005068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cui Y, et al. Historical variations in mutation rate in an epidemic pathogen, Yersinia pestis. Proc Natl Acad Sci USA. 2012;110:577–582. doi: 10.1073/pnas.1205750110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1
Supplementary Table 5
Supplementary Table 6
Supplementary Table 8
Supplementary Table 9
Supplementary Tables 2-4, 7, 10-11