Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis (original) (raw)
. Author manuscript; available in PMC: 2015 Sep 23.
Published in final edited form as: Nat Cell Biol. 2010 Jan 17;12(2):153–163. doi: 10.1038/ncb2015
Abstract
Efficient tissue regeneration is dependent on the coordinated responses of multiple cell types. Here, we describe a new subpopulation of fibro/adipogenic progenitors (FAPs) resident in muscle tissue but arising from a distinct developmental lineage. Transplantation of purified FAPs results in the generation of ectopic white fat when delivered subcutaneously or intramuscularly in a model of fatty infiltration, but not in healthy muscle, suggesting that the environment controls their engraftment. These cells are quiescent in intact muscle but proliferate efficiently in response to damage. FAPs do not generate myofibres, but enhance the rate of differentiation of primary myogenic progenitors in co-cultivation experiments. In summary, FAPs expand upon damage to provide a transient source of pro-differentiation signals for proliferating myogenic progenitors.
Skeletal muscle consists of postmitotic multinucleated myofibres. Satellite cells, which are found juxtaposed to myofibres beneath the basal lamina, contribute to the growth and regeneration of postnatal muscle by fusing with damaged myofibres or by producing new myofibres1–7. Progression from quiescent satellite cell to mature postmitotic myofibre proceeds through discrete cellular intermediates, collectively called myogenic progenitors (MPs), which can be distinguished using molecular markers8.
In vivo, this stepwise process is coordinated by specific extracellular signals. After satellite cell activation, paracrine or autocrine engagement of Notch results in myoblast expansion, partly by direct inhibition of terminal differentiation9. Following expansion, pro-differentiation signals are required to induce myoblast maturation. The precise nature of these signals is the object of intense debate in the field, and several candidate molecules have been proposed for this role, including Wnt family members, IL-6 and insulin-like growth factors (IGFs)10–13, suggesting that multiple factors may be involved in promoting myogenic differentiation. Importantly, the sources of these factors within healing muscle tissue have yet to be determined.
In the context of healing muscle tissue, expanding myoblasts interact with inflammatory and stromal cells. It seems likely that these interactions are important in regulating their activity. The role of inflammatory cells in muscle regeneration has been explored recently14–16; however, nothing is known about the contributions of other tissue-resident populations. After chronic injury, muscle is often replaced by a mix of fibrous tissue and white adipocytes in a process termed fatty degeneration. This fibro/adipogenic infiltration compromises muscle function and alters the tissue environment, potentially limiting the success of regenerative approaches17. Thus, the cells responsible for fibro/adipogenic infiltration probably constitute an important part of the physiological context in which satellite cells function.
It has been suggested that fibrocytes and adipocytes develop from myogenic cells due to alternative lineage choice, which takes place when myogenic differentiation is impaired by cell-autonomous defects or by environmental changes associated with ageing and disease12,18–20. Here, we report the identification of a new population of bipotent FAPs present in skeletal muscle, but developmentally distinct from myogenic progenitors. We show that FAPs are abundant in healthy muscle, and that they rapidly enter the cell cycle in response to acute muscle damage, suggesting a role for this population during regeneration. Indeed, we found that FAPs are a source of signals inducing the differentiation of primary myoblasts. Our results suggest that FAPs proliferate in response to muscle injury to transiently establish an environment that enhances myogenic differentiation.
RESULTS
Purification of MPs with distinct developmental potential from skeletal muscle
To identify progenitor lineages from dissociated skeletal muscle, we used flow cytometry to detect CD34, which is expressed by satellite cells21, and Sca-1, which is expressed by progenitor populations of multiple tissues and whose expression in skeletal muscle progenitors is controversial22–24 (Fig. 1; Supplementary Information, Fig. S1). Although it has been suggested that haematopoietic (CD45+) and/or endothelial (CD31+) cells might be induced to acquire myogenic potential, their role in physiological muscle regeneration is unclear23,25. Therefore, we focused on the following CD45−CD31− lineage-negative (lin−) cell populations: Sca-1−CD34−, Sca-1−CD34+ and Sca-1+CD34+ (Fig. 1b). The proliferative and developmental potentials of cells sorted from undamaged muscle were analysed invitro under growth, osteogenic, chondrogenic myogenic and adipogenic conditions (Figs 1c–e and 2; Supplementary Informtion, Fig. S2 and Table S1) and invivo after transplantation (Fig. 3).
Figure 1.

Prospective isolation of progenitor populations from skeletal muscle. (a) Viable cells were identified based on forward/side scatter, Hoechst staining to exclude anuclear debris and low propidium iodide (PI) staining to exclude dead cells. Haematopoietic (CD45) and endothelial (CD31) cells were also excluded from analysis. (b) Expression of CD34 and Sca-1 in Hoechstmid PIlo CD45−CD31−(lin−) cells. Sca-1− CD34−, Sca-1−CD34+ (MP) and Sca-1+CD34+ cells were sorted and characterized. PE, phycoerythrin. (c) Lin−Sca-1−CD34− cells contain osteogenic and chondrogenic activity. Mineralized, multilayered nodules in cultures grown in osteogenic conditions for 10 weeks, and stained with alizarin red (left, scale bar, 100 μm). Alcian blue-positive cartilage in cryosections of cell pellets grown in chondrogenic conditions (right; scale bar, 25 μm). (d) Lin−Sca-1+CD34+ cells contain adipogenic progenitors. Sorted cells spontaneously gave rise to multilocular adipocytes (centre). Triglycerides were detected by oil red O staining in unilocular mature adipocytes after 30 days (right). Scale bars, 50 μm (left and centre) and 100 μm (right). (e) MP cells spontaneously differentiate in culture. MyHC-expressing myotubes were observed after 15 days in culture (centre and right). Scale bars, 50 μm (left) and 100 μm (centre and right). (f) Sca-1−CD34+ cells (MP; red), but not Sca-1+CD34+ adipogenic cells (blue), express α7 integrin. The specificity of the α7 staining was confirmed by the ‘fluorescence minus one’ (FMO) control.
Figure 2.
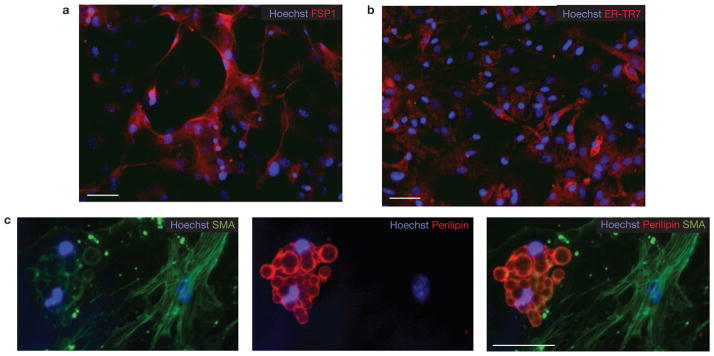
Lin−Sca-1+CD34+ cells generate both fibroblasts and adipocytes. (a–c) Lin−α7−Sca-1+ cultures were grown for three weeks in growth media and then immunostained using antibodies against fibroblast markers FSP-1 (a) and ER-TR7 (b). Scale bars, 50 μm (a) and 100 μm (b). (c) Single lin−Sca-1+α7− cells were deposited into individual wells of a 96-well plate directly from the sorter. After three weeks culture in growth medium, cells were immunostained for smooth muscle actin (SMA) and perilipin (a mature adipocyte marker). Scale bar, 50 μm.
Figure 3.

Developmental potential of sorted progenitor populations in vivo. (a) Lin−Sca-1−CD34+ (MP; red) and Lin−Sca-1+CD34+ (FAP; blue) cells were isolated from transgenic mice ubiquitously expressing GFP. (b) MP cells engraft in skeletal muscle. Freshly isolated MP cells (5 × 104) from GFP-expressing mice were injected into the tibialis anterior muscle of syngeneic hosts. Three weeks later, we observed GFP-expressing myofibres along the needle tract (n = 6). Laminin staining identified the basement membranes of myofibres. (c) Subcutaneous transplantation of FAP cells. Freshly isolated FAP cells (4 × 104) from GFP-expressing mice were injected subcutaneously into syngeneic GFP− recipients (n = 6). Three weeks later, confocal microscopy revealed GFP+, perilipin-expressing adipocytes located between the skeletal muscle (muscle) and dermis. GFP expression was detected by immunostaining. (d) High magnification image of transplanted GFP+ FAPs shows colocalization of GFP with the mature adipocyte marker perilipin. A maximum intensity projection image from a confocal optical stack is shown. (e) FAPs from a donor ubiquitously expressing membrane-bound human alkaline phosphatase (hAP) were transplanted in skeletal muscle that was previously injected with glycerol to induce adipocytic infiltration (n = 4). In these conditions FAPs efficiently engrafted and gave rise to differentiated adipocytes. All scale bars, 50 μm.
Cultures of double-sorted lin−Sca-1−CD34− cells failed to generate myosin heavy chain (MyHC)-positive structures under any of the conditions tested, but generated mineralized bone nodules and alcian blue-positive cartilage when cultured under osteogenic or chondrogenic conditions, respectively (Fig. 1c). In addition, they generated adipocytes when cultured in adipogenic medium containing PPARγ (peroxisome proliferator-activated receptor γ) activators (Supplementary Information, Fig. S2). Due to the rarity of clonogenic cells within this subset (1 in 757 sorted cells, Supplementary Information, Fig. S3), we excluded lin−Sca-1−CD34− cells from further analysis.
The lin−Sca-1+CD34+ cell subset spontaneously generated adipocytes under all culture conditions, but failed to give rise to mineralized nodules or cartilage (Fig. 1d; Supplementary information, Figs S2, S4 and Table S1). These cells were abundant, representing up to 15% of lin− cells in undamaged muscle, and upregulated the expression of mature adipocyte markers upon differentiation (Supplementary Information, Fig. S5). Adipogenic progenitors expressing identical surface markers were recently isolated from white adipose depots26–28, suggesting that similar cells may be present in multiple tissues.
Finally, lin−Sca-1−CD34+ cells gave rise to cultures containing both differentiated myofibres and proliferating mononuclear cells, as expected29. Despite being cultured under conditions that efficiently inhibit the differentiation of pre-plating-enriched primary myoblasts30, sorted lin−Sca-1−CD34+ cells spontaneously generated myofibres in vitro (Fig. 1e; Supplementary Information, Fig. S5).
CD34 surface expression is rapidly lost after MP activation, preventing the use of this marker to sort myoblasts from damaged tissue21. However, myogenic cells can be isolated with an alternative marker combination based on α7 integrin5,31. Indeed, we found that all myogenic activity in both undamaged and damaged muscle originates from lin−α7+Sca-1− cells (Supplementary Information, Fig. S6). Similarly, we found that lin−α7−Sca-1+ cells were as efficient as lin−Sca-1+CD34+ cells at generating adipocytes invitro (Supplementary Information, Fig. S6). Thus, we developed two strategies, using CD34 or α7 integrin, to prospectively identify identical myogenic progenitors and muscle-resident adipogenic cells, in both undamaged and damaged muscle (Fig. 1f).
Single lin−α7−Sca-1+ cells produce both fibroblasts and adipocytes in vitro
We used limiting dilution assays to measure the frequency of adipogenic and myogenic progenitors, defined as cells capable of clonal expansion and terminal differentiation into adipocytes or myofibres, in sorted cell populations. We found that 1 in 16 (95% confidence interval 12.0–21.4) double-sorted lin−α7+Sca-1− MPs were capable of forming colonies, and that over 95% of these colonies contained MyHC+ myofibres, suggesting that all clonogenic cells within this population were myogenic. Lin−α7−Sca-1+ cells formed colonies with similar efficiency (1 in 19; 95% confidence interval 14.2–22.3) but only 35% of these colonies contained adipocytes, whereas the remaining 65% contained cells that did not express myogenic or adipogenic markers, but were positive for the fibroblast markers ER-TR7 (ref. 12), FSP1 (ref. 32) and α-smooth muscle actin (SMA; Fig. 2; Supplementary Information, Fig. S7)33. The frequency of adipogenic colonies did not significantly change after exposure to PPARγ activators during the last week of culture.
When single lin−α7−Sca-1+ cells were directly sorted into individual wells, some of the resulting colonies contained both perilipin-expressing adipocytes and SMA-expressing fibroblasts, suggesting that this subset contains progenitors capable of producing both cell types (Fig. 2c). This notion is consistent with the fact that adipocytic infiltration and fibrosis are invariably linked during fatty degeneration of diseased skeletal muscle.
A recent report describes the development of fibrosis in skeletal muscle of adult mice in which PDGFRα signalling has been constitutively activated34, strongly suggesting that fibrocyte precursors express this receptor. Consistent with the notion that adipocyte infiltration and fibrosis are invariably linked in the fatty degeneration of diseased skeletal muscle, we found that lin−α7−Sca-1+ cells were uniformly positive for surface PDGFRα (Supplementary Information, Fig. S8) and that they represented over 85% of muscle-resident PDGFRα-expressing cells in undamaged muscle and up to 98% in damaged muscle (Supplementary Information, Fig. S9). Hence, we will hereafter refer to these cells as FAPs.
FAP engraftment is dictated by the environment
We tested the developmental potential of MPs and FAPs in vivo by transplantation (Fig. 3; Table 1). For each subset, 5 × 104 cells were purified from EGFP-expressing donors and immediately injected in the undamaged tibialis anterior muscle of syngeneic recipient animals (n = 6; Fig. 3a, b). As expected, MP cells engrafted by fusing to myofibres (Fig. 3b). In contrast, no donor cells were found in the muscles of animals injected with FAPs three weeks after transplant, despite the fact that subcutaneous delivery of cells from the same preparations yielded clusters of donor-derived adipocytes (n = 6; Fig. 3c, d). These data suggest that healthy muscle does not support FAP engraftment or adipocytic differentiation, consistent with the fact that fibro/adipocytic infiltration is not observed following healing of young healthy muscle, but only when myogenic regeneration fails due to ageing or disease. Next, we transplanted FAPs in tibialis anterior muscle injected with glycerol, a model of adipocytic infiltration35. In all transplanted animals (n = 4), we readily detected areas containing numerous donor-derived, perilipin-positive adipocytes (Fig. 3e). Overall, our data support the notion that FAP engraftment and differentiation into adipocytes is modulated by the environment of the recipient tissue.
Table 1.
FAP cells do not contribute to skeletal muscle in vivo
| Donor cells | Recipient TA | Animal ID | Number of hPLAP+ myofibres detected |
|---|---|---|---|
| 2 × 104 hPLAP+ MP | Undamaged | 1 | 77 |
| 2 | 62 | ||
| 3 | 55 | ||
| 4 | 35 | ||
| 2 × 104 hPLAP+ MP | NTX-damaged | 1 | 202 |
| 2 | 129 | ||
| 3 | 192 | ||
| 2 × 104 hPLAP+ FAP | Undamaged | 1 | 0 |
| 2 | 0 | ||
| 3 | 0 | ||
| 2 × 104 hPLAP+ FAP | NTX-damaged | 1 | 0 |
| 2 | 0 | ||
| 3 | 0 |
FAPs and MPs have distinct developmental potentials and do not arise from a common progenitor
Our data suggests that FAPs and MPs are distinct, lineage-specific progenitor populations present in undamaged, young adult muscle. Recently, adipogenic progenitors isolated from white fat depots were shown to fuse with differentiating C2C12 myoblasts in vitro26. To test whether muscle-derived FAPs can be recruited to the myogenic lineage in this context, we isolated them from GFP+ animals and cultured them together with freshly isolated GFP− MPs for ten days (Fig. 4a; Supplementary Information, Fig. S10). Immunostaining revealed that none of the GFP+ FAPs acquired expression of MyHC despite being in direct contact with terminally differentiated GFP− myotubes (Fig. 4a). In fact, a subset of these GFP+ cells had clearly initiated lipid accumulation, confirming their commitment to adipogenic differentiation.
Figure 4.

Skeletal muscle-derived FAPs and MPs have distinct developmental potentials and do not arise from a common progenitor. (a) FAP and MP co-cultivation confirms that their developmental potentials are non-overlapping. FAPs (a) or MPs (Supplementary Information, Fig. S6) sorted from transgenic GFP+ animals were co-cultivated for 14 days with equal numbers of GFP− MPs. Confocal microscopy revealed no contribution of GFP+ FAPs to MyHC+ cells, and no contribution of GFP− MPs to lipid-laden adipocytes (n = 6; a). Scale bar, 100 μm. (b) Schematic of the lineage tracing strategy. Myf5–Cre–R26R3–YFP mice were generated by crossing Myf5-Cre mice with a reporter strain carrying YFP integrated into the ROSA26 locus downstream of a floxed transcriptional stop sequence. CRE expression results in the heritable and irreversible expression of YFP under control of the ROSA26 locus. (c) MPs, but not FAPs, arise from a Myf-5-expressing precursor cell. Analysis of MPs and FAPs from Myf5–Cre–R26R3–YFP mice revealed YFP expression in a large proportion of MPs (top), whereas over 99% of FAPs were YFP-negative (bottom). (d, e) YFP expression in sorted MPs and FAPs from Myf5–Cre–R26R3–YFP mice after five days in culture. Over 85% of cultured MPs expressed YFP (d); in contrast, no YFP was detected in FAP cultures (e). Scale bars, 50 μm
To directly test whether FAPs can respond to damage by generating myofibres in vivo, we injected 2 × 104 double-sorted MPs or FAPs from mice ubiquitously expressing hPLAP (human placental alkaline phosphatase) into the tibialis anterior muscle of either undamaged or notexin-damaged recipients. After three weeks, we detected donor-derived myofibres in all animals receiving MPs, with an approximate fourfold increase in engraftment in the notexin-damaged group (Table 1). Conversely, no donor-derived myofibres were detected in any of the animals receiving FAPs (Table 1). Thus, FAPs do not undergo myogenesis or fuse to differentiating myogenic cells in vitro or invivo.
These findings suggest that FAPs originate from a separate developmental source rather than arising from myogenic cells in response to pathological alterations of the environment. To test this hypothesis directly, we bred knockin mice carrying Cre recombinase under the control of Myf5 regulatory elements with mice expressing YFP in a Cre-dependent fashion. In this approach, transient Myf5 expression during development leads to the heritable activation of YFP transcription from the ROSA locus, allowing the identification of the progeny of the original Myf5-expressing cell (Fig. 4b). The same strategy was recently used to show that brown fat and muscle share developmental origins36. Fluorescence-activated cell sorting (FACS) analysis of skeletal muscle preparations from these mice revealed that 60% of MPs expressed YFP (Fig. 4c). Although the frequency of YFP+ MPs in our experiments was lower than the previously reported 90% (ref. 4), it increased to > 85% after five days of culture (Fig. 4d). Conversely, Sca-1+CD34+ FAPs did not express YFP, and remained YFP− after culture (Fig. 4c, e). Thus, FAPs are unlikely to arise from myogenic cells.
FAPs are rapidly induced to proliferate upon muscle damage
In skeletal muscle, fibrosis and adipocyte infiltration in response to damage are only observed in ageing or disease. Yet, FAPs are abundant in young, healthy muscle. To investigate a potential physiological role of FAPs in the regeneration of healthy muscle, we analysed their response to notexin (NTX), a myotoxin which acts on terminally differentiated myofibres without damaging mononuclear progenitors37,38. Four days after NTX injection, all myogenic progenitors had downregulated CD34 (data not shown), confirming that they had been uniformly exposed to damage signals.
To determine whether FAPs respond to damage by entering the cell cycle, BrdU (5-bromodeoxyuridine) was administered either continuously or as a pulse during the 24 h immediately before collection, and animals were collected at time 0 (no damage) and at 24-h intervals thereafter. We observed significant proliferation of both FAPs and MPs (Fig. 5a, b), with more FAPs than MPs incorporating BrdU at early time points (Fig. 5b). Pulse delivery of BrdU confirmed that a higher fraction of FAPs than MPs proliferated during the first 72 h after damage (Fig. 5c). Thus, the ratio between the two cell types should change substantially during the first few days after damage induction. Indeed, between 48 and 72 h after damage, FAPs dramatically increased, nearly equalling MPs in number (Fig. 5d). Between four and five days after damage, the FAP:MP ratio rapidly returned to pre-damage levels in the absence of differences between the proliferation rates of either population, suggesting that FAPs may be actively removed from the tissue. Further work will be required to verify this hypothesis.
Figure 5.

FAPs proliferate in response to muscle damage. (a) Detection of BrdU incorporation in endogenous FAPs. Myofibre damage was induced by intramuscular injection of notexin into the tibialis anterior muscle. Starting 12 h before damage, BrdU was administered by intraperitoneal (IP) injection (100 mg kg−1 every 12 h) and in drinking water (0.8 mg ml−1 in 2% sucrose). After two days, BrdU incorporation in FAPs was detected by flow cytometry. Graphs show overlaying of BrdU+ events (blue dots) onto Sca-1/CD34 plots (right). The specificity of BrdU staining was confirmed by staining cells from non-BrdU-treated animals (left). (b) Comparison of FAP and MP proliferation kinetics after damage. BrdU was administered as in a. Tibialis anteriors were collected every 24 h after damage induction, up to 96 h. The data are presented as BrdU+ events (blue) overlaid onto Sca-1/α7 integrin plots. Percentages indicate the frequency of BrdU+ cells within the gate. PE, phycoerythrin. (c) Daily analysis of damage-induced progenitor cell proliferation. Damage was induced at day 0 in all animals. BrdU was administered by IP injection (100 mg kg−1, 24 h pulse) at 24 and 12 h before collection. Time after damage indicates the period over which BrdU was administered. Data were analysed by flow cytometry as in b. (d) Dynamic changes in the ratio of FAP to MP cells after muscle damage. Ratios were determined by comparing the number of events falling into FAP or MP gates using flow cytometric analysis. Day 0 represents undamaged animals. Error bars in c and d represent the mean ± s.e.m., n = 45 (c) or 18 (d). (e) Regression analysis of limiting dilution data from FAPs and MPs sorted from undamaged and damaged tibialis anteriors. The indicated numbers of cells were deposited in replicate wells of 96-well plates directly from the FACS sorter. After 21 days, wells were scored for the presence of colonies. Data from damaged tibialis anteriors are highlighted in red. Data were analysed based on the single-hit Poisson model for limiting dilution analysis. 95% confidence intervals are represented by dashed lines and are located in brackets in the legend. *P < 0.05, **P < 0.01 (Student’s _t_-test).
Limiting dilution analysis confirmed that MP proliferation is limited at early time points, as the frequencies of myogenic colony-forming cells in undamaged muscle and in muscle 48 h after damage were not significantly different. In contrast, FAP clonogenicity doubled after damage (Fig. 5e). We did not observe any MyHC-positive structures in FAP-seeded wells, nor any perilipin-positive adipocytes in MP-seeded wells, confirming that muscle damage does not alter FAP cell fate or surface phenotype.
To gain further insight in the role of FAPs in regeneration, we investigated their location within tissue. Previously published invitro analysis of individual myofibres conclusively shows that adipogenic cells are fibre-associated in undamaged muscle18. Furthermore, adipogenic progenitors have been localized to the vasculature in fat depots, suggesting that they may occupy a similar position in muscle27. We identified FAPs in situ based on their expression of PDGFRα as well as using CD31 and Sca-1 staining, as FACS analysis indicated that only FAPs and CD31+ cells are positive for Sca-1 in skeletal muscle (Fig. 1a, b)39. Confocal microscopy of damaged muscle indeed revealed abundant mononuclear FAPs associated with blood vessels and damaged myofibres (Fig. 6; Supplementary Information, Fig. S11). Thus, FAPs proliferate efficiently after damage and are found in close proximity to ailing myofibres, suggesting they may have a role in modulating myogenesis.
Figure 6.

FAPs are in close proximity to myofibres after damage. (a) CD31−Sca-1+ cells are found adjacent to both regenerating myofibres and blood vessels after NTX damage. Tissues were collected three days after damage induction, fixed, cryosectioned and then immunostained for CD31 and Sca-1. A single optical slice from a confocal _z_-stack is shown. Background autofluorescence from myofibres is shown in the blue channel. White arrowheads indicate Sca-1+CD31− cells. Scale bar, 25 μm. (b) PDGFRα-positive cells surround the myofibres three days after damage. Scale bar, 50 μm. (c) Detail of the boxed area in b. (d) Single confocal optical section clearly confirms that PDGFRα (white arrowheads) is expressed on the surface of myofibre (m)-associated cells. Scale bar, 25 μm.
FAPs provide an environment favouring myogenic differentiation
The activation of FAPs in healthy muscle after damage suggests a role for these cells in regeneration that is independent of the formation of fibrocytes or adipocytes. To determine whether FAPs may be a source of signals known to enhance myogenic differentiation, we investigated the expression of IGF-1 (ref. 11), IL-6 (ref. 13), Wnt1, Wnt3A and Wnt5A (ref. 12) from FAPs and MPs. We found that, compared with MPs, FAPs expressed similar or greater levels of all transcripts at all timepoints tested (data not shown). In particular, after NTX damage, IL-6 expression remained constant in MPs but increased nearly tenfold in FAPs, suggesting that FAPs represent an inducible source of IL-6 (Supplementary Information, Fig. S12). Thus, FAPs produce several factors known to favour myogenic differentiation.
To test whether FAPs regulate the differentiation of myogenic progenitors, we exposed MPs to GFP+ FAPs invitro and measured the effect of co-culture on the spontaneous generation of myofibres ten days later (Fig. 7a). The frequency of differentiated cells was calculated as the percentage of GFP+ myonuclei (nuclei present in terminally differentiated MyHC+GFP+ structures) relative to total GFP+ cell nuclei. As shown in Fig. 4a, FAPs do not fuse with differentiating primary myofibres, and therefore should not contribute directly to the number of myonuclei. GFP− MPs fuse with GFP+ myotubes as syncitial myofibres are generated, thus potentially biasing our analysis toward an overestimation of the number of GFP+ myonuclei in control co-cultures. Nevertheless, the frequency of GFP+ myonuclei was drastically increased in the presence of FAPs (Fig. 7a). Thus, FAPs increase the terminal differentiation of myogenic progenitors.
Figure 7.

FAPs enhance myogenic differentiation. (a) Immunohistological analysis of co-cultures revealed increased MP differentiation in the presence of FAPs. MPs (5,000) were isolated from GFP+ mice and co-cultivated with 5,000 GFP− FAPs or GFP− MPs. After ten days, cultures were fixed and stained for MyHC. Data are expressed as the ratio of nuclei in GFP+MyHC+ cells (myonuclei) to total nuclei in GFP+ cells. (b) FAPs induced MP differentiation in a dose-dependent manner. GFP+ MPs were co-cultivated with increasing numbers of GFP− FAPs and re-isolated by FACS after ten days. The expressions of markers of myogenic differentiation were analysed by qRT-PCR using Taqman probe and primer sets spanning exon–exon boundaries. TBP, TATA-binding protein. P values were determined using ANOVA. (c) FAPs increase MP commitment to terminal differentiation. GFP+ MPs were co-cultivated with equal numbers of GFP− FAPs or GFP− MPs and re-isolated by FACS after seven days. qRT-PCR analysis was performed as described in b. Gene expression is presented in arbitrary units. (d) MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) proliferation assay revealed little difference in proliferation between MP-only or MP-FAP co-cultures. Assays were performed on freshly sorted cell cultures containing either 5,000 MPs, or 2,500 MPs co-cultured with 2,500 FAPs for seven days. Data are expressed as absorbance at 570 nm (A570). (e) No difference in BrdU incorporation in MPs exposed to equal numbers of GFP+ MPs or GFP+ FAPs. GFP− MPs (5,000) were co-cultivated with either 5,000 GFP+ MPs or 5,000 GFP+ FAPs for a total of seven days. BrdU (10 μM) was applied during the last 24 h of co-culture. GFP+ cells were removed using FACS and immunostaining for BrdU was performed on the remaining GFP− MPs. Error bars in a–e represent the mean ± s.e.m., n = 3 (b, c and e), 4 (d) or 6 (a). *P < 0.05, ***P < 0.001 (Student’s _t_-test).
One striking observation from our invivo studies was that the FAP:MP ratio changed considerably, with FAP numbers peaking 2–3 days after damage, and rapidly falling thereafter. We reasoned that such a change in progenitor numbers might be important if the effects of FAP-derived signals on myogenic cells were dose dependent. To test this hypothesis, we repeated the co-cultivation experiments using 0.1:1, 0.5:1 and 1:1 ratios of GFP− FAP to GFP+ MP. We used flow cytometry to separate GFP+ MP cells from co-cultured FAP or control cells after ten days in culture and measured myogenic differentiation markers by quantitative reverse transcription-PCR (qRT-PCR). Increasing numbers of FAPs correlated with higher expression of late markers Myogenin, Mrf4 and MyHC, revealing a dose-dependent effect of FAPs on MP differentiation (Fig. 7b).
Next, we tested whether FAPs may influence the commitment of proliferating myoblasts to terminal differentiation. To this end, we analysed changes in expression of myogenic markers by MPs after seven days of co-culture, a time point that precedes the appearance of myofibres. We observed a reduction in the expression of early markers Pax3 and Pax7, and an increase in myoblast markers MyoD and Myogenin in the presence of FAPs (Fig. 7c). These results suggest that signals originating from FAPs increase the frequency of proliferating primary myoblasts that commit to terminal differentiation.
Potentially, an increase in progenitor differentiation could be secondary to an increase in culture density and thus to an effect on proliferation. However, no significant difference in proliferation was observed between test and control samples during the first five days of co-culture (Fig. 7d). Furthermore, despite a small but significant difference in overall co-culture proliferation detected at day 7, the frequency of MPs engaged in a cell cycle was comparable in the presence or absence of FAPs (Fig. 7d, e). Overall, these data suggest that the increase in myogenic differentiation caused by FAPs is not secondary to an effect on MP proliferation.
Together, these results suggest that FAPs generate a transient pro-differentiation niche for MPs. Our findings strongly support a key role of these cells in muscle healing in vivo, suggesting that successful regeneration requires the concerted action of multiple cell types, forming a complex biological system.
DISCUSSION
Here we describe a new muscle-resident progenitor with adipogenic potential, and propose that in skeletal muscle the chondro-osteogenic, adipogenic and myogenic potentials are readily attributable to distinct lineage-committed cell types.
The production of cartilage and bone within muscle only occurs in the context of rare diseases such as fibrodysplasia ossificans progressiva40; however, fibro/adipogenic infiltration is a common outcome of chronic muscle disease41. It is not surprising then, that quiescent FAPs represent a significant fraction of the mononuclear mesenchymal cells found in adult skeletal muscle.
Recent literature suggests that both adipogenic and fibrogenic cells originate from myogenic cells through alternative lineage choice dictated by a pathological environment12,18–20. A previous study used single myofibre cultures to demonstrate that myogenic and adipogenic cells are associated with the same fibre but stopped short of formally proving that fat and muscle can arise from the same cell18. Another group has recently reported that, in ageing mice, fibrogenic cells can arise from Pax7+ myogenic progenitors in response to altered Wnt signalling12. We cannot formally exclude that myogenic cells may become an additional source of fibroblasts when exposed to an ageing environment. However, the abundance and proliferative capacity of FAPs strongly suggests that they are the main source of fibroblasts and adipocytes in adult tissue. Thus, these cells are excellent candidates for progenitors involved in muscle repair, which restores tissue continuity but, unlike regeneration, does not restore function42. As such, their study may lead to therapeutic strategies to reduce scarring and fibrosis in chronic disease.
Adipogenic progenitors with a surface phenotype identical to that of FAPs were recently isolated from adipose depots26–28, but it is currently unclear whether FAPs represent the same cells. Unlike adipogenic progenitors, single FAPs efficiently generate both fibroblasts and adipocytes, but fail to generate other mesenchymal lineages. Furthermore, PPARγ2 expression was very low in freshly isolated FAPs from undamaged muscle (Supplementary Information, Fig. S5B). This seems to be consistent with data from a previous study, where a transgenic PPARγ reporter was used to identify adipogenic progenitors from adipose vasculature, but this study was unable to detect similar PPARγ+ cells in muscle. Thus, FAPs may represent an earlier bi-potent progenitor that precedes the initiation of PPARγ expression.
Specialized environments required for differentiation have been described in tissues characterized by high cellular turnover. Within the thymus, for example, progression along the T-cell developmental cascade is spatially regulated, requiring the migration of progenitors to specific locations43. Here, we report an example of a transient pro-differentiation niche. It is tempting to speculate that in tissues with very low homeostatic cell replacement, such as muscle, pro-differentiation signals may only be required for a short time after acute damage, and thus be provided by short-lived, specialized cells. Ascertaining the prevalence of such temporally regulated niches in low turnover tissues will require further investigation.
Our work describes a new population of FAPs resident in muscle tissue, and assigns to these cells a role in successful muscle regeneration. These findings introduce the concept of muscle healing as a system where the main components involved in regeneration and repair communicate with one another to coordinate their output. If the additional part played by inflammatory cells in directing these processes is considered, a picture of healing as a complex biological system emerges. We can only hope to understand such complex systems by studying them in their entirety.
METHODS
Animals
All mice were maintained in a pathogen-free facility, and all experiments were performed in accordance with the University of British Columbia Animal Care Committee regulations. Adult C57BL/6 mice (> 8 weeks) were used in this study. C57BL/6-CMV-β actin-EGFP transgenic mice were a gift from I. Weissman (Stanford University, CA, USA). Invivo lineage tracing was performed in Myf5–Cre–R26R3–YFP mice, generated by breeding heterozygous Myf5–Cre (P. Soriano, Department of Developmental and Regenerative Biology, Mount Sinai School of Medicine, NY, USA) with ROSA26–YFP44 reporter mice. Muscle damage was induced by intramuscular injection of 0.15 μg notexin snake venom (Latoxan), into the tibialis anterior muscle.
Tissue preparation
Skeletal muscle from both hindlimbs was carefully dissected and then gently torn with tissue forceps until homogeneous. Collagenase type 2 (Sigma; 2ml of 2.5 U ml−1), in 10 mM CaCl2, was added to every two hindlimbs, and the preparation was placed at 37 C for 30 min. After washing, a second enzymatic digestion was performed with Collagenase D (Roche Biochemicals; 1.5 U ml−1) and Dispase II (Roche Biochemicals; 2.4 U ml−1), in a total volume of 1 ml per mouse, at 37 C for 60 min. Preparations were passed through a 40-μm cell strainer (Becton Dickenson), and washed. Resulting single cells were collected by centrifugation at 400_g_ for 5 min.
Flow cytometry/FACS
Cell preparations were incubated with primary antibodies for 30 min at 4 °C in supplemented PBS containing 2 mM EDTA and 2% FBS at ~3 × 107 cells per ml. We used the following monoclonal primary antibodies: anti-CD31 (clones MEC13.3, Becton Dickenson, and 390, Cedarlane Laboratories), anti-CD34 (clone RAM34, eBioscience), anti-CD45 (clone 30-F11, Becton Dickenson), anti-CD45.1 (clone A20, Becton Dickenson), anti-CD45.2 (clone 104, eBiosciences), anti-Sca-1 (clone D7, eBiosciences) and anti-α7 integrin (produced in-house). Typical antibody dilutions used were: anti-CD31, 1:100–400; anti-CD34, 1:50–200; anti-CD45, 1:200–400; anti-CD45.1, 1:200–400; anti-CD45.2, 1:200–400; anti-Sca-1, 1:2,000–5,000; anti-α7 integrin, 1:100–400. For all antibodies we performed fluorescence minus one controls by staining with appropriate isotype antibodies. Where necessary, biotinylated primary antibodies were detected using streptavidin coupled to phycoerythrin, allophycocyanin or FITC (Caltag). To assess viability, cells were stained with propidium iodide (1 μg ml−1) and Hoechst 33342 (2.5 μg ml−1) and resuspended at ~1 × 107 cellsml −1 immediately before sorting or analysis.
Analysis was performed on LSRII (Becton Dickenson) equipped with three lasers. Data were collected using FacsDIVA software. Sorts were performed on a FACS Vantage SE (Becton Dickenson) or FACS Aria (Becton Dickenson), both equipped with three lasers, using a 100-μm nozzle at 18 psi to minimize the effects of pressure on the cells. Sorting gates were strictly defined based on isotype control (fluorescence minus one) stains. Biexponential analysis was performed using FlowJo 8.7 (Treestar) software.
Cell culture
Cells were grown in high-glucose Dulbecco’s modified eagle medium (DMEM), supplemented with 2.5 ng ml−1 bFGF (Invitrogen) 20% FBS and 10% heat-inactivated horse serum. This medium is referred to in text as ‘growth medium’. Cells were seeded in tissue-culture-treated plates coated with Matrigel (Becton Dickenson) or Type 1 collagen (Sigma). After sorting, cells were allowed to adhere for three days, after which half the medium was changed. Media was changed every 2–4 days thereafter. For mesenchymal differentiation, we used reported conditions. Briefly, we cultured cells in DMEM with 20% FBS under the following conditions: for osteogenic differentiation, we supplemented media with 10 nM dexamethasone, 5 mM β-glycerophosphate and 50 μg ml−1 ascorbic acid; for adipogenic differentiation, we supplemented media with 0.25 μM dexamethasone and 0.5 mM isobutylmethylxanthine, 1 μg ml−1 insulin and 5 μM troglitazone; for chondrogenic differentiation we pelleted cells by centrifugation at 400_g_ for 5 min, and grew them in media supplemented with 1 ng ml−1 TGFβ1 and 50 μg ml−1 ascorbic acid. For myogenic differentiation, we used DMEM medium supplemented with 5% horse serum.
Limiting dilution analysis
From each test population, 1–100 cells were sorted into individual wells of a Matrigel-coated 96-well plate directly from the sorter. Cells were grown as described in the text. After three weeks, cultures were fixed and stained for MyHC and nuclei. Wells were scored for the presence of colonies (> 8 cells), cells undergoing terminal myogenic differentiation (MyHC-positive) and lipid laden adipocytes. A minimum of 30 replicate wells was generated for each cell dose. Limiting dilution analysis calculations were based on the single hit Poisson model (see Statistics).
Gene expression analysis
RNA isolation was performed using RNeasy mini kits (Qiagen), and RNA quantification was performed using a ND1000 spectrophotometer (Nanodrop). Reverse transcription was performed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Gene expression analysis was performed using Taqman Gene Expression Assays (Applied Biosystems), on a 7900HT Real Time PCR system (Applied Biosystems). Sequence information for the primers contained in the Taqman assays are not in the public domain, but ordering information is provided in Supplementary Information, Table S3. Data were acquired and analysed using SDS 2.0 and SDS RQ Manager software (Applied Biosystems).
Transplantation
Host mice (wild type) were anesthetized using isoflurane. Donor cells were isolated from transgenic mice ubiquitously expressing GFP+. Cells were sorted into cold DMEM and collected by centrifugation at 450_g_ for 5 min. For subcutaneous transplantation, cells were resuspended in 25 μl Matrigel and loaded into an ice-cold needle and syringe immediately before injection. Cells were injected into the subscapular region, with control cells injected on the contralateral side. For intramuscular transplantation, sorted cells were resuspended in 20 μl sterile PBS and injected into the tibialis anterior. Tissues were collected after three weeks.
Histology and imaging
Before tissue collection, animals were perfused transcardially with 20 ml PBS/2 mM EDTA, followed by 20 ml 4% paraformaldehyde (PFA). Tissues were processed for cryosectioning or paraffin-embedding using standard methods. Immunostaining was performed using monoclonal antibodies against perilipin (Sigma), α smooth muscle actin (Sigma), ER-TR7 (Novus), FSP1 (Novus) myosin heavy chain (in-house) or BrdU (Becton Dickenson). Briefly, samples were permeabilized in 0.3% Triton X-100 (Sigma) in PBS, and blocked for 2 h at room temperature in PBS containing 25% normal goat serum (NGS), 0.3% Triton X-100, 3% bovine serum albumin (BSA) and 0.1% NaN3. Cells were stained overnight at 4 °C using antibody diluted in 10% NGS, 0.3% Triton X-100, 3% BSA and 0.1% NaN3. Primary antibody was detected using goat anti-mouse IgG antibodies conjugated to Alexa 568 or 488 (Molecular Probes), or using the anti-Mouse IgG HRP detection kit (Becton Dickenson).
Cells were visualized using a microscope (Axiovert 200 for inverted microscopy, Axioplan2 for conventional microscopy; Carl Zeiss Microimaging) and images were acquired using a charge-coupled device camera (Retiga Exi, Axiovert 200 or Retiga Ex, Axioplan2; qImaging) and OpenLab4 software (Improvision). To confirm the specificity of the GFP signal, we compared the signal with non-specific autofluorescent background in all other channels45. Confocal microscopy was performed using a Nikon C1 laser scanning confocal microscope equipped with lasers at 488 nm, 568 nm and 633 nm. Images were captured using the least exposure time possible, and manipulation of brightness and contrast, colouring adjustments and assembly into figures were performed using ImageJ, OpenLab4 (Improvision), Illustrator CS3 (Adobe) and Photoshop CS3 (Adobe).
BrdU-labelling studies
For in vivo studies, BrdU was administered in drinking water (0.8 g l−1 in 2% sucrose) and by intraperitoneal injection (100 mg kg−1). In cell culture experiments, 10 μM BrdU was added to culture medium. BrdU treatment regimens are described in the text. For flow cytometric analysis, cells were stained for surface markers as indicated. Samples were fixed in 2% PFA for 20 min, and membranes were permeablized in 0.2% saponin for 10 min. Cells were treated with 30 μg DNAse I (Sigma) for 1 h at 37 °C, after which a 1:25 dilution of anti-BrdU (clone PRB-1, Invitrogen) was applied to the samples for 30 min at room temperature.
MTT assay
Assays were performed in 96-well cell cultures. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) solution (50 μl of 5 mg ml−1 dissolved in PBS) was added to each well and samples were incubated at 37 °C for 4 h. The solution was removed, and the purple formazan salt product resulting from the reduction of the yellow MTT was solublized in 100 μl DMSO and quantified spectrophotometrically at 570 nm (SpectraMax 190, Molecular Devices).
Statistics
Preliminary analysis and data collation was performed using Microsoft Excel. Statistical tests, including Student’s _t_-test, ANOVA and regression analyses were performed using Prism 4 (GraphPad Software). Analysis of limiting dilution data were performed using a web application made available by the Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia (http://bioinf.wehi.edu.au/software/limdil/)46. This software tests departures from the single-hit Poisson model using a generalized linear model. Error bars in all figures, including Supplementary Information, represent the mean ± s.e.m., with the exception of Fig. 7a, which represent the mean ± s.d. Raw data points for all experiments presented as averages in the main figures are available in Supplementary Information, Table S2.
Supplementary Material
Acknowledgments
We thank J. Qiao, C. Chang, J. M. Joe, the BRC Animal Facility, the BRC Core Staff and A. Johnson and J. Duenas of the UBC FACS Facility for their expert technical assistance. We are also grateful to A. Uezumi for advice on modelling fatty degeneration in murine skeletal muscle. This research was supported by grants from The Foundation for Cell therapy (Jesse’s Journey) and The Canadian Institute for Health Research (CIHR MOP-97856; CSB-94219; MUS-94019) to F.M.V.R. who holds a Canada Research Chair in Regenerative medicine and is a fellow of the Michael Smith Foundation for Health Research. A.W.B.J. is supported by a fellowship from CIHR, A.N. from the Heart and Stroke Foundation of Canada and L.S. from NSERC (PGSD2-362406-2008).
Footnotes
AUTHOR CONTRIBUTIONS
A.W.B.J. designed and performed experiments, analysed data, interpreted results and wrote the manuscript. L.Y. and A.N. designed and performed experiments and analysed data. L.S. and J.W. performed experiments. F.L. and M.A.R. provided new reagents and performed experiments. F.M.V.R. designed and performed experiments, interpreted results and wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Charge SB, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol Rev. 2004;84:209–238. doi: 10.1152/physrev.00019.2003. [DOI] [PubMed] [Google Scholar]
- 2.Dhawan J, Rando TA. Stem cells in postnatal myogenesis: molecular mechanisms of satellite cell quiescence, activation and replenishment. Trends Cell Biol. 2005;15:666–673. doi: 10.1016/j.tcb.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 3.Collins CA, et al. Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell. 2005;122:289–301. doi: 10.1016/j.cell.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 4.Kuang S, Kuroda K, Le Grand F, Rudnicki MA. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell. 2007;129:999–1010. doi: 10.1016/j.cell.2007.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sacco A, Doyonnas R, Kraft P, Vitorovic S, Blau HM. Self-renewal and expansion of single transplanted muscle stem cells. Nature. 2008;456:502–506. doi: 10.1038/nature07384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mauro A. Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol. 1961;9:493–495. doi: 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morgan JE, Partridge TA. Muscle satellite cells. Int J Biochem Cell Biol. 2003;35:1151–1156. doi: 10.1016/s1357-2725(03)00042-6. [DOI] [PubMed] [Google Scholar]
- 8.Buckingham M. Myogenic progenitor cells and skeletal myogenesis in vertebrates. Curr Opin Genet Dev. 2006;16:525–532. doi: 10.1016/j.gde.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 9.Conboy IM, Rando TA. The regulation of Notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev Cell. 2002;3:397–409. doi: 10.1016/s1534-5807(02)00254-x. [DOI] [PubMed] [Google Scholar]
- 10.Otto A, et al. Canonical Wnt signalling induces satellite-cell proliferation during adult skeletal muscle regeneration. J Cell Sci. 2008;121:2939–2950. doi: 10.1242/jcs.026534. [DOI] [PubMed] [Google Scholar]
- 11.Bodine SC, et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nature Cell Biol. 2001;3:1014–1019. doi: 10.1038/ncb1101-1014. [DOI] [PubMed] [Google Scholar]
- 12.Brack AS, et al. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science. 2007;317:807–810. doi: 10.1126/science.1144090. [DOI] [PubMed] [Google Scholar]
- 13.Serrano AL, Baeza-Raja B, Perdiguero E, Jardi M, Munoz-Canoves P. Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metab. 2008;7:33–44. doi: 10.1016/j.cmet.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 14.Arnold L, et al. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med. 2007;204:1057–1069. doi: 10.1084/jem.20070075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sonnet C, et al. Human macrophages rescue myoblasts and myotubes from apoptosis through a set of adhesion molecular systems. J Cell Sci. 2006;119:2497–2507. doi: 10.1242/jcs.02988. [DOI] [PubMed] [Google Scholar]
- 16.Contreras-Shannon V, et al. Fat accumulation with altered inflammation and regeneration in skeletal muscle of CCR2−/− mice following ischemic injury. Am J Physiol Cell Physiol. 2007;292:C953–967. doi: 10.1152/ajpcell.00154.2006. [DOI] [PubMed] [Google Scholar]
- 17.Lipton B. Skeletal muscle regneration in muscular dystrophy. In: Mauro A, editor. Muscle Regeneration. Raven Press; 1979. pp. 31–40. [Google Scholar]
- 18.Shefer G, Wleklinski-Lee M, Yablonka-Reuveni Z. Skeletal muscle satellite cells can spontaneously enter an alternative mesenchymal pathway. J Cell Sci. 2004;117:5393–5404. doi: 10.1242/jcs.01419. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, et al. Transforming growth factor-β1 induces the differentiation of myogenic cells into fibrotic cells in injured skeletal muscle: a key event in muscle fibrogenesis. Am J Pathol. 2004;164:1007–1019. doi: 10.1016/s0002-9440(10)63188-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Huard J. Differentiation of muscle-derived cells into myofibroblasts in injured skeletal muscle. Am J Pathol. 2002;161:895–907. doi: 10.1016/S0002-9440(10)64250-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beauchamp JR, et al. Expression of CD34 and Myf5 defines the majority of quiescent adult skeletal muscle satellite cells. J Cell Biol. 2000;151:1221–1234. doi: 10.1083/jcb.151.6.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mitchell PO, et al. Sca-1 negatively regulates proliferation and differentiation of muscle cells. Dev Biol. 2005;283:240–252. doi: 10.1016/j.ydbio.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 23.Polesskaya A, Seale P, Rudnicki MA. Wnt signaling induces the myogenic specification of resident CD45+ adult stem cells during muscle regeneration. Cell. 2003;113:841–852. doi: 10.1016/s0092-8674(03)00437-9. [DOI] [PubMed] [Google Scholar]
- 24.Sherwood RI, et al. Isolation of adult mouse myogenic progenitors: functional heterogeneity of cells within and engrafting skeletal muscle. Cell. 2004;119:543–554. doi: 10.1016/j.cell.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 25.De Angelis L, et al. Skeletal myogenic progenitors originating from embryonic dorsal aorta coexpress endothelial and myogenic markers and contribute to postnatal muscle growth and regeneration. J Cell Biol. 1999;147:869–878. doi: 10.1083/jcb.147.4.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodeheffer MS, Birsoy K, Friedman JM. Identification of white adipocyte progenitor cells in vivo. Cell. 2008;135:240–249. doi: 10.1016/j.cell.2008.09.036. [DOI] [PubMed] [Google Scholar]
- 27.Tang W, et al. White fat progenitor cells reside in the adipose vasculature. Science. 2008;322:583–586. doi: 10.1126/science.1156232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joe AW, Yi L, Even Y, Vogl AW, Rossi FM. Depot-specific differences in adipogenic progenitor abundance and proliferative response to high-fat diet. Stem Cells. 2009;27:2563–2570. doi: 10.1002/stem.190. [DOI] [PubMed] [Google Scholar]
- 29.Montarras D, et al. Direct isolation of satellite cells for skeletal muscle regeneration. Science. 2005;309:2064–2067. doi: 10.1126/science.1114758. [DOI] [PubMed] [Google Scholar]
- 30.Rando TA, Blau HM. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J Cell Biol. 1994;125:1275–1287. doi: 10.1083/jcb.125.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blanco-Bose WE, Yao CC, Kramer RH, Blau HM. Purification of mouse primary myoblasts based on α 7 integrin expression. Exp Cell Res. 2001;265:212–220. doi: 10.1006/excr.2001.5191. [DOI] [PubMed] [Google Scholar]
- 32.Strutz F, et al. Identification and characterization of a fibroblast marker: FSP1. J Cell Biol. 1995;130:393–405. doi: 10.1083/jcb.130.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nature Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 34.Olson LE, Soriano P. Increased PDGFRα activation disrupts connective tissue development and drives systemic fibrosis. Dev Cell. 2009;16:303–313. doi: 10.1016/j.devcel.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arsic N, et al. Vascular endothelial growth factor stimulates skeletal muscle regeneration in vivo. Mol Ther. 2004;10:844–854. doi: 10.1016/j.ymthe.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 36.Seale P, et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454:961–967. doi: 10.1038/nature07182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harris JB, Vater R, Wilson M, Cullen MJ. Muscle fibre breakdown in venom-induced muscle degeneration. J Anat. 2003;202:363–372. doi: 10.1046/j.1469-7580.2003.00171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harris JB. Myotoxic phospholipases A2 and the regeneration of skeletal muscles. Toxicon. 2003;42:933–945. doi: 10.1016/j.toxicon.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 39.Kafadar KA, et al. Sca-1 expression is required for efficient remodeling of the extracellular matrix during skeletal muscle regeneration. Dev Biol. 2009;326:47–59. doi: 10.1016/j.ydbio.2008.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shore EM, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nature Genet. 2006;38:525–527. doi: 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- 41.Wallace GQ, McNally EM. Mechanisms of muscle degeneration, regeneration, and repair in the muscular dystrophies. Annu Rev Physiol. 2008;71:37–57. doi: 10.1146/annurev.physiol.010908.163216. [DOI] [PubMed] [Google Scholar]
- 42.Goss RJ. Regeneration versus repair. In: Cohen IK, Diegelmann RF, Lindblad WJ, editors. Wound Healing: Biochemical and Clinical Aspects. W. B. Saunders Co; 1992. pp. 20–39. [Google Scholar]
- 43.Ladi E, Yin X, Chtanova T, Robey EA. Thymic microenvironments for T cell differentiation and selection. Nature Immunol. 2006;7:338–343. doi: 10.1038/ni1323. [DOI] [PubMed] [Google Scholar]
- 44.Srinivas S, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brazelton TR, Blau HM. Optimizing techniques for tracking transplanted stem cells in vivo. Stem Cells. 2005;23:1251–1265. doi: 10.1634/stemcells.2005-0149. [DOI] [PubMed] [Google Scholar]
- 46.Shackleton M, et al. Generation of a functional mammary gland from a single stem cell. Nature. 2006;439:84–88. doi: 10.1038/nature04372. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.