The Clinical Phenotypes of the Juvenile Idiopathic Inflammatory Myopathies (original) (raw)
Abstract
The juvenile idiopathic inflammatory myopathies (JIIM) are systemic autoimmune diseases characterized by skeletal muscle weakness, characteristic rashes, and other systemic features. Although juvenile dermatomyositis (JDM), the most common form of JIIM, has been well studied, the other major clinical subgroups of JIIM, including juvenile polymyositis (JPM) and juvenile myositis overlapping with another autoimmune or connective tissue disease (JCTM), have not been well characterized, and their similarity to the adult clinical subgroups is unknown. We enrolled 436 patients with JIIM, including 354 classified as JDM, 33 as JPM, and 49 as JCTM, in a nationwide registry study. The aim of the study was to compare demographics; clinical features; laboratory measures, including myositis autoantibodies; and outcomes among these clinical subgroups, as well as with published data on adult patients with idiopathic inflammatory myopathies (IIM) enrolled in a separate natural history study.
We used random forest classification and logistic regression modeling to compare clinical subgroups, following univariate analysis. JDM was characterized by typical rashes, including Gottron papules, heliotrope rash, malar rash, periungual capillary changes, and other photosensitive and vasculopathic skin rashes. JPM was characterized by more severe weakness, higher creatine kinase levels, falling episodes, and more frequent cardiac disease. JCTM had more frequent interstitial lung disease, Raynaud phenomenon, arthralgia, and malar rash. Differences in autoantibody frequency were also evident, with anti-p155/140, anti-MJ, and anti-Mi-2 seen more frequently in patients with JDM, anti-signal recognition particle and anti-Jo-1 in JPM, and anti-U1-RNP, PM-Scl, and other myositis-associated autoantibodies more commonly present in JCTM. Mortality was highest in patients with JCTM, whereas hospitalizations and wheelchair use were highest in JPM patients. Several demographic and clinical features were shared between juvenile and adult IIM subgroups. However, JDM and JPM patients had a lower frequency of interstitial lung disease, Raynaud phenomenon, “mechanic’s hands” and carpal tunnel syndrome, and lower mortality than their adult counterparts. We conclude that juvenile myositis is a heterogeneous group of illnesses with distinct clinical subgroups, defined by varying clinical and demographic characteristics, laboratory features, and outcomes.
INTRODUCTION
The juvenile idiopathic inflammatory myopathies (JIIM) are acquired inflammatory disorders of skeletal muscle of unknown etiology. They are systemic autoimmune diseases characterized by symmetric proximal weakness, rashes, and other systemic features.9,38 The JIIM, like the adult idiopathic inflammatory myopathies (IIM) and other autoimmune disorders, appear to be composed of a number of clinical and serologic phenotypes, each of which defines more homogeneous subsets of patients in terms of demographic and clinical features, the presence of certain associated autoantibodies, outcomes, and responses to therapy.37 Such homogeneous phenotypes may share unique combinations of environmental and genetic risk factors that result in a discrete disorder.37 (For more about classifying the adult IIM, see the article in this issue by Fernandez et al.9a)
Of the various clinical forms of JIIM, juvenile dermatomyositis (JDM) is the most frequent and best characterized.13,25,26,29,42 JDM is defined by the presence of Gottron papules, raised red patches overlying the interphalangeal joints or other joint extensor surfaces, or the heliotrope rash, a red or purple discoloration over the eyelids.5,20 Relatively little has been described about the distinct features of the other major clinical subgroups of JIIM, in part due to inadequate numbers of patients, including juvenile polymyositis (JPM), which is characterized by weakness and muscle inflammation in the absence of the distinctive rashes of JDM, and overlap myositis—that is, myositis in which patients meet criteria for either JDM or JPM, as well as for another connective tissue disease.19,32,42,57 Juvenile and adult IIM are often considered to be separate disorders, but the degree of similarity has not been completely assessed.34,38
We conducted the current study to develop classification methods, using demographic, clinical, and laboratory features, to better define the major clinical subgroup phenotypes of JIIM. We also compared the JIIM and adult subgroup phenotypes to see if these illnesses differed clinically, as they have important pathophysiologic differences.39
PATIENTS AND METHODS
Patients
Four hundred thirty-six patients with probable or definite JDM or JPM5 were enrolled in the National Institutes of Health Clinical Center or the Food and Drug Administration’s investigational review board-approved natural history protocols from March 1989 through August 2010; patients were diagnosed with myositis between May 1957 and March 2010.4,23,37,56 Patients were recruited for enrollment through myositis patient support groups, through an advertisement in medical journals, and by writing to pediatricians and pediatric and adult rheumatologists, neurologists, and dermatologists. Patients provided a blood sample for autoantibody testing after written consent/assent was obtained according to the Declaration of Helsinki. The treating physician completed a questionnaire that included clinical, demographic, and laboratory data, and outcomes. A pediatric rheumatologist (LGR or GM) reviewed available medical records for 69% of the patients to confirm the questionnaire material and complete missing and follow-up data. Twenty-four additional patients referred to the study were excluded, as they did not have an idiopathic inflammatory myopathy. They included 10 patients with a dystrophy, 6 with viral myositis (including 1 associated with human immunodeficiency virus [HIV] infection), 4 with undifferentiated connective tissue disease, and 4 with an undefined noninflammatory myopathy.
Sixty percent of patients met the Bohan and Peter criteria5 for definite dermatomyositis (DM) or polymyositis (PM), and 40% for probable DM or PM, with a careful evaluation to exclude other disorders; all were diagnosed before 18 years of age. JDM was diagnosed in patients with myositis if they had either heliotrope rash or Gottron papules as well as 2 other criteria from Bohan and Peter;5,27 354 of the 436 patients were classified as having JDM. All 33 patients classified with JPM and 8 JPM patients classified with juvenile myositis overlapping with another autoimmune or connective tissue disease (JCTM) had a muscle biopsy consistent with an inflammatory myopathy; the majority of biopsies were reviewed by 2 authors (LGR and FWM), often in consultation with pathologists from the Division of Neuropathology of the Armed Forces Institute of Pathology, Washington, DC. Patients were classified as having JCTM if they met criteria for DM or PM as well as another autoimmune disease. Of 49 JCTM patients, 41 had JDM and 8 had JPM. The overlapping conditions included systemic lupus erythematosus in 29% of the JCTM patients; juvenile idiopathic arthritis in 22%; systemic sclerosis in 16%; juvenile localized scleroderma (including linear scleroderma, morphea, or eosinophilic fasciitis) in 16%; Sjögren syndrome and insulin-dependent diabetes in 6% each; and psoriasis, ulcerative colitis, and discoid lupus in 2% each. Two patients met criteria for more than 1 associated autoimmune disease. Twenty-six additional patients were excluded: 9 had possible JDM, 3 possible JPM, 6 possible JCTM, 3 had amyopathic or hypomyopathic JDM, and 1 each had orbital, focal, eosinophilic, granulomatous, or graft-versus-host myositis.
Progression of the first symptoms of myositis to full disease presentation was characterized as acute if it occurred in less than 1 month, subacute if it occurred in 1–3 months, slow if it occurred over 3–6 months, and insidious if the time to full illness presentation was more than 6 months. Severity of illness at onset, up to the time of diagnosis, was graded on a 4-point Likert scale as determined by the enrolling physician, and graded from mild to extremely severe disease activity. Muscle enzyme values were adjusted to a common upper limit of normal, with the highest value recorded.
Disease course was classified as monocyclic if the patient achieved remission without evidence of active disease, based on clinical features and laboratory testing, within 2 years of diagnosis; as polycyclic if the patient had recurrence of active disease after a definite remission; as chronic continuous if disease activity persisted more than 2 years; and as undefined if less than 2 years of follow-up after diagnosis was available.18 Mortality was identified through report of death by a relative, but for the majority of cases, the Social Security Death Index was used to determine a patient’s status, and this was last examined in March 2011. Patients for whom the social security number was not available were searched by name and date of birth. Three hundred eight patients had follow-up information about final outcomes obtained ≥3 months after the date of enrollment at a median of 1.6 years later.
The childhood cohort was compared to an adult IIM study population, which included 70 adult DM, 48 adult PM, and 28 adult connective tissue myopathy (CTM) patients with probable or definite IIM5 enrolled at the National Institutes of Health Clinical Center in Bethesda, MD, between 1983 and 1990; adult IIM patients were diagnosed between January 1965 and January 1990.21 The pediatric physician questionnaire was expanded from the adult IIM version.
Methods
Patient sera were tested for myositis autoantibodies by validated methods, including protein and RNA immunoprecipitation (IP) using radiolabeled HeLa or K562 cell extracts and double immunodiffusion.1,48,51 For anti-p155/140 and anti-MJ autoantibodies, serum samples were screened by IP, and results were confirmed by IP-blotting.51 Sera were considered positive if they blotted the antigen in immunoprecipitates prepared using reference serum (direct) or if reference serum blotted the antigen in immunoprecipitates prepared using patient serum (reverse). Since some IP-positive sera do not react by immunoblotting, reverse IP-blotting was used for most sera.51
Statistical analysis was performed using SAS for Windows, version 9.1.3 (SAS Institute, Cary, NC). Median and interquartile ranges (IQRs) were used to describe continuous data, and frequency distributions were used to assess binomial, ordinal, and categorical variables. For continuous variables, the distribution patterns, such as median and dispersion, were assessed using SAS. To test for significance, the Kruskal-Wallis test was used to compare JDM versus JPM versus JCTM, and the Wilcoxon rank sum test was used to compare JPM versus JDM, JCTM versus JDM, and JCTM versus JPM. The chi-square test and, for small sample sizes, the Fisher exact test were conducted to determine significant differences between variables of interest and clinical subgroups. Missing data were noted in the tables only when >5% of cases were missing. We compared JIIM and adult IIM clinical groups to a National Institutes of Health-based adult IIM study population.21 For univariate analysis, p < 0.05 was considered significant to carry into multivariable analysis, as described below. However, variables with >10% missing data were not further considered, and for more complete variables, cases with missing data were also deleted in order to perform the multivariable analysis.
We used a 2-staged approach to further identify variables important in the classification of JIIM clinical groups. First, we used random forests to identify and validate the predictors from among a large group of candidate variables, and second, we used logistic regression to determine the strength of association of these predictors with clinical subgroups. Based on the results of the univariate analysis, variables that differed significantly between clinical groups were studied using the learning machine Random Forest23 (further details are available at http://www.stat.berkeley.edu/users/\~breiman/RandomForests/cc\_home.htm). For code see the R statistical package version 2.8.1 (The R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0, http://www.r-project.org).[23](#R23) Random forest analysis is a nonlinear, nonparametric algorithm that can be used for prediction and classification. It generates estimates of ranking for predictive importance. Random forest analysis was used to determine which characteristics are potentially informative within each clinical subgroup.
Variables that were clinically significant based on previous literature but were not statistically significant in univariable analyses were also entered into the model.19,20,32,52 Unordered discrete variables were recoded into binomial factors, and ordered discrete variables and continuous variables were entered into the model as is. Since the number of patients differed in each clinical subgroup, the data were resampled in the random forest model to ensure balance.23 Random forests works by generating a large number of classification trees. Each tree is developed on a random sample of patients (sample with replacement) and uses a randomly sampled subset of independent variables (sampled without replacement) as candidate predictors at each node in the tree.
The relative importance of independent variables is determined by first counting the number of test patients correctly classified by each tree, then randomly altering the value of 1 independent variable of the test patients, and determining if the tree correctly classifies these patients. A large difference in the number of patients correctly classified when the independent variable was altered indicates that the variable is important. This difference is averaged over all trees and repeated for each independent variable. We used the mean decrease in accuracy (MDA) measure to rank the relative importance of predictors. MDA is the importance score for each variable; it quantifies the relative contribution of that variable to the prediction accuracy of the model. To get the estimate for error, each tree is constructed using a different bootstrap sample from the original data. About one-third of the cases are left out of the bootstrap sample and not used in the construction of the _k_th tree. The proportion of times that a test patient was misclassified by each tree, averaged over all trees, is a relatively unbiased estimate of classification error, known as the out-of-bag error estimate. Out-of-bag error rates <30% were considered acceptable.23 Out-of-bag error rates were similar for 500, 1000, and 5000 trees. Due to the stability of the error rates, we present the results of 500 forests and 1000 trees. To identify the most important features, using the top variables from the full model, we re-ran a random forest model using the 7–10 strongest predictors based on the MDA scores (>10%) and also based on significance from prior publications. The predictive models using the most important features had error rates similar to those using all the features. Little predictive power was lost using the smaller set.
Multivariable logistic regression analysis was performed as an independent method to confirm the random forest classification features and to determine the effect size. The independent variables that were ranked the highest in relative importance in the pruned random forest analyses were entered into the logistic regression model as independent variables. The dependent variable was a particular clinical subgroup. Odds ratios, 95% confidence intervals, and p values to test for significance were calculated. For this analysis, a c statistic ≥90% was considered very good, 90% >c ≥80% was good, 80% >c ≥70% was fair, and c <70% was considered poor.
Certain variables were recoded before they were entered in the model. We recoded creatine kinase (CK) levels to kilounits/liter because the odds ratio was easier to interpret. For other continuous variables, such as age at onset or delay in diagnosis, we recoded the data into a dichotomous variable using the median value of the reference group as the cutoff.
RESULTS
Demographics and Disease Onset
The demographic characteristics and features of illness onset for the total JIIM population and the clinical subgroups are presented in Table 1. The majority of the patients had JDM (81.2%), with 7.6% of patients classified as JPM, and 11.2% as JCTM. The median age at diagnosis for all JIIM patients was 7.9 years (IQR, 5.2–12.0 yr). Both JPM and JCTM patients were significantly older at diagnosis than JDM patients. The median age at diagnosis for JDM was 7.4 years, whereas the median age at diagnosis for JPM was 12.1 years and for JCTM was 10.2 years. Age at onset of first symptom related to myositis followed similar trends as age at diagnosis. Delay in diagnosis, that is, the time from onset of first symptom to diagnosis, was longest in JCTM patients (median, 7.1 mo) and significantly longer compared with JDM patients (median, 4.0 mo). Disease duration (time from diagnosis to last follow-up available) did not significantly differ between the 3 subgroups; the median disease duration for all JIIM patients was 4.3 years (IQR, 2.2–7.7 yr; range, 0–41.3 yr).
TABLE 1.
Demographic and Disease Onset Data for Patients With Juvenile Myositis Categorized Clinically
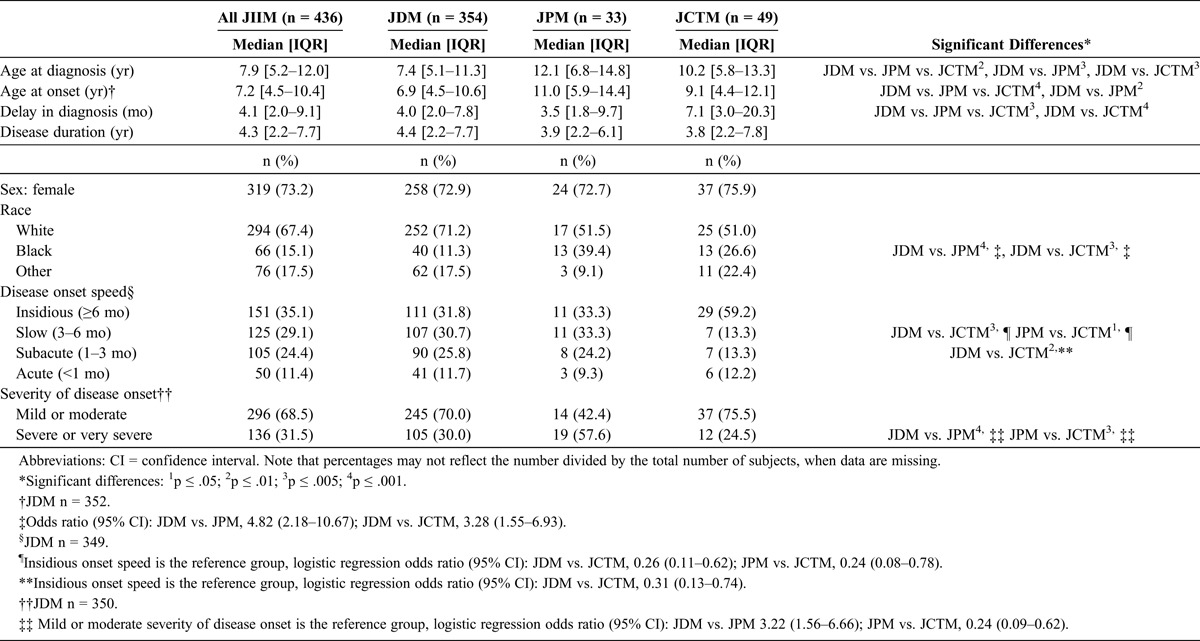
There was a higher proportion of girls than boys in all 3 clinical subgroups; about three-fourths (73.2%) of the study population was female. This female predilection was evident both pre- and post-puberty (data not shown). There were more white patients (67.4%) in the study population than other racial and ethnic groups. However, the racial distribution differed in JPM and JCTM compared with JDM. White patients comprised 71.2% of the JDM population, compared to approximately half of the JPM and JCTM populations (51.5% and 51.0%, respectively). There was a significantly higher frequency of black patients with JPM and JCTM compared with JDM (see Table 1).
The majority of patients had either a slow or insidious onset (29.0% and 35.0%, respectively). JCTM patients were less likely to have slow or subacute onset (13.3% each) compared with JDM patients (30.7% and 25.8%, respectively), which was confirmed by logistic regression analysis. Approximately one-third (31.5%) of all JIIM cases had a severe or very severe disease onset. JPM patients (57.6%) were significantly more likely to have a severe or very severe disease onset, compared to 30.0% of JDM patients and 24.5% of JCTM patients, which was confirmed by logistic regression.
Signs and Symptoms Among JIIM Clinical Groups
Analysis of frequencies of signs and symptoms (Table 2) showed a number of differences among the clinical groups. Almost every patient showed some musculoskeletal system involvement, with proximal muscle weakness (99.8%) being almost universally present, as expected, as a cardinal manifestation of the disease. Arthralgia (61.3%), myalgia (59.9%), and joint contractures (54.2%) were also present in most JIIM patients. When clinical groups were compared, arthralgia (81.6%) and arthritis (69.4%) were more common in JCTM patients than in JDM patients. Compared with JCTM patients, JPM patients were more likely to report falling episodes (59.4%) as a sign of more severe weakness, and less likely to have arthritis (45.5%).
TABLE 2.
Symptoms and Signs by System Involvement for Patients With Juvenile Myositis Categorized Clinically
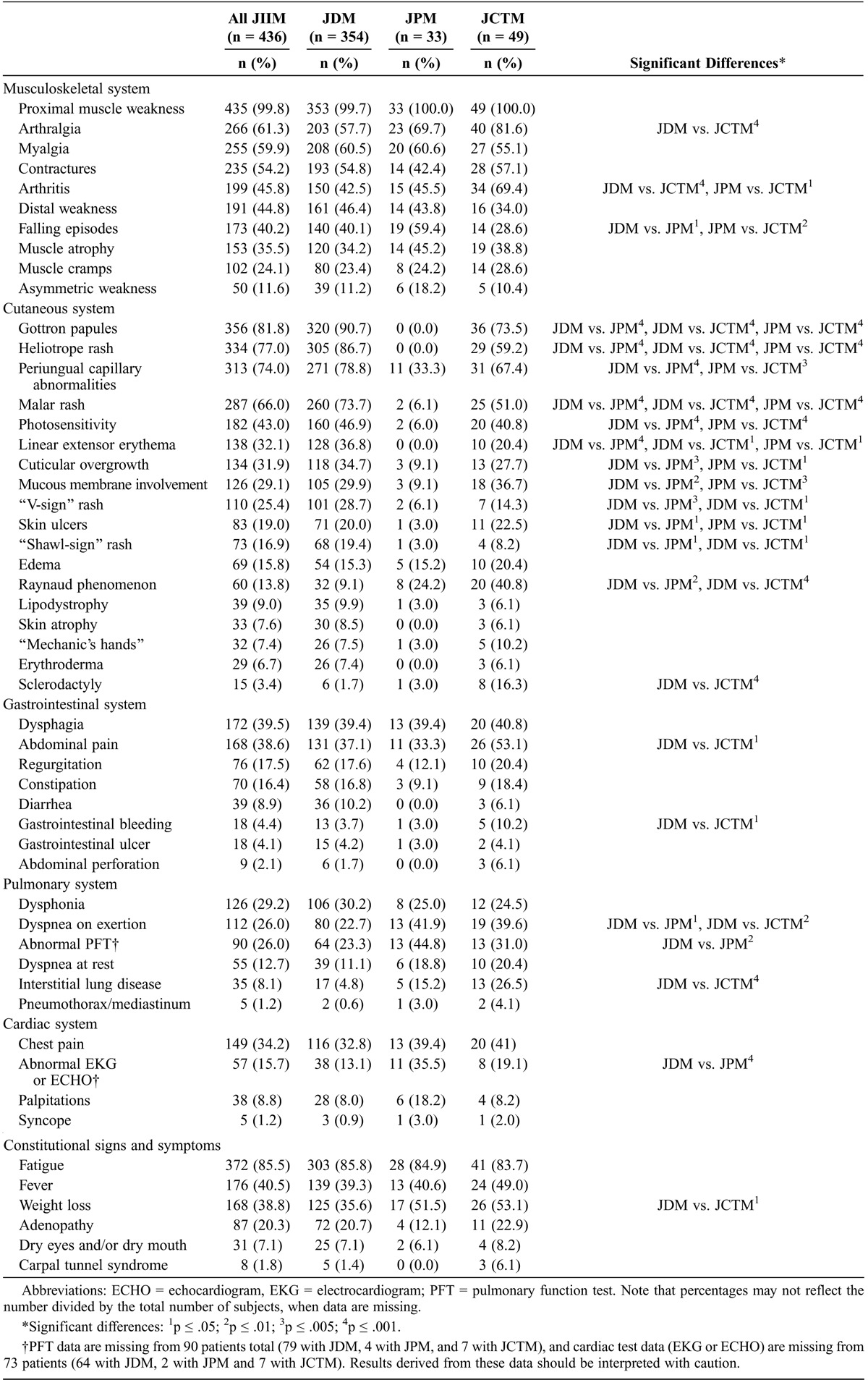
As expected, cutaneous involvement was common in JDM and JCTM patients, most of whom had JDM. The most common cutaneous findings in JDM and JCTM patients included Gottron papules and heliotrope rash, which are part of the Bohan and Peter classification criteria,5 as well as periungual capillary abnormalities and malar rash, which were present in most JDM and JCTM patients; both were present significantly more frequently than in JPM patients. Raynaud phenomenon was more common in JCTM (40.8%) and JPM (24.2%) than in JDM patients (9.1%), and sclerodactyly was more common in JCTM (16.3%) than in JDM (1.7%) patients. Cuticular overgrowth, mucous membrane involvement (including gingival capillary changes), and cutaneous ulcers were more common in JDM and JCTM than JPM, and linear extensor erythema and “V-sign” and “shawl-sign” rashes were more common in JDM than JPM or JCTM. Cutaneous involvement was not unexpectedly less frequent in JPM patients; however, 33.3% of patients did have periungual capillary abnormalities.
Generally, gastrointestinal involvement did not differ among the 3 subgroups. Dysphagia and abdominal pain (unrelated to medication usage) were each present in almost 40% of patients, but abdominal pain was more frequent in JCTM patients (53.1%). Gastrointestinal bleeding was significantly more common in JCTM compared to JDM patients (10.2% vs. 3.7%).
Dysphonia was reported in almost 30% of all JIIM patients. Dyspnea on exertion was seen in 26% of patients, and more frequently in JPM (41.9%) and JCTM (39.6%) than in JDM (22.7%). Primary interstitial lung disease (ILD) and dyspnea at rest were present in only 8% and 13% of patients, respectively. ILD was significantly more common in JCTM (26.5%) than in JDM (4.8%) patients. Chest pain was present in approximately 30% of patients. Electrocardiogram or echocardiogram abnormalities were present more frequently in JPM than JDM patients, although a large percentage of JDM patients did not undergo those tests. In support of increased cardiac disease in JPM, however, there was a nonsignificant trend of more frequent palpitations in JPM (35.5%) than in JDM patients (13.1%).
Fatigue was a commonly reported symptom in all clinical groups, with 85% of patients having documentation of this symptom. Weight loss was more common in JCTM than in JDM patients, being present in more than half of JCTM and JPM patients.
Autoantibodies Among JIIM Clinical Groups
Autoantibodies were frequent in all 3 clinical subgroups, but their distribution differed (Table 3). Antinuclear antibodies (ANA) were present in 72.3% of patients overall. The median ANA titer for JCTM patients was 1:1280, which was higher than in JDM and JPM patients; both JDM and JPM had median ANA titers of 1:320.
TABLE 3.
Autoantibody Data for Patients With Juvenile Myositis Categorized Clinically∗
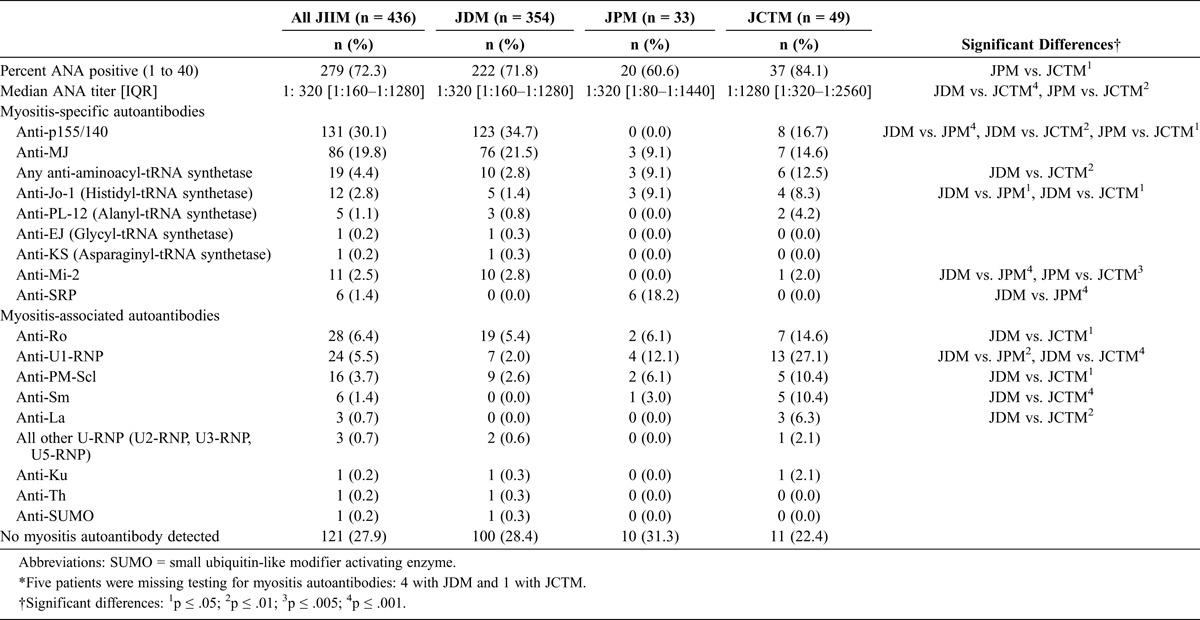
Approximately 70% of patients whose sera was tested by IP had at least 1 myositis-specific or myositis-associated autoantibody present. In JIIM overall, the most common autoantibodies were anti-p155/140 (30.1%) and anti-MJ (19.8%). Autoantibodies to p155/140 were significantly more frequent in JDM patients than in JPM and JCTM patients. Of the JCTM patients with anti-p155/140 autoantibodies, all 8 had JDM. The MJ autoantibodies were detected in the serum of patients from each of the clinical subgroups; of the patients with JCTM who had anti-MJ, 6 had JDM and 1 had JPM.
Among myositis-specific autoantibodies, 4.4% of all JIIM patients had autoantibodies in the anti-tRNA synthetase family, with anti-Jo-1 being the most common. Anti-Jo-1 was more common in JPM (9.1%) and JCTM (8.3%) than in JDM (1.4%). Anti-Mi-2 was seen only in JDM (2.8%) and JCTM patients (2.0%, all of whom had JDM), but not in JPM. Anti-signal recognition particle (SRP) autoantibodies were seen only in JPM cases and were the most frequent autoantibodies present: 18.2% of JPM patients had SRP autoantibodies.
Several myositis-associated autoantibodies were more frequent in patients with JCTM. The most common myositis-associated autoantibodies in JIIM were anti-Ro autoantibodies, which occurred in 6.4% of patients overall. Anti-Ro autoantibodies were more common in JCTM than in JDM patients (14.6% vs. 5.4%, respectively). Anti-U1-ribonucleoprotein (RNP) autoantibodies were also present in all clinical groups, with over one-quarter (27.1%) of JCTM patients being positive for anti-U1-RNP autoantibodies, which was significantly higher than in JDM patients (2.0%). Anti-polymyositis-scleroderma (PM-Scl) autoantibodies were more common in JCTM than JDM patients (10.4% vs. 2.6%, respectively). Anti-Sm and anti-La autoantibodies were also more likely to be present in JCTM patients compared with JDM patients; anti-Sm autoantibodies were present in 10.4% and anti-La were present in 6.3% of JCTM patients, whereas neither was present in the serum of JDM patients. Other less common myositis-associated autoantibodies included other anti-U-RNP autoantibodies (U2, U3, and U5-RNP), anti-Ku, anti-Th, and anti-small ubiquitin-like modifier autoantibodies.
Serum Muscle Enzyme Levels Among JIIM Clinical Groups
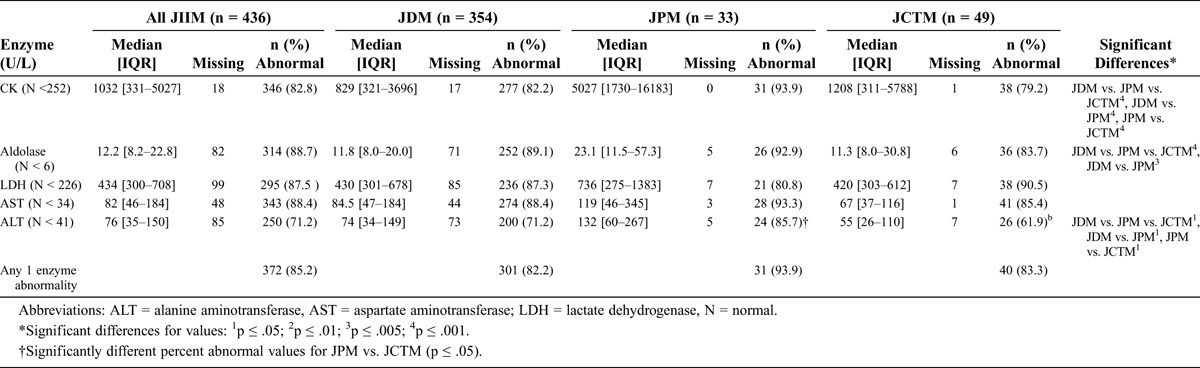
Most JIIM patients, but not all, had elevated serum muscle enzyme levels for their most-abnormal recorded value (Table 4). JPM patients had significantly higher median levels of CK and alanine aminotransferase than JDM and JCTM patients, which was also confirmed in white patients alone. Aldolase values were higher in JPM compared with JDM patients.
TABLE 4.
Most Abnormal Enzyme Levels in Patients With Juvenile Myositis Categorized Clinically
Disease Outcomes Among JIIM Clinical Groups
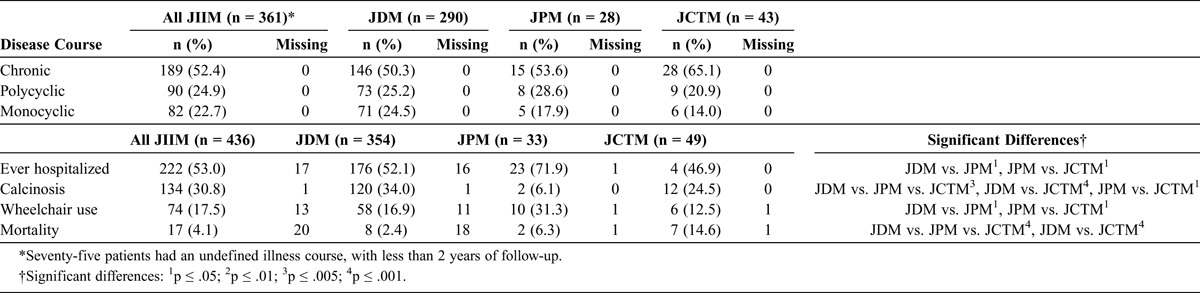
Most patients had a chronic disease course (52.4%), and there was similar distribution of polycyclic and monocyclic disease courses (24.9% and 22.7%, respectively). Logistic regression analysis showed no differences among the clinical groups and disease courses. Hospitalization was common in all 3 clinical subgroups; 53% of all JIIM cases were hospitalized at least once (Table 5). JPM patients were hospitalized more frequently (71.9%) than JDM (52.1%) or JCTM patients (46.9%). Calcinosis was most common in JDM patients (34.0%) and JCTM patients (24.5%) and was uncommon in JPM patients (6.1%). The overall mortality during the follow-up period was 4.1%; it was highest in JCTM patients (14.6%), intermediate (6.3%) in JPM, and lowest (2.4%) in JDM patients.
TABLE 5.
Outcomes in Patients With Juvenile Myositis Categorized Clinically
Comparison of JDM and JPM Patients in the JCTM Clinical Group
To examine whether JCTM is a homogeneous subgroup, we compared the JDM and JPM patients within this subgroup to each other and observed that they were similar, except for the following differences: the JPM patients in the JCTM subgroup were frequently black (62.5%) compared to the JDM patients (19.5%, p = 0.007). The rashes associated with JDM, including Gottron papules, heliotrope and malar rashes, periungual capillary abnormalities, and photosensitivity, were more frequent in JDM patients in the JCTM subgroup than in the JPM patients, as expected. The autoantibody distribution was similar, except that U1-RNP was more frequent in the JPM/JCTM patients (62.5%) vs. JDM/JCTM (20%), and p155/140 was associated only with the JDM/JCTM patients. Median alanine aminotransferase values were higher in JPM/JCTM (228 U/L) than JDM/JCTM patients (45 U/L).
Comparison of Juvenile and Adult IIM Clinical Groups
The frequency distribution of the clinical subgroups differed between juvenile and adult IIM patients, as expected. The majority (81.2%) of JIIM patients had JDM, whereas less than half (47.9%) of adult IIM patients were in that subgroup (p < 0.0001). PM and CTM were present in fewer juvenile than adult IIM patients (7.6% vs. 32.9% of PM patients, p < 0.0001 and 11.2% vs. 19.2% of CTM patients, p = 0.021, respectively).21
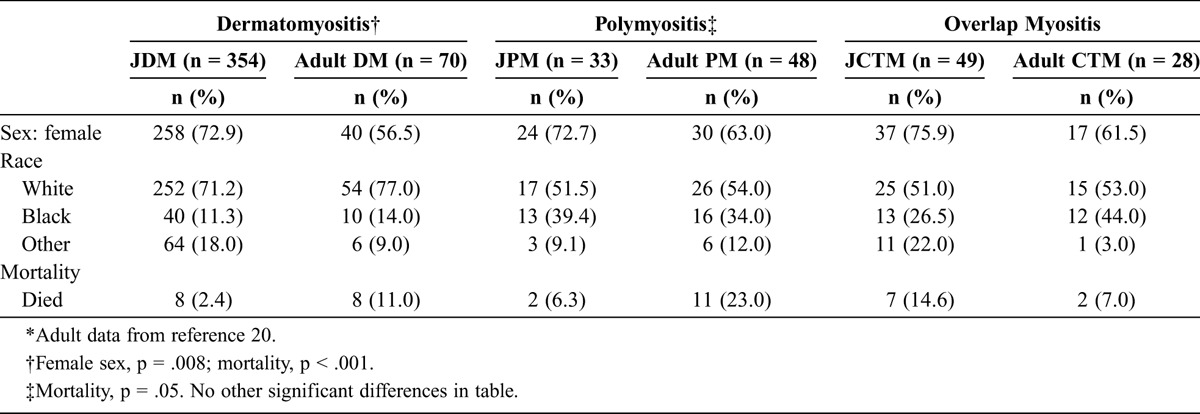
The female predilection of all 3 clinical subgroups was similar to that seen in adult IIM clinical subgroups, but a greater proportion of female patients with DM were juvenile (72.9%) compared with adult (56.5%) (Table 6). The racial distributions were similar for adults and children within each clinical subgroup. Mortality was higher for adults than for children in the DM clinical subgroup (11.0% vs. 2.4%), although the duration of follow-up was slightly longer in the adult patients (mean, 5.1 yr). Almost 1 in 4 (23.0%) adult PM patients died during the study period, in contrast to only 6.3% of the JPM patients. The JCTM patients had a significantly higher mortality frequency than adult CTM patients (14.6% vs. 6.0%), despite a shorter duration of follow-up.
TABLE 6.
Demographic and Mortality Data for 436 Juvenile vs. 146 Adult Myositis Patients∗
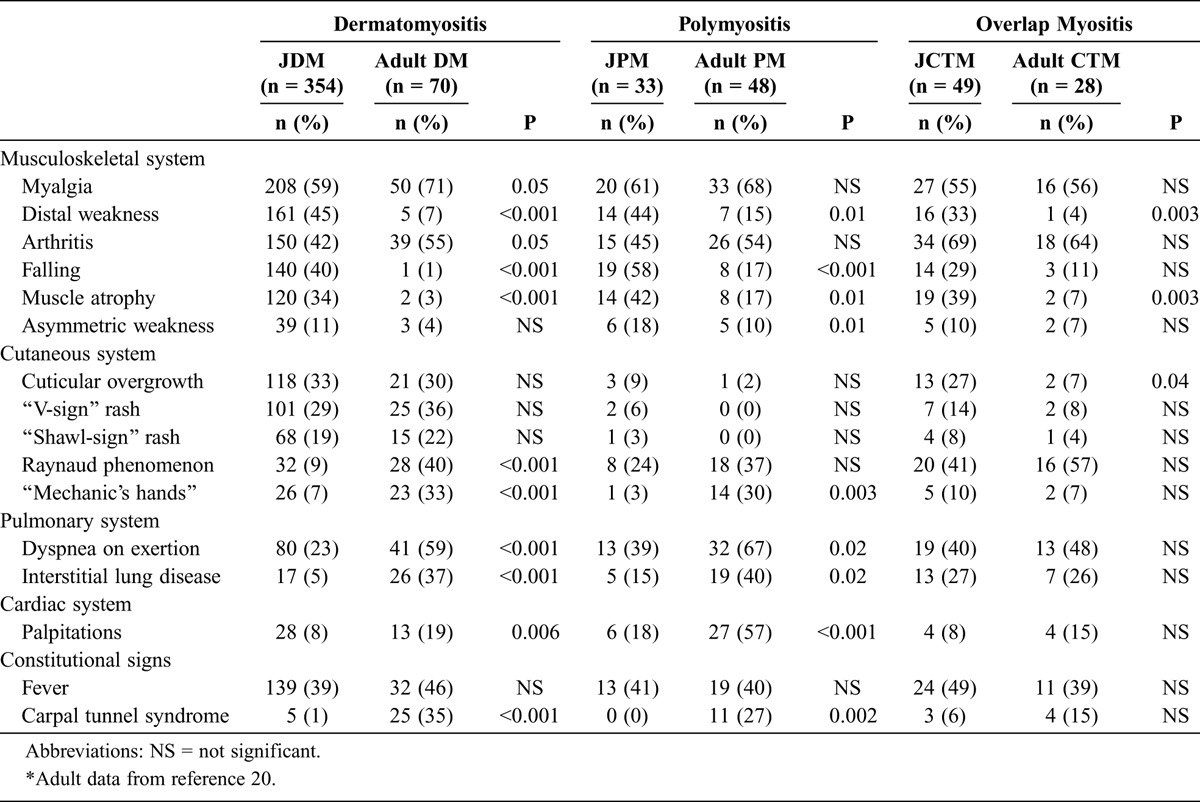
In terms of clinical features, V-sign and shawl-sign rashes, cuticular overgrowth, fever, and a low frequency of asymmetric weakness were seen at similar frequencies in juvenile and adult myositis clinical subgroups (Table 7). Distal weakness, falling episodes, and muscle atrophy were generally more frequent in JIIM patients than their adult counterparts in all clinical subgroups. “Mechanic’s hands” and carpal tunnel syndrome were more common in adult than in JDM and JPM patients. Raynaud phenomenon was more prevalent in adult DM patients compared with JDM patients. Dyspnea on exertion, ILD, and palpitations were also more common in adult patients compared with juvenile patients with DM and PM.
TABLE 7.
Symptoms and Signs of Patients With Juvenile vs. Adult Myositis∗
Multivariable Analysis of JPM versus JDM
Thirty-two variables were used as potential predictors in the random forest classification model of JPM versus JDM based on their significance in the univariable analysis or based on previous literature. In addition, clinically significant variables that were based on published literature but were not statistically significant in the univariable analyses, including calcinosis, muscle atrophy, and arthritis, were entered in the model.19,20,32 Because Gottron papules and heliotrope rash are part of the definition of JDM,5 they were not included in the random forest model, as they are 100% predictive of JDM versus other clinical subgroups.
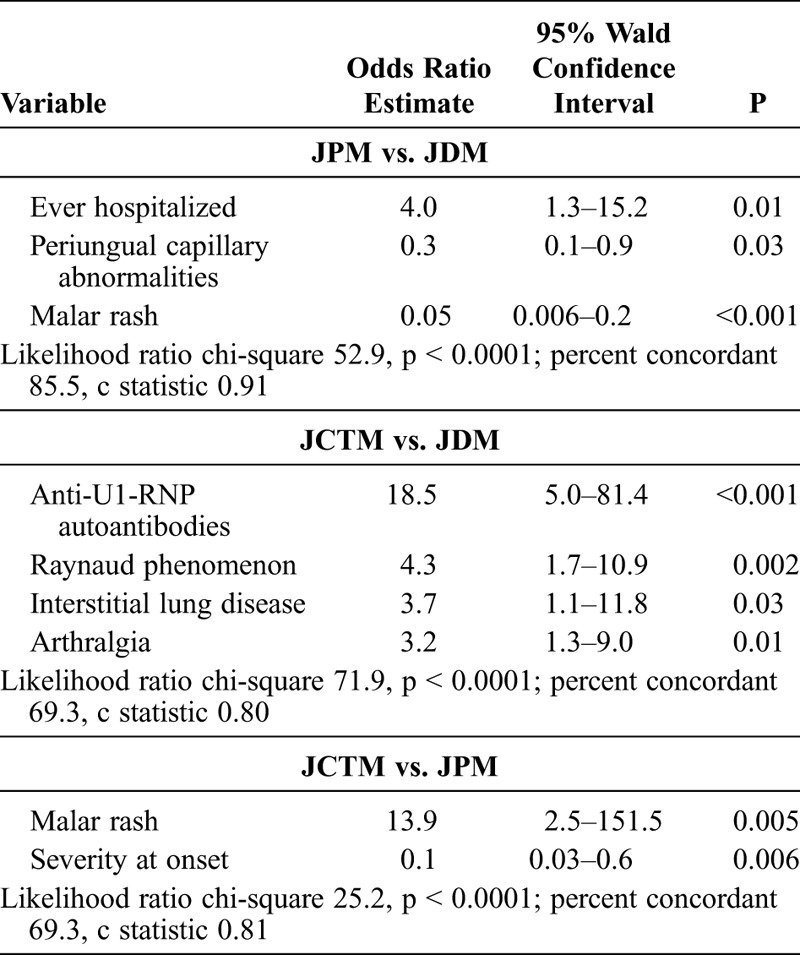
In a random forest classification model of 500 forests and 1000 trees using 266 patients with JDM and 24 with JPM who had complete data, the most important predictors of JPM compared with JDM were malar rash (MDA, 97.0), periungual capillary abnormalities (MDA, 40.3), anti-p155/140 autoantibodies (MDA, 36.5), CK level (MDA, 36.1), linear extensor erythema (MDA, 33.4), whether the patient was ever hospitalized (MDA, 26.1), and a history of photosensitivity (MDA, 23.2), with an out-of-bag error rate of 20.3%. The data were reanalyzed selecting the top variables from the random forest analysis based on the MDA and clinical significance. In the pruned model, malar rash had the highest MDA at 93.1, CK level was second in importance with an MDA of 48.4, and periungual capillary changes was third in importance with an MDA of 24. The out-of-bag error rate for this pruned model was 17.6%.
Multivariable logistic regression results, selecting the top variables from the pruned random forest analysis, are shown in Table 8. The variables anti-p155/140 autoantibodies and linear extensor erythema were removed because no cases of JPM were positive for either variable; therefore, the logistic regression model failed to converge when either variable was included in the model. In the final model, the odds of ever being hospitalized were 4-fold greater for JPM patients than JDM patients. The odds of having malar rash were 20 times lower, and the odds of having periungual capillary abnormalities were 3 times lower for JPM than for JDM patients. The c statistic was 0.91, indicating a very good fit in discriminating between these 2 clinical subgroups.
TABLE 8.
Final Multivariable Logistic Regression Analyses
Multivariable Analysis of JCTM versus JDM
Twenty-six variables were used as potential predictors in the random forest classification model of JCTM versus JDM based on their significance in the univariable analysis. Muscle atrophy and palpitations were also included in the model as clinically significant variables based on prior publications, but they were not statistically significant in the univariable analyses.20,52 Gottron papules and heliotrope rash were removed because they are part of the definition of JDM.5
In a random forest classification model of 500 forests and 1000 trees using 314 patients with JDM and 46 with JCTM who had complete data, the most important predictors of the JCTM group compared to JDM were the anti-U1-RNP autoantibodies, Raynaud phenomenon, arthralgia, and ILD, with an out-of-bag error rate of 31.7%. A pruned random forest model examining these top predictor variables resulted in similar MDA scores and variable order, as well as a similar out-of-bag error rate (33.3%). In the pruned model, the MDA scores were 80.0 for anti-U1-RNP autoantibody, 77.4 for Raynaud phenomenon, 45.4 for arthralgia, 26.5 for ILD, 26.4 for anti-p155/140 autoantibody, and 11.0 for weight loss.
Multivariable logistic regression analysis results are shown in Table 8, using the top predictor variables from the pruned random forest analysis. The final multivariable logistic regression model included anti-U1-RNP autoantibodies, Raynaud phenomenon, ILD, and arthralgia. The c statistic was 0.80 with a likelihood ratio p value < 0.0001, indicating a good fit for discriminating between these 2 subgroups. Anti-U1-RNP autoantibodies were 18 times more likely in JCTM versus JDM patients. Raynaud phenomenon, ILD, and arthralgia were also 3 to 4 times more common in JCTM versus JDM patients.
Multivariable Analysis of JCTM versus JPM
Twenty-three variables were used as potential predictors in the random forest model of JCTM versus JPM based on their significance in the univariable analysis using 40 patients with JCTM and 28 with JPM with complete data. Presence of anti-U1-RNP autoantibodies was also included because of its prior clinical significance.52,54,57 Gottron papules and heliotrope rash were removed from the analysis because they are part of the definition of JDM.5 In a random forest classification model of 500 forests and 1000 trees, the most important predictor of JCTM compared with JPM was presence of malar rash (MDA, 100). The other important variables in the random forest model include arthritis (MDA, 54.4), serum CK level (MDA, 52.8), severity at illness onset (MDA, 41.4), the presence of mucous membrane involvement (including gingival capillary changes) (MDA, 41.1), photosensitivity (MDA, 32.3), whether the patient was ever hospitalized (MDA, 24.3), linear extensor erythema (MDA, 18.9), periungual capillary abnormalities (MDA, 18.7), and sclerodactyly (MDA, 18.2).
To further analyze the most clinically important variables, the top 10 variables from the random forest model were entered into a pruned random forest model, with variable selection based on MDA score and prior literature.52,54,57 Using fewer variables, the out-of-bag error rate decreased slightly to 20.5%. The most important variables were malar rash (MDA, 100), CK level (MDA, 62.4), severity at onset (MDA, 55.6), arthritis (MDA, 45.4), mucous membrane involvement (MDA, 40.8), photosensitivity (MDA, 16.8), linear extensor erythema (MDA, 16.1), ever hospitalized (MDA, 13.4), and sclerodactyly (MDA, 8.9). The presence of periungual capillary abnormalities was no longer an important predictor.
Five variables (malar rash, CK level, severity at onset, arthritis, and mucous membrane involvement) were entered into the logistic regression model for JCTM versus JPM. In the final model, malar rash and severity at onset were significant predictors: the odds of having malar rash were almost 14 times greater for a JCTM patient than for a JPM patient, and the odds of having a more severe onset were 6.25 less for JCTM compared to JPM (see Table 8). The c statistic was 0.81, which indicated a good fit for discriminating between these clinical subgroups.
DISCUSSION
To our knowledge, the current study is the largest and most comprehensive analysis to date of demographic, clinical, and laboratory characteristics, as well as outcomes for each of the major clinical subgroups of juvenile myositis. The analysis demonstrates that childhood myositis is a heterogeneous group of illnesses with different clinical and demographic characteristics, laboratory features, and outcomes.
Characteristics of JIIM Clinical Subgroup Phenotypes
We discovered a number of distinct features associated with the 3 JIIM clinical subgroups, with the most essential findings reported in Table 9. The most important distinguishing features in our analysis, based on the multivariable logistic regression models, included periungual capillary abnormalities and malar rash for JDM; severe disease onset, frequent hospitalizations, and high CK levels for JPM; and malar rash, Raynaud phenomenon, ILD, arthralgia, and the presence of U1-RNP autoantibodies for JCTM.
TABLE 9.
Phenotypic Characteristics of the Clinical Subgroups of Juvenile Myositis∗
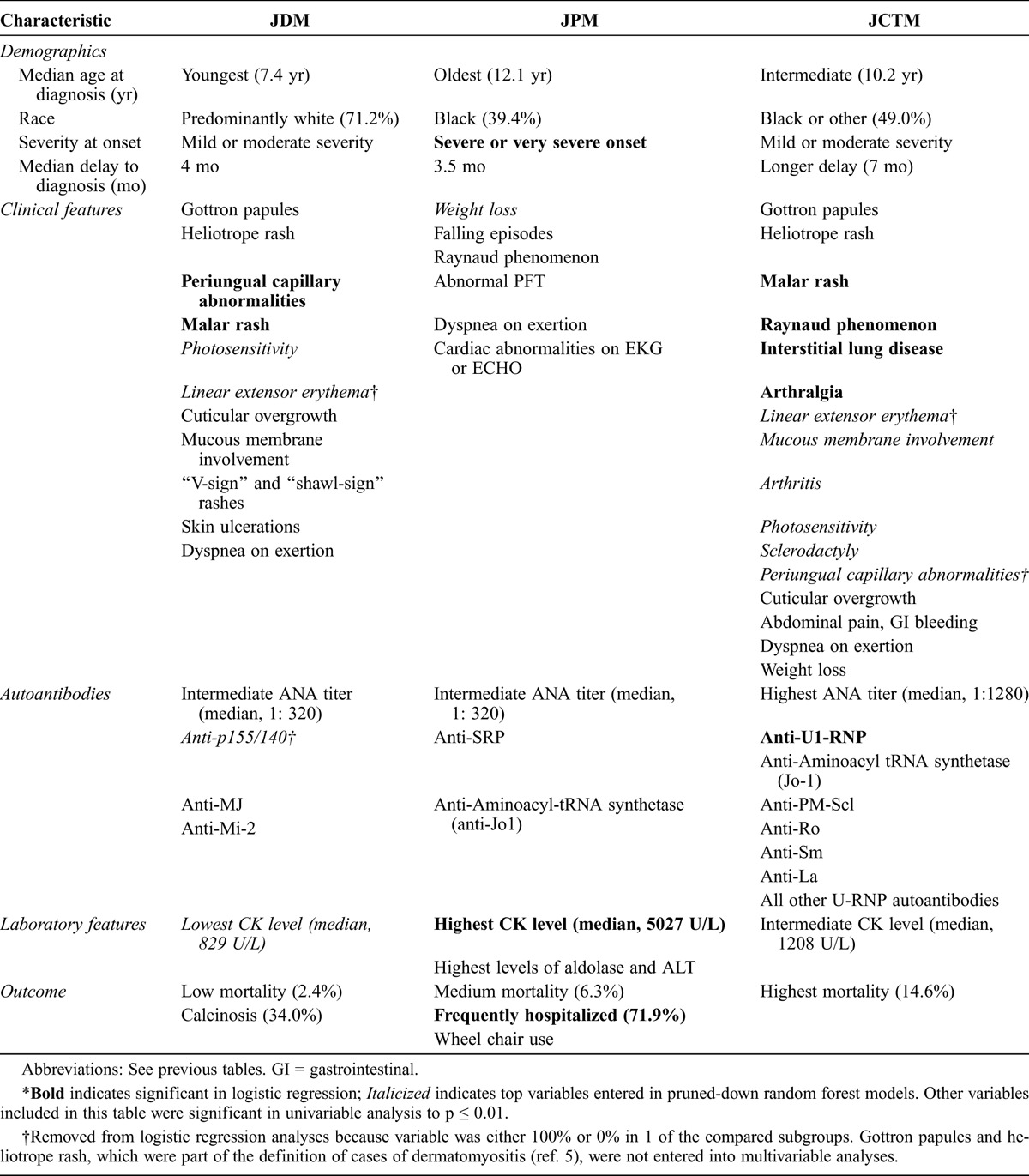
JDM was the most common IIM in children. Important features of JDM included a younger age at onset and diagnosis compared with JCTM and JPM, which is consistent with the limited literature.20 For JDM, the female predominance, median age at onset (7.4 yr), and median delay to diagnosis (4.1 mo) were also consistent with previous reports.30,47 JDM had a larger proportion of white patients than the other clinical subgroups.31
In terms of the clinical features of JDM, we found that cutaneous findings (presence of malar rash and periungual capillary changes) were major characteristics that differentiated this group from the other 2 clinical subgroups. Gottron papules and heliotrope rash are pathognomonic and, by definition, assist in confirming the diagnosis of JDM; they were present in 90.7% and 86.7% of our JDM patients, respectively. Photosensitivity and linear extensor erythema were also important in distinguishing JDM versus JPM patients, and V-sign and shawl-sign rashes were helpful in distinguishing JDM from both JPM and JCTM. Skin ulceration was also more common in JDM compared to JPM patients (20% and 3%, respectively), as was mucous membrane involvement, consisting primarily of gingival capillary changes. Ulcers, as well as periungual and gingival capillary abnormalities, reflect significant vasculopathy in the skin or other organs and are considered a serious manifestation of JDM.6,7,43 Calcinosis was a characteristic feature of JDM, present in 34% of patients and comparable to the reported prevalence of 20%–50%.17,33,40–42 These cutaneous findings are important distinguishing features between JDM and JPM patients and were not present in the latter group.
Over 70% of JDM patients had a positive ANA titer, which is consistent with the findings of previous studies.55,57 A positive ANA was present slightly less frequently in JPM patients (60.6%) and more frequently in JCTM patients (84.1%), similar to the report of Wedderburn et al.57 Also, ANA titers were significantly higher in JCTM patients than JDM or JPM patients. Two autoantibodies, anti-p155/140 and anti-MJ, were the most frequently present in JDM patients.3,12,50 In JDM, anti-Mi-2 was also detected. These autoantibodies were associated with JDM or JDM in the presence of JCTM, but not with JPM. JDM patients had the lowest CK level compared to JPM and JCTM. The frequency of elevation of serum levels of muscle-derived enzymes, which is an important laboratory feature to assist in the diagnosis and assessment of patients, was similar to findings in other JDM registry studies, in which 80%–92% of JDM patients had an abnormality in any 1 muscle enzyme and 54%–87% of patients had specific enzyme abnormalities.26,28,41,42
In terms of outcomes, only 2.4% of JDM patients died, which is consistent with other recent JDM registries in which mortality ranged from 0.5%–2.5%.17,26,42 The majority of JDM patients in this database had either chronic or polycyclic disease, consistent with other studies.13,17,41,45 Wheelchair use was higher for JDM patients in the present study than previously reported,18 but lower than in patients with JPM.
There has been debate about the existence of JPM as an entity distinct from muscular dystrophy and other noninflammatory myopathies.8 The current study demonstrates that JPM is distinct from JDM and JCTM. JPM patients were older than JDM patients at diagnosis of myositis; the median age at onset was 11 years for JPM patients, which is older than the age at onset reported by others.19,44 Approximately 40% of JPM cases were black. The key characteristics of JPM patients were severe illness onset and higher frequency of hospitalization. JPM patients had more frequent falling episodes and higher CK levels, suggestive of more severe weakness. Other clinical manifestations of JPM included frequent cardiac abnormalities, dyspnea on exertion, abnormal pulmonary function tests, Raynaud phenomenon, and weight loss, particularly compared to patients with JDM. JPM patients had poorer outcomes than JDM patients, including higher mortality and more frequent wheelchair use. Certain features of JPM that are more characteristic of JDM, including the presence of malar rash, photosensitivity, and calcinosis, were infrequently present in up to 6% of JPM patients. This may in part be the result of defining JDM versus JPM based on the presence of Gottron papules and heliotrope rash, rather than on the basis of muscle biopsy features.5
JPM patients were more likely to have anti-SRP autoantibodies. The current study supports previous literature that the anti-SRP autoantibodies are myositis-specific autoantibodies linked to severe PM in adult patients who are frequently black,18,20,49 as well as in children with JPM. Rouster-Stevens and Pachman40 described 3 black patients with anti-SRP autoantibodies who had severe PM with severe weakness, very high CK levels, and frequent cardiac involvement.
The current analysis demonstrates clear clinical and serologic differences between JCTM patients and the other 2 clinical subgroups. JCTM patients were more likely to have Raynaud phenomenon, sclerodactyly, ILD, and arthralgia. Approximately half of the JCTM cases were not white. JCTM patients had a longer interval from first symptom to diagnosis than JDM or JPM patients. Our JCTM patients were older than JDM patients, similar to those reported by Wedderburn et al.57
Because 83.7% of JCTM patients had JDM overlap, most of the patients also had rashes similar to JDM patients, including Gottron papules, heliotrope rash, periungual capillary abnormalities, malar rash, photosensitivity, and linear extensor erythema. Calcinosis, which is a characteristic feature of JDM, is also common in JCTM patients, present in 24.5% of patients, although it was not as common as in JDM patients.
JCTM patients had a higher prevalence of an elevated ANA titer, which is consistent with other studies.54,57 JCTM was associated with anti-U1-RNP, anti-PM-Scl, anti-Ro, anti-La, and anti-Sm autoantibodies, consistent with other reports in children and adults with overlap myositis.52,54,57 JCTM patients were significantly more likely to report a chronic disease course and have a higher mortality rate than JDM patients, which may in part result from the longer delay in diagnosis and the higher frequency of ILD. Another explanation for the higher mortality is that the overlapping autoimmune diseases, including systemic lupus erythematosus and scleroderma, have higher mortality rates.2,15,24
Comparing JIIM to Adult IIM Clinical Subgroups
This study suggests that the clinical subgroups in juvenile myositis share some similarities with the corresponding clinical subgroups of adult IIM patients, but also have some important differences.
The racial distribution was similar for DM and PM in adults and children, and the sex distribution was similar between the PM and CTM groups. There was no difference in mortality between juvenile and adult CTM patients with a comparable length of follow-up. However, adult CTM cases had the lowest mortality compared with the other adult groups, whereas JCTM cases had the highest mortality compared with the other juvenile clinical subgroups. This may be due, in part, to differences in the definition of JCTM versus adult CTM. The JCTM patients did not have any predominant overlapping autoimmune disease, whereas 50% of the adult CTM patients had systemic lupus erythematosus.
Adult and juvenile DM patients were also similar with regard to the high frequency of certain clinical manifestations, including fever, cuticular overgrowth, V-sign rash, and shawl-sign rash. Adult and juvenile PM cases were similar with respect to the clinical presentation of myalgia, arthritis, Raynaud phenomenon, and fever. CTM patients generally shared similar clinical manifestations in children and adults.
Juvenile and adult IIM differed substantially in frequency distribution among the clinical subgroups of IIM, with JDM being much more frequent than adult DM, and JPM and JCTM being less frequent than their adult counterparts. In fact, 2 common adult subgroups, inclusion body myositis and cancer-associated myositis,21 were not represented in this very large juvenile cohort and have been reported only rarely.36 This difference in the frequency distribution of juvenile and adult IIM clinical subgroups may be a result of differences in genetic and environmental risk factors between children and adults with myositis,11,23 or it may be related to differences in the frequencies of the myositis autoantibodies in children compared to adults, which will be further examined in a subsequent report.
Distal weakness, falling, and muscle atrophy were surprisingly more common in JIIM patients in each of the clinical subgroups than their adult counterparts, indicating that muscular symptoms were more pronounced in children. It is unclear whether this relates to developmental differences in the muscle manifestations of myositis in children or to other factors, such as variations in enrollment. All adult cases were enrolled at 1 center with clinical expertise in myositis, whereas the children were enrolled by multiple physicians at different locations. A natural history study of muscle strength formally assessed by comparative methods in children versus adult IIM patients suggests that adult DM and PM patients have more severe weakness and a greater frequency of distal weakness than JDM patients.15
Raynaud phenomenon, mechanic’s hands, and carpal tunnel syndrome, features that are associated with the anti-synthetase syndrome, were more common in adult DM patients than in JDM patients. The higher frequency of these manifestations in adults may be due to the fact that anti-synthetase autoantibodies are more common in adult myositis patients.21 Dyspnea on exertion, ILD, and palpitations were also more common in adult patients than juvenile patients with DM, which is indicative of more frequent cardiac and pulmonary involvement in adults.
Although JPM, like adult PM, patients frequently had associated cardiac and pulmonary disease, these manifestations were less common in children with myositis overall. Raynaud phenomenon and carpal tunnel syndrome were more prevalent in adult PM than in JPM, which is consistent with the findings of other large registries of adult myositis patients.16,53
The adult mortality rate is greatly impacted by cardiac and lung involvement,22 and these manifestations were more frequent in adult than juvenile DM and PM. Adult DM and PM patients were 5 and 3 times more likely to die than JDM and JPM patients, respectively. This is consistent with previous literature suggesting higher mortality in adult DM/PM patients than JDM patients.16,31,46,53 Some of the mortality differences between children and adults with DM and PM could also be explained by recent changes in treatment, which would have improved the outcomes for children, as the children were enrolled up to 1 decade later. We found comparable mortality in juvenile and adult CTM patients, which is consistent with a higher mortality in some reports of overlap myositis in adults.10,52 The higher mortality in JCTM may be due to a higher prevalence of ILD.
Methodology and Limitations
In the present study, we utilized several predictive methods, including machine learning and logistic regression modeling approaches, for analyzing clinical features to discover unique phenotypes. Machine learning can be used to recognize hidden patterns and make accurate predictions based on models derived from complex data.23 In this study, random forests provided a robust method to assess the accuracy and importance of phenotypic characteristics in JIIM. Application of the random forest procedure to these data proved to be successful in classifying novel and important features for each of the 3 JIIM subgroups. The results suggested that the use of relevant biological features for classifier construction can improve the classifier performance. Some variables, however, such as ANA, muscle enzyme levels, and specific cardiac and pulmonary testing, were missing too much data to be further examined in random forest models. This study also did not examine how certain features were co-associated.
Although the current study is one of the largest registry studies of juvenile myositis, the number of JPM and JCTM cases was relatively low, which limited the power to formally test for interactions. In fact, in multivariable logistic regression analyses, only a few variables were significant. This analysis also focused on the clinical subgroup phenotypes, and additional differences may be explained by myositis-autoantibody phenotypes,20,36 which were not examined in this study, but will be further assessed in a subsequent report. Another limitation was in comparing the JIIM data, which were from a nationwide registry, to the adult IIM data, which were largely drawn from patients referred to the National Institutes of Health primarily from the mid-Atlantic region. Also, the adult dataset was slightly older in historical time than the juvenile dataset, so differences between these populations may be related to differences in delay to diagnosis or in recognition of these syndromes, as well as treatment differences during these disparate time periods, which could impact outcomes, particularly mortality. Another limitation was that treatment data were not available for this analysis. Consequently, clinical features, outcomes (particularly calcinosis, disease course, and mortality), and serum enzyme levels could have been impacted by differences in therapy among patients and between clinical subgroups. Many physicians, who may have different levels of experience with myositis, referred patients for the study, which also could have impacted data quality. However, medical records were reviewed for approximately 70% of the enrolled patients to confirm the information contained in the physician questionnaire. Finally, we recognize that this study is subject to referral bias, including the fact that a blood sample was required for enrollment. The fact that the features of JDM, including demographic, clinical, laboratory, and outcomes, agreed with a number of published JDM registry studies13,17,26,29,41,42,45 provided additional confidence in the detailed information provided in this report, including the clinical subgroup findings and the comparison with adult patients.
Conclusions
We conducted the current study to develop classification methods, using demographic, clinical, and laboratory features, to better identify the clinical subgroup phenotypes of JIIM. The Childhood Myositis Heterogeneity Study is one of the largest and among the most in-depth interdisciplinary registries of juvenile myositis to date. This is the first study, to our knowledge, that compares the 3 major clinical subgroups within JIIM. This is also the first study that compares juvenile and adult IIM patients. The findings indicate that JDM, JPM, and JCTM are distinct subgroups that have unique characteristics, with some similarities and yet some differences from the comparable adult subgroups. Although past studies have established that certain clinical, laboratory, immunologic, outcome, and genetic features are associated with particular phenotypes, there has not been a comprehensive evaluation of all these features together to assess the possibility that new clusters of these variables may define new clinically important subgroups. The current study may aid physicians clinically in diagnosing patients with myositis, interpreting patients’ diverse symptoms and signs, managing disease, and predicting prognosis. The findings of this analysis will help establish improved classification criteria and aid in understanding the characteristics of these rare and heterogeneous diseases.
ACKNOWLEDGMENTS
We thank Drs. Karyl Barron and Michael Ward for valuable comments after their critical reading of the manuscript. We thank Mona Shah’s PhD thesis committee of the George Washington University Department of Epidemiology and Biostatistics for their valuable feedback on this work, including committee members Drs. Heather Young and Yinglei Lai and examiners Drs. Mark Gourley and Margaret Ulfers.
APPENDIX
Childhood Myositis Heterogeneity Collaborative Study Group
Members of the Childhood Myositis Heterogeneity Collaborative Study Group who contributed to this study:
Leslie S. Abramson, Barbara Adams, Daniel A. Albert, Kathy Amoroso, Bita Arabshahi, Eugene R. Arthur, Balu H. Athreya, Alan N. Baer, Imelda M. Balboni, C. April Bingham, William P. Blocker, John F. Bohnsack, Susan Ballinger, Gilles Boire, Michael Borzy, Gary R. Botstein, Suzanne Bowyer†, Jon M. Burnham, Ruy Carrasco, Victoria W. Cartwright, Gail D. Cawkwell, Chun Peng T. Chao, E. Darryl Crisp, Randy Q. Cron, Marietta M. DeGuzman, Michael H. Dietz, Anne Eberhart, Barbara S. Edelheit, John F. Eggert, Andrew H. Eichenfield, Melissa E. Elder, Janet E. Ellsworth, Kathleen A. Fearn, Terri H. Finkel, Irene Flato, Robert C. Fuhlbrigge, Christos A. Gabriel, Vernon F. Garwood, Abraham Gedalia, Natalie L. Gehringer, Stephen W. George, Harry L. Gewanter, Ellen A. Goldmuntz, Donald P. Goldsmith, Phillip Gorden, Gary V. Gordon, Alexia C. Gospodinoff, Beth Gottlieb, Thomas A. Griffin, Brandt P. Groh, Hillary M. Haftel, Melissa Hawkins-Holt, Michael Henrickson, Gloria C. Higgins, George Ho, Mark F. Hoeltzel, J. Roger Hollister, Russel J. Hopp, Norman T. Ilowite, Lisa Imundo, Jerry C. Jacobs†, Laura James-Newton, Anna Jansen, James Jarvis, Rita Jerath, Courtney R. Johnson, Olcay Y. Jones, Lawrence K. Jung, Lawrence J. Kagen, Thomas V. Kantor, Ildy M. Katona, James D. Katz, Yukiko Kimura, Daniel J. Kingsbury, Steven J. Klein, C. Michael Knee, W. Patrick Knibbe, David K. Kurahara, Bianca A. Lang, Andrew Lasky, Alexander R. Lawton, Julia Lee, Johanan Levine, Carol B. Lindsley, Robert N. Lipnick, Seth H. Lourie, Katherine L. Madson, Harold G. Marks, Paul L. McCarthy, John J. Miller III, Stephen R. Mitchell, Hamid Jack Moallem, Chihiro Morishima, Frederick T. Murphy, Terrance O’Hanlon, Kiem G. Oen, Judyann C. Olson, Elif A. Oral, Barbara E. Ostrov, Lauren M. Pachman, Ramesh Pappu, Murray H. Passo, Maria D. Perez, Donald A. Person, Karin S. Peterson, Paul H. Plotz, Marilyn G. Punaro, C. Egla Rabinovich, Charles D. Radis, Linda I. Ray, Ann M. Reed, Robert M. Rennebohm, Peter D. Reuman, Rafael F. Rivas-Chacon, Deborah Rothman, Kenneth N. Schikler, Donald W. Scott, Bracha Shaham, Robert M. Sheets, David D. Sherry, Edward Sills, Sara H. Sinal, Abigail Smukler, Amy J. Starr, Sangeeta H. Sule, Robert P. Sundel, Ilona S. Szer, Simeon I. Taylor, Elizabeth S. Taylor-Albert, Richard K. Vehe, Scott A. Vogelgesang, Larry B. Vogler, Emily Von Scheven, Steven Wall, Carol A. Wallace, Jennifer C. Wargula, Patience H. White, M. Jack Wilkenfeld, Andrew P. Wilking, Lan Wu, Christianne M. Yung, Lawrence S. Zemel.
†Deceased.
Footnotes
Abbreviations: ANA = antinuclear antibodies, CK = creatine kinase, CTM = connective tissue myopathy, DM = dermatomyositis, IIM = idiopathic inflammatory myopathies, ILD = interstitial lung disease, IP = immunoprecipitation, IQR = interquartile range, JCTM = juvenile myositis overlapping with another autoimmune or connective tissue disease, JDM = juvenile dermatomyositis, JIIM = juvenile idiopathic inflammatory myopathies, JPM = juvenile polymyositis, MDA = mean decrease in accuracy, PM = polymyositis, PM-Scl = polymyositis-scleroderma, RNP = ribonucleoprotein, SRP = signal recognition particle
The contributions of Drs. Shah, Mamyrova, Malley, Miller, and Rider are in the public domain.
This work was supported in part by the Intramural Research Programs of NIEHS, NIH (project number ES101074), NIAMS, NIH, and CBER, Food and Drug Administration. Gulnara Mamyrova was supported by the Cure JM Foundation and previously by The Myositis Association. Ira Targoff is a consultant to the Oklahoma Medical Research Foundation Clinical Immunology Laboratory.
The authors do not have any conflicts of interest related to this work.
Contributor Information
Lisa G. Rider, From the Environmental Autoimmunity Group (MS, GM, FWM, LGR), Program of Clinical Research, National Institute of Environmental Health Sciences; Center for Information Technology (JDM), National Institutes of Health, DHHS, Bethesda, Maryland; Department of Epidemiology and Biostatistics (MS, MMR) and Division of Rheumatology, Department of Medicine(GM), George Washington University School of Medicine, Washington, DC; IWK Health Center and Dalhousie University (AMH), Halifax, Nova Scotia, Canada; and Veteran’s Affairs Medical Center (INT), University of Oklahoma Health Sciences Center, and Oklahoma Medical Research Foundation, Oklahoma City, Oklahoma..
with the Childhood Myositis Heterogeneity Collaborative Study Group∗†, ∗Contributing members listed in the Appendix.; †Current address for Dr. Mamyrova: Division of Rheumatology, Department of Medicine, George Washington University School of Medicine, Washington, DC.
REFERENCES
- 1.Arnett FC, Targoff IN, Mimori T, Goldstein R, Warner NB, Reveille JD. Interrelationship of major histocompatibility complex class II alleles and autoantibodies in four ethnic groups with various forms of myositis. Arthritis Rheum. 1996; 39: 1507–1518. [DOI] [PubMed] [Google Scholar]
- 2.Barron KS, Silverman ED, Gonzales J, Reveille JD. Clinical, serologic, and immunogenetic studies in childhood- onset systemic lupus erythematosus. Arthritis Rheum. 1993; 36: 348–354. [DOI] [PubMed] [Google Scholar]
- 3.Betteridge ZE, Gunawardena H, McHugh NJ. Novel autoantibodies and clinical phenotypes in adult and juvenile myositis. Arthritis Res Ther. 2011; 13: 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bingham A, Mamyrova G, Rother KI, Oral E, Cochran E, Premkumar A, Kleiner D, James-Newton L, Targoff IN, Pandey JP, Carrick DM, Sebring N, O’Hanlon TP, Ruiz-Hidalgo M, Turner M, Gordon LB, Laborda J, Bauer SR, Blackshear PJ, Imundo L, Miller FW, Rider LG. Childhood Myositis Heterogeneity Study Group. Predictors of acquired lipodystrophy in juvenile-onset dermatomyositis and a gradient of severity. Medicine (Baltimore). 2008; 87: 70–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bohan A, Peter JB. Polymyositis and dermatomyositis. Parts 1 and 2. N Engl J Med. 1975; 292: 344–347, 3403–3407. [DOI] [PubMed] [Google Scholar]
- 6.Christen-Zaech S, Seshadri R, Sundberg J, Paller AS, Pachman LM. Persistent association of nailfold capillaroscopy changes and skin involvement over thirty-six months with duration of untreated disease in patients with juvenile dermatomyositis. Arthritis Rheum. 2008; 58: 571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crowe WE, Bove KE, Levinson JE, Hilton PK. Clinical and pathogenetic implications of histopathology in childhood polydermatomyositis. Arthritis Rheum. 1982; 25: 126–139. [DOI] [PubMed] [Google Scholar]
- 8.Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003; 362: 971–982. [DOI] [PubMed] [Google Scholar]
- 9.Feldman BM, Rider LG, Reed AM, Pachman LM. Juvenile dermatomyositis and other idiopathic inflammatory myopathies of childhood. Lancet. 2008; 2201–2212. [DOI] [PubMed] [Google Scholar]
- 9a.Fernandez C, Bardin N, Maues de Paula A, Salort-Campana E, Benyamine A, Franques J, Schleinitz N, Weiller PJ, Pouget J, Pellissier JF, Figarella-Branger D. Correlation of clinicoserologic and pathologic classifications of inflammatory myopathies: study of 178 cases and guidelines for diagnosis. Medicine (Baltimore). 2013; 92: 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garton MJ, Isenberg DA. Clinical features of lupus myositis versus idiopathic myositis: a review of 30 cases. Br J Rheumatol. 1997; 36: 1067–1074. [DOI] [PubMed] [Google Scholar]
- 11.Gourley M, Miller FW. Mechanisms of disease: environmental factors in the pathogenesis of rheumatic disease. Nat Clin Pract Rheumatol. 2007; 3: 172–180. [DOI] [PubMed] [Google Scholar]
- 12.Gunawardena H, Betteridge ZE, McHugh NJ. Myositis-specific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatology (Oxford). 2009; 48: 607–612. [DOI] [PubMed] [Google Scholar]
- 13.Guseinova D, Consolaro A, Trail L, Ferrari C, Pistorio A, Ruperto N, Buoncompagni A, Pilkington C, Maillard S, Oliveira SK, Sztajnbok F, Cuttica R, Corona F, Katsicas MM, Russo R, Ferriani V, Burgos-Vargas R, Solis-Vallejo E, Bandeira M, Baca V, Saad-Magalhaes C, Silva CA, Barcellona R, Breda L, Cimaz R, Gallizzi R, Garozzo R, Martino S, Meini A, Stabile A, Martini A, Ravelli A. Comparison of clinical features and drug therapies among European and Latin American patients with juvenile dermatomyositis. Clin Exp Rheumatol. 2011; 29: 117–124. [PubMed] [Google Scholar]
- 14.Harris-Love MO, Shrader JA, Koziol D, Pahlajani N, Jain M, Smith M, Cintas HL, McGarvey CL, James-Newton L, Pokrovnichka A, Moini B, Cabalar I, Lovell DJ, Wesley R, Plotz PH, Miller FW, Hicks JE, Rider LG. Distribution and severity of weakness among patients with polymyositis, dermatomyositis and juvenile dermatomyositis. Rheumatology (Oxford). 2009; 48: 134–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hashkes PJ, Wright BM, Lauer MS, Worley SE, Tang AS, Roettcher PA, Bowyer SL. Mortality outcomes in pediatric rheumatology in the US. Arthritis Rheum. 2010; 62: 599–608. [DOI] [PubMed] [Google Scholar]
- 16.Hochberg MC, Feldman D, Stevens MB. Adult onset polymyositis/dermatomyositis: an analysis of clinical and laboratory features and survival in 76 patients with a review of the literature. Semin Arthritis Rheum. 1986; 15: 168–178. [DOI] [PubMed] [Google Scholar]
- 17.Huber AM, Lang B, LeBlanc CM, Birdi N, Bolaria RK, Malleson P, MacNeil I, Momy JA, Avery G, Feldman BM. Medium- and long-term functional outcomes in a multicenter cohort of children with juvenile dermatomyositis. Arthritis Rheum. 2000; 43: 541–549. [DOI] [PubMed] [Google Scholar]
- 18.Kao AH, Lacomis D, Lucas M, Fertig N, Oddis CV. Anti-signal recognition particle autoantibody in patients with and patients without idiopathic inflammatory myopathy. Arthritis Rheum. 2004; 50: 209–215. [DOI] [PubMed] [Google Scholar]
- 19.Lorenzoni PJ, Scola RH, Kay CS, Prevedello PG, Espindola G, Werneck LC. Idiopathic inflammatory myopathies in childhood: a brief review of 27 cases. Pediatr Neurol. 2011; 45: 17–22. [DOI] [PubMed] [Google Scholar]
- 20.Love LA, Leff RL, Fraser DD, Targoff IN, Dalakas M, Plotz PH, Miller FW. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore). 1991; 70: 360–374. [DOI] [PubMed] [Google Scholar]
- 21.Lundberg IE, Forbess CJ. Mortality in idiopathic inflammatory myopathies. Clin Exp Rheumatol. 2008; 26 (Suppl. 51): S109–S114. [PubMed] [Google Scholar]
- 22.Malley JD, Malley KG, Pajevic S.Statistical Learning for Biomedical Data. Cambridge: Cambridge University Press; 2011. [Google Scholar]
- 23.Mamyrova G, O’Hanlon TP, Monroe JB, Carrick DM, Malley JD, Adams S, Reed AM, Shamim EA, James-Newton L, Miller FW, Rider LG. Immunogenetic risk and protective factors for juvenile dermatomyositis in Caucasians. Arthritis Rheum. 2006; 54: 3979–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martini G, Vittadello F, Kasapcopur O, Magni MS, Corona F, Duarte-Salazar C, Nemcova D, Len CA, Garay SM, Ullman S, Zulian F. Factors affecting survival in juvenile systemic sclerosis. Rheumatology (Oxford). 2009; 48: 119–122. [DOI] [PubMed] [Google Scholar]
- 25.Mathiesen PR, Zak M, Herlin T, Nielsen SM. Clinical features and outcome in a Danish cohort of juvenile dermatomyositis patients. Clin Exp Rheumatol. 2010; 28: 782–789. [PubMed] [Google Scholar]
- 26.McCann LJ, Juggins AD, Maillard SM, Wedderburn LR, Davidson JE, Murray KJ, Pilkington CA. The Juvenile Dermatomyositis National Registry and Repository (UK and Ireland)—clinical characteristics of children recruited within the first 5 yr. Rheumatology (Oxford). 2006; 45: 1255–1260. [DOI] [PubMed] [Google Scholar]
- 27.Oddis CV, Rider LG, Reed AM, Ruperto N, Brunner HI, Koneru B, Feldman BM, Giannini EH, Miller FW. International consensus guidelines for trials of therapies in the idiopathic inflammatory myopathies. Arthritis Rheum. 2005; 52: 2607–2615. [DOI] [PubMed] [Google Scholar]
- 28.Pachman LM, Abbott K, Sinacore JM, Amoruso L, Dyer A, Lipton R, Ilowite N, Hom C, Cawkwell G, White A, Rivas-Chacon R, Kimura Y, Ray L, Ramsey-Goldman R. Duration of illness is an important variable for untreated children with juvenile dermatomyositis. J Pediatr. 2006; 148: 247–253. [DOI] [PubMed] [Google Scholar]
- 29.Pachman LM, Hayford JR, Chung A, Daugherty CA, Pallansch MA, Fink CW, Gewanter HL, Jerath R, Lang BA, Sinacore J, Szer IS, Dyer AR, Hochberg MC. Juvenile dermatomyositis at diagnosis: clinical characteristics of 79 children. J Rheumatol. 1998; 25: 1198–1204. [PubMed] [Google Scholar]
- 30.Pachman LM, Lipton R, Ramsey-Goldman R, Shamiyeh E, Abbott K, Mendez EP, Dyer A, Curdy DM, Vogler L, Reed A, Cawkwell G, Zemel L, Sandborg C, Rivas-Chacon R, Hom C, Ilowite N, Gedalia A, Gitlin J, Borzy M. History of infection before the onset of juvenile dermatomyositis: results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Research Registry. Arthritis Rheum. 2005; 53: 166–172. [DOI] [PubMed] [Google Scholar]
- 31.Ponyi A, Constantin T, Balogh Z, Szalai Z, Borgulya G, Molnar K, Tefner I, Garami M, Fekete G, Danko K. Disease course, frequency of relapses and survival of 73 patients with juvenile or adult dermatomyositis. Clin Exp Rheumatol. 2005; 23: 50–56. [PubMed] [Google Scholar]
- 32.Ramanan AV, Feldman BM. Clinical features and outcomes of juvenile dermatomyositis and other childhood onset myositis syndromes. Rheum Dis Clin North Am. 2002; 28: 833–858. [DOI] [PubMed] [Google Scholar]
- 33.Ravelli A, Trail L, Ferrari C, Ruperto N, Pistorio A, Pilkington C, Maillard S, Oliveira SK, Sztajnbok F, Cuttica R, Beltramelli M, Corona F, Katsicas MM, Russo R, Ferriani V, Burgos-Vargas R, Magni-Manzoni S, Solis-Valleoj E, Bandeira M, Zulian F, Baca V, Cortis E, Falcini F, Alessio M, Alpigiani MG, Gerloni V, Saad-Magalhaes C, Podda R, Silva CA, Lepore L, Felici E, Rossi F, Sala E, Martini A. Long-term outcome and prognostic factors of juvenile dermatomyositis: a multinational, multicenter study of 490 patients. Arthritis Care Res (Hoboken). 2010; 62: 63–72. [DOI] [PubMed] [Google Scholar]
- 34.Rider LG, Lindsley C, Cassidy J. Juvenile dermatomyositis. In: Cassidy J, Petty R, Laxer R, Lindsley C, eds. Textbook of Pediatric Rheumatology. New York: Saunders Elsevier; 2011: 375–413. [Google Scholar]
- 35.Rider LG, Miller FW. Classification and treatment of the juvenile idiopathic inflammatory myopathies. Rheum Dis Clin North Am. 1997; 23: 619–655. [DOI] [PubMed] [Google Scholar]
- 36.Rider LG, Miller FW. Deciphering the clinical presentations, pathogenesis, and treatment of the idiopathic inflammatory myopathies. JAMA. 2011; 305: 183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rider LG, Wu L, Mamyrova G, Targoff IN, Miller FW. Environmental factors preceding illness onset differ in phenotypes of the juvenile idiopathic inflammatory myopathies. Rheumatology (Oxford). 2010; 49: 2381–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robinson AB, Reed AM. Clinical features, pathogenesis and treatment of juvenile and adult dermatomyositis. Nat Rev Rheumatol. 2011; 7: 664–675. [DOI] [PubMed] [Google Scholar]
- 39.Rouster-Stevens KA, Pachman LM. Autoantibody to signal recognition particle in African American girls with juvenile polymyositis. J Rheumatol. 2008; 35: 927–929. [PubMed] [Google Scholar]
- 40.Sallum AM, Pivato FC, Doria-Filho U, Aikawa NE, Liphaus BL, Marie SK, Silva CA. Risk factors associated with calcinosis of juvenile dermatomyositis. J Pediatr (Rio J). 2008; 84: 68–74. [DOI] [PubMed] [Google Scholar]
- 41.Sanner H, Kirkhus E, Merckoll E, Tollisen A, Roisland M, Lie BA, Taraldsrud E, Gran JT, Flato B. Long-term muscular outcome and predisposing and prognostic factors in juvenile dermatomyositis: a case-control study. Arthritis Care Res (Hoboken). 2010; 62: 1103–1111. [DOI] [PubMed] [Google Scholar]
- 42.Sato JO, Sallum AM, Ferriani VP, Marini R, Sacchetti SB, Okuda EM, Carvalho JF, Pereira RM, Len CA, Terreri MT, Lotufo SA, Romanelli PR, Ramos VC, Hilario MO, Silva CA, Corrente JE, Saad-Magalhaes C. A Brazilian registry of juvenile dermatomyositis: onset features and classification of 189 cases. Clin Exp Rheumatol. 2009; 27: 1031–1038. [PubMed] [Google Scholar]
- 43.Savioli C, Silva CA, Fabri GM, Kozu K, Campos LM, Bonfa E, Sallum AM, de Siqueira JT. Gingival capillary changes and oral motor weakness in juvenile dermatomyositis. Rheumatology (Oxford). 2010; 49: 1962–1970. [DOI] [PubMed] [Google Scholar]
- 44.Serratrice G, Schiano A, Pellissier JF, Desnuelle C. Les expressions anatomocliniques des polysyosites chez l’enfant: vingt-trois cas. Ann Pediatr (Paris). 1989; 36: 237–243. [PubMed] [Google Scholar]
- 45.Stringer E, Singh-Grewal D, Feldman BM. Predicting the course of juvenile dermatomyositis: significance of early clinical and laboratory features. Arthritis Rheum. 2008; 58: 3585–3592. [DOI] [PubMed] [Google Scholar]
- 46.Sultan SM, Ioannou Y, Moss K, Isenberg DA. Outcome in patients with idiopathic inflammatory myositis: morbidity and mortality. Rheumatology. 2002; 41: 22–26. [DOI] [PubMed] [Google Scholar]
- 47.Symmons DP, Sills JA, Davis SM. The incidence of juvenile dermatomyositis: results from a nation-wide study. Br J Rheumatol. 1995; 34: 732–736. [DOI] [PubMed] [Google Scholar]
- 48.Targoff IN. Autoantibodies to aminoacyl-transfer RNA synthetases for isoleucine and glycine. Two additional synthetases are antigenic in myositis. J Immunol. 1990; 144: 1737–1743. [PubMed] [Google Scholar]
- 49.Targoff IN, Johnson AE, Miller FW. Antibody to signal recognition particle in polymyositis. Arthritis Rheum. 1990; 33: 1361–1370. [DOI] [PubMed] [Google Scholar]
- 50.Targoff IN, Mamyrova G, Trieu EP, Perurena O, Koneru B, O’Hanlon TP, Miller FW, Rider LG. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum. 2006; 54: 3682–3689. [DOI] [PubMed] [Google Scholar]
- 51.Targoff IN, Trieu EP, Miller FW. Reaction of anti-OJ autoantibodies with components of the multi-enzyme complex of aminoacyl-tRNA synthetases in addition to isoleucyl-tRNA synthetase. J Clin Invest. 1993; 91: 2556–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Troyanov Y, Targoff IN, Tremblay JL, Goulet JR, Raymond Y, Senecal JL. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore). 2005; 84: 231–249. [DOI] [PubMed] [Google Scholar]
- 53.Uthman I, Vazquez-Abad D, Senecal JL. Distinctive features of idiopathic inflammatory myopathies in French Canadians. Semin Arthritis Rheum. 1996; 26: 447–458. [DOI] [PubMed] [Google Scholar]
- 54.Vancsa A, Gergely L, Ponyi A, Lakos G, Nemeth J, Szodoray P, Danko K. Myositis-specific and myositis-associated antibodies in overlap myositis in comparison to primary dermatopolymyositis: Relevance for clinical classification: retrospective study of 169 patients. Joint Bone Spine. 2010; 77: 125–130. [DOI] [PubMed] [Google Scholar]
- 55.Van Rossum MA, Hiemstra I, Prieur AM, Rijkers GT, Kuis W. Juvenile dermato/polymyositis: a retrospective analysis of 33 cases with special focus on initial CPK levels. Clin Exp Rheumatol. 1994; 12: 339–342. [PubMed] [Google Scholar]
- 56.Vegosen LJ, Weinberg CR, O’Hanlon TP, Targoff IN, Miller FW, Rider LG. Seasonal birth patterns in myositis subgroups suggest an etiologic role of early environmental exposures. Arthritis Rheum. 2007; 56: 2719–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wedderburn LR, McHugh NJ, Chinoy H, Cooper RG, Salway F, Ollier WE, McCann LJ, Varsani H, Dunphy J, North J, Davidson JE. HLA class II haplotype and autoantibody associations in children with juvenile dermatomyositis and juvenile dermatomyositis-scleroderma overlap. Rheumatology (Oxford). 2007; 46: 1786–1791. [DOI] [PubMed] [Google Scholar]