Induced expression of expanded CGG RNA causes mitochondrial dysfunction in vivo (original) (raw)
Abstract
Fragile X-associated tremor/ataxia syndrome (FXTAS) is a late-onset neurodegenerative disorder affecting carriers of premutation forms of the FMR1 gene, resulting in a progressive development of tremor, ataxia and neuropsychological problems. The disease is caused by an expanded CGG repeat in the FMR1 gene, leading to an RNA gain-of-function toxicity mechanism. In order to study the pathogenesis of FXTAS, new inducible transgenic mouse models have been developed that expresses either 11CGGs or 90CGGs at the RNA level under control of a Tet-On promoter. When bred to an hnRNP-rtTA driver line, doxycycline (dox) induced expression of the transgene could be found in almost all tissues. Dox exposure resulted in loss of weight and death within 5 d for the 90CGG RNA expressing mice. Immunohistochemical examination of tissues of these mice revealed steatosis and apoptosis in the liver. Decreased expression of GPX1 and increased expression of cytochrome C is found. These effects were not seen in mice expressing a normal sized 11CGG repeat. In conclusion, we were able to show in vivo that expression of an expanded CGG-repeat rather than overexpression of a normal CGG-repeat causes pathology. In addition, we have shown that expanded CGG RNA expression can cause mitochondrial dysfunction by regulating expression levels of several markers. Although FTXAS patients do not display liver abnormalities, our findings contribute to understanding of the molecular mechanisms underlying toxicity of CGG repeat RNA expression in an animal model. In addition, the dox inducible mouse lines offer new opportunities to study therapeutic interventions for FXTAS.
Keywords: apoptosis, caspase 3, CGG repeat, cytochrome C, FXTAS, gpx-1, inducible mouse model, mitochondria, RNA gain-of-function, Tet-On
Abbreviations
dox
doxycycline
eGFP
enhanced green fluorescent protein
FXTAS
Fragile X-associated tremor/ataxia syndrome
gpx
gluthation peroxidase
rtTA
reverse tetracycline transactivator
TRE
Tet Responsive Element
Introduction
Fragile X-associated tremor/ataxia syndrome (FXTAS) is a late-onset neurodegenerative disorder affecting carriers of the Fragile X premutation forms of the FMR1 gene located on the X-chromosome.1 The chances of developing FXTAS increase dramatically with age, with approximately 30% of male and 8–11% of female PM carriers over the age of 50 y.2 Premutation alleles of the FMR1 gene contain 55–200 CGG repeats in the 5′UTR compared to less than 54 for most individuals in the general population. FXTAS results in progressive development of tremor, ataxia and neuropsychological problems, including anxiety, memory impairment and dementia. Medical co-morbidities may include thyroid disease, fibromyalgia, gastro-intestinal symptoms, hypertension, migraine, auto-immune disease, impotence and neuropathy.3,4 The prevalence of premutation carriers in the general population is approximately 1 in 200 females and 1 in 400 males.5,6 Premutation carriers irrespective of showing signs of FXTAS, have been shown to have up to 8-fold elevated FMR1 mRNA levels in peripheral blood leukocytes, despite close to normal or slightly lowered FMRP protein levels.7 Neurohistological examination of brains of both male and female premutation carriers who displayed the neurological phenotype revealed the presence of eosinophilic, intranuclear inclusions in neurons and astrocytes, and Purkinje cell dropout.8,9 The inclusions were seen in various regions throughout the brain. The intranuclear inclusions stain positively with antibodies against several proteins, including ubiquitin, molecular chaperones, components of the proteasome, Sam68 and Drosha.10-12 Based on the toxic RNA gain-of-function model proposed for FXTAS, it was predicted that FMR1 mRNA would be present within the intranuclear inclusions. Indeed, FMR1 mRNA could be detected in inclusions from post-mortem human FXTAS brain tissue, although FMRP has not been localized in the inclusions.13 Recently, an additional mechanism of toxicity that is triggered by CGG repeat associated non-AUG initiated (RAN) translation has been proposed to underlie pathology in FXTAS.14 This is based on evidence that trinucleotide repeats can be translated into protein even if they do not reside in an AUG-initiated open reading frame (ORF), and such translation can occur in all 3 possible ORF’s of a transcript generating multiple potentially toxic products from a single repeat. In the case of FXTAS it has been proposed that RAN translation initiated in the 5′-UTR of FMR1 mRNA results in the production of a cytotoxic polyglycine- and polyalanine-containing protein named FMRpolyG and FMRpolyA, respectively. Indeed, the presence of FMRpolyG could be demonstrated in brain tissue from patients with FXTAS and the expanded CGG knock-in (KI) mouse models. The mechanisms underlying RAN translation are as yet unknown.
The development of mouse models of FXTAS has facilitated studies on the underlying cellular and molecular bases of this neurodegenerative disease. Knock-in mouse lines have been generated with either an expanded human CGG98 or CGG118 repeat, which exhibit most of the symptoms observed in humans with FXTAS, including ubiquitin-positive intranuclear neuronal inclusions, elevated levels of Fmr1 mRNA and reduced Fmrp expression.15-18 In addition, neuropsychological and cognitive deficits, including poor motor function, impaired memory, progressive spatial processing deficits and evidence of increased anxiety could be demonstrated using various behavioral tests. 19-23 Such studies have provided critical information about the molecular events that occur with the onset and progression of the disorder, including new insights into the role of RNA toxicity in the pathophysiology of FXTAS and the relationship between the number of CGG repeats and disease progression.
However, the pathological consequences of the intranuclear inclusions as well as the exact mechanisms involved in the RNA gain-of-function toxicity model remain to be elucidated. Therefore we developed a new inducible transgenic mouse model with RNA overexpression of a normal length 11CGG repeat or an expanded 90CGG repeat in order to study their effects in vivo. Our model is a Tet-On system using the hnRNP-rtTA driver to regulate expression of the CGG repeat RNA (i.e., either 11CGG or 90CGG) in almost all tissues of the bigenic mice by adding doxycycline (dox) to the drinking water or to the food. In this study we show that overexpression of a 90CGG repeat, but not an 11CGG repeat, at the RNA level can induce toxicity in vivo. Expression of expanded 90CGG RNA induced dysfunction of the liver and eventually death of the animals. We found increased steatosis, apoptosis and disrupted expression of several enzymes involved in the processing of reactive oxygen species (ROS).
Results
A new dox inducible mouse model to study the role of expanded CGG repeat RNA
Although a leading hypothesis for the cause of FXTAS is a toxic RNA gain-of-function mechanism, it has not been clear whether overexpression of a CGG-repeat containing RNA per se is sufficient to cause toxicity in vivo, even when the repeat size is within the normal size range, or whether toxicity depends on the presence of an expanded CGG repeat. Therefore, we generated new dox inducible mouse lines in which a normal sized (i.e., for mouse) 11CGG or an expanded 90CGG repeat was overexpressed, and examined the mice for pathology (Fig. 1A). We never observed any repeat instability when breeding the TRE-nCGG-eGFP mice (data not shown). The repeat size remained equal during all breedings, with an estimated 90CGGs for the expanded transgene, corresponding to a 630 bp band by agarose gel electrophoresis (Fig. 1B).
Figure 1.
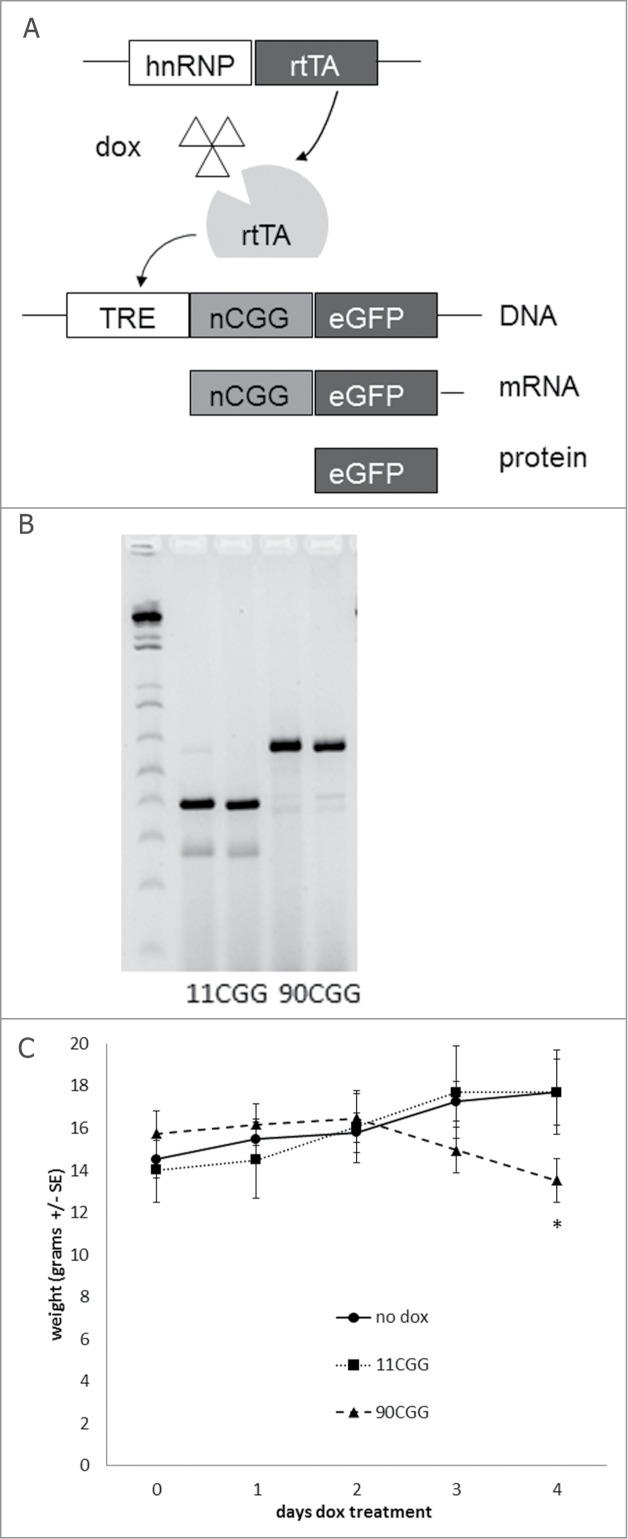
New inducible transgenic mouse model. (A) The Tet-On system was used to generate bigenic mice expressing a 11CGG or a 90CGG repeat at the RNA level in all tissues. Expression of rtTA is controlled by the hnRNP promoter on a transgene. Upon dox administration rtTA will be activated and can bind the Tet Responsive Element (TRE) on another transgene, this induces expression of the nCGG repeat at the RNA level and eGFP at the protein level. (B) Genotyping PCR showing the repeat size of 2 × 11 and 2 × 90 CGGs at approximately 390 bp and 630 bp, respectively. (C) 90CGG expressing mice lose weight when compared to 11CGG expressing mice or bigenic mice (TRE-90CGG-eGFP/hnRNP-rtTA) without dox treatment. The straight line with circles shows the weight of mice not treated with dox (n = 7), the dotted line with squares shows the weight of 11CGG expressing mice after 2 mg/ml dox-water treatment (n = 7), and the striped line with triangles show the weight of 90CGG expressing mice after 2 mg/ml dox-water treatment (n = 6). Error bars are +/− SE; * = p < 0.05.
We first generated mouse lines bearing a TRE-nCGG-eGFP transgene. We cross-bred this line with an existing hnRNP-rtTA mouse line that expresses a reverse tetracycline-controlled transactivator protein (rtTA) that is activated when mice are treated with dox in drinking water or food.24 Bigenic offspring with the expanded CGG RNA (TRE-90CGG-eGFP/hnRNP-rtTA) treated with dox in their drinking water (2 mg/ml) or food (1mg/kg) starting at 3 weeks of age, began to lose weight after 2 d of dox treatment. Therefore we stopped dox treatment after 4 d since the mice died after 5 d of treatment (Fig. 1C). In contrast, TRE-11CGG-eGFP/hnRNP-rtTA mice did not show this weight loss, steadily gained weight, and remained viable. In addition, monogenic mice with only the hnRNP-rtTA or only the TRE-90CGG-eGFP transgene treated with dox, and untreated bigenic TRE-90CGG-eGFP/hnRNP-rtTA mice did not show any weight loss or loss of viability (Fig. 1C; data not shown). These results suggest that the weight loss and subsequent deaths observed in TRE-90CGG-eGFP/hnRNP-rtTA are caused by expression of the 90CGG transgene and are not due to dox treatment or overexpression of mRNA per se (without a CGG repeat expansion). Indeed, we were able to treat TRE-11CGG-eGFP/hnRNP-rtTA mice with a higher dose of dox-water (4mg/ml) or for as long as 25 d without evidence of weight loss (data not shown).
Expression of expanded CGG repeat RNA is toxic in vivo
Dox treatment of bigenic TRE-nCGG-eGFP/hnRNP-rtTA mice resulted in eGFP expression in all tissues examined (Fig. 2). Immunological staining revealed eGFP expression in heart, lung, intestine, liver, kidney, spleen, and brain. The level of transgene expression was compared in different tissues. Quantitative RT-PCR showed that the level op nCGG-eGFP RNA expression was higher in liver when compared to lung, kindey and brain. In liver, the overexpression of 99CGG RNA was 600 ± 80 times higher when compared to endogenous Fmr1 RNA levels.
Figure 2.
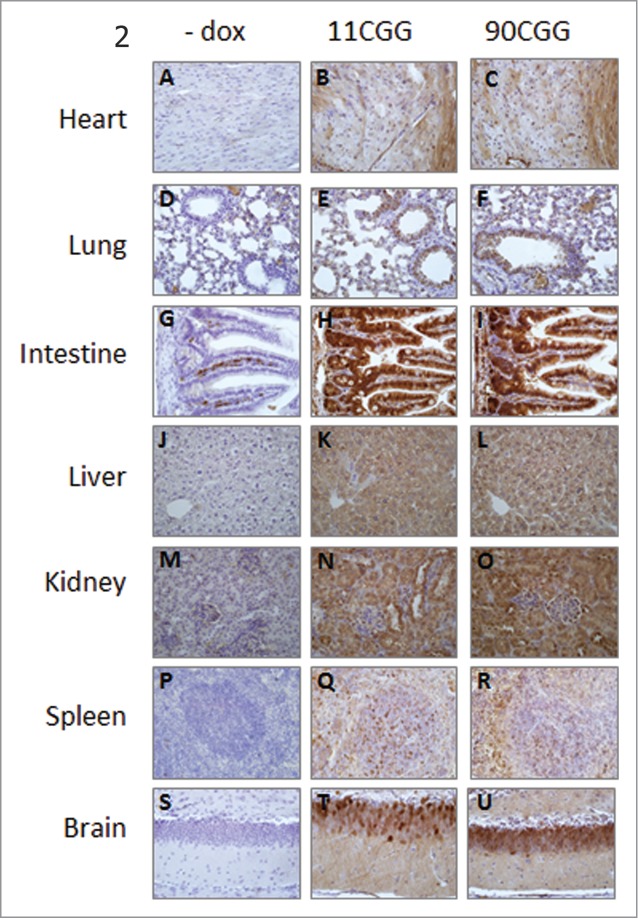
eGFP expression in different tissues. Representative photomicrographs (40x) of immunohistochemical staining with GFP antibody on heart (A–C), lung (D–F), intestine (G–I), liver (J–L), kidney (M–O), spleen (P–R), and brain (S–U) from no dox control mice (A, D, G, J, M, P, S), TRE-11CCG-eGFP mice (B, E, H, K, N, Q, T), and TRE-90CGG-eGFP mice (C, F, I, L, O, R, U).
No eGFP expression was found after dox treatment of monogenic TRE-nCGG-eGFP mice (data not shown). Also, no eGFP expression was seen in bigenic TRE-nCGG-eGFP/hnRNP-rtTA mice not treated with dox (Fig. 2, 3B and 3C), indicating that our Tet-On system does not show any leakage of expression.
Figure 3.
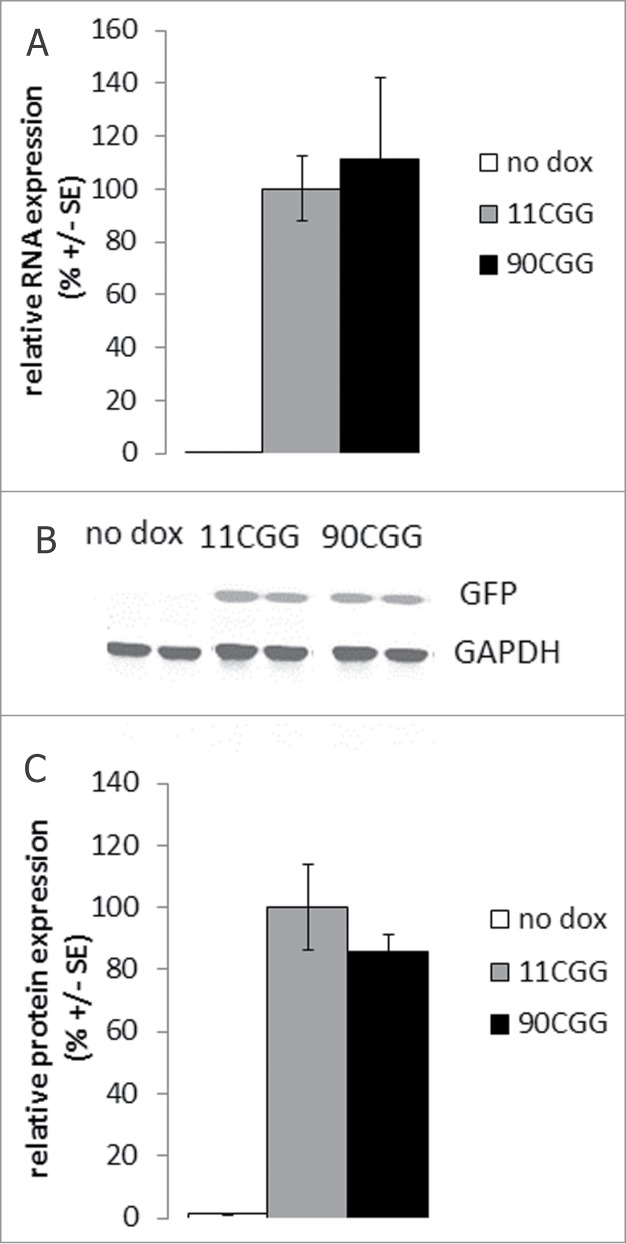
Comparable eGFP RNA and protein expression in the liver of 11CGG and 90CGG mice after 4 d dox treatment. (A) Quantitative RT-PCR on RNA isolated from livers of no dox control mice (white bars; n = 7), TRE-11CGG-eGFP/hnRNP-rtTA mice treated 4 d with dox (gray bars; n = 7), and TRE-90CGG-eGFP/hnRNP-rtTA mice after 4 d dox treatment (black bars; n = 6). 90CGG-eGFP levels are not significantly different from 11CGG-eGFP (p = 0.7) (B) Representative Western blot for eGFP on liver homogenates of no dox control mice and 11CGG or 90CGG expressing mice after 4 d dox treatment with GAPDH as a loading control. (C) Quantification of eGFP protein expression after Western blot on liver homogenates from no dox control mice (white bars; n = 4), TRE-11CGG-eGFP/hnRNP-rtTA mice treated 4 d with dafter 4 d dox treatment (gray bars; n = 8), and TRE-90CGG-eGFP/hnRNP-rtTA mice after 4 d dox treatment (black bars; n = 7). 90CGG-eGFP levels do not significantly differ from the 11CGG-eGFP levels (p = 0.3). Error bars are +/− SE.
We compared the expression levels of CGG RNA and eGFP protein of the TRE-11CGG-eGFP/hnRNP-rtTA and TRE-90CGG-eGFP/hnRNP-rtTA mice after 4 d of dox treatment (2 mg/ml in drinking water). No significant differences were found in the expression levels of 11CGG and 90CGG RNA (Fig. 3A; data not shown). In addition, there were no significant differences in eGFP protein levels between 11CGG and 90CGP expressing mice as determined by Quantitative Western blot (Fig. 3B, 3C; data not shown). Because there were no differences in expression of 90CGG RNA in mice treated with doxycycline in water when compared to mice treated with doxycycline in their food (data not shown), the 2 dox exposed groups were combined into one group.
The brains of all mice were evaluated for the presence of ubiquitin positive intranuclear inclusions, the major hallmark of FXTAS. Although the brains of dox treated bigenic TRE-90CGG-eGFP/hnRNP-rtTA mice did show expression of eGFP and thus the expanded CGG RNA (Fig. 2C), we did not find any ubiquitin positive inclusions. Also all the other organs that did (over)express the expanded CGG RNA did not form any ubiquitin positive inclusions (data not shown). Apparently, the dox treatment of only 4 d was not long enough to induce the formation of the inclusions.
Expression of expanded repeat RNA causes mitochondrial dysfunction in the liver
When sacrificing the mice it was evident that the liver was affected by the dox induced expression of the 90CGG RNA. The livers in the TRE-90CGG-eGFP/hnRNP-rtTA mice treated with dox were pale and pink in color, compared to the dark reddish-brown color of a normal, healthy liver (Fig. 4A, and B). All control mice, including TRE-11CGG-eGFP/hnRNP-rtTA or monogenic mice treated with dox, showed livers with normal appearance. We performed routine histological examination of a large number of tissues, including the brain, liver, kidney, spleen, heart, lung and the intestinal tract. The only pathology found was in the liver, with all other tissues appearing normal. Closer examination of the liver using various histological stainings revealed mild steatosis limited to the livers of the TRE-90CGG-eGFP/hnRNP-rtTA mice treated with dox (Fig. 4D–F).
Figure 4.
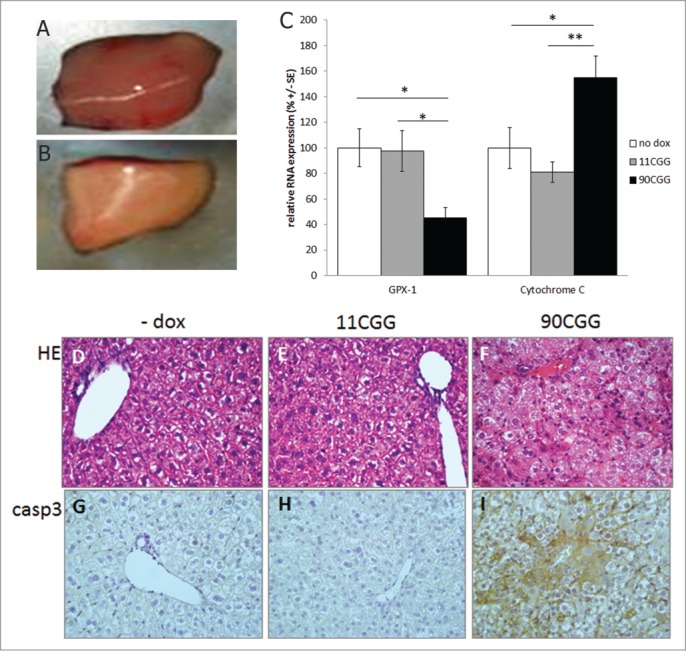
Mitochondrial dysfunction in the liver of 90CGG expressing mice. The color of 90CGG expressing livers is pink and pale (A) compared to the dark reddish-brown color of the liver of control mice (B). Quantitative RT-PCR on RNA isolated from liver of no dox control mice (white bars; n = 7), TRE-11CGG-eGFP/hnRNP-rttA mice treated 4 d with dox (gray bars; n = 7), and TRE-90CGGeGFP/hnRNP-rtTA mice treated 4 d with dox (black bars; n = 6) for oxidative stress markers GPX1 and cytochrome C (C); Error bars are +/− SE; * = p < 0.05; ** = p < 0.01. Representative photomicrographs (40x) of hamatoxylin and eosin (HE) staining (D–F) and cleaved caspase 3 (casp3) immunostaining (G–I) on the livers of no dox controls mice and 11 or 90CGG expressing mice.
The presence of steatosis of the liver is suggestive of mitochondrial dysfunction, which can in turn lead to apoptosis.25 Furthermore, evidence has been found for mitochondrial dysfunction and abnormalities in several neurodegenerative diseases, including FXTAS, and in cultured primary neurons from the exCGG-KI mouse model.26,27 Thus, we hypothesized that mitochondrial dysfunction might underlie the abnormalities we found in the livers of mice that expressed the 90CGG RNA and consequent early demice of these mice.
RNA expression levels of several markers for mitochondrial stress were tested using qRT-PCR. Expression of SOD2, NPUFS4, and ATPB1 was normal (supplemental data). We did find altered RNA expression levels in the livers for CYP2E1, SOD1, and Catalase, both in TRE-11CGG-GFP/hnRNP-rtTA and TRE-90CGG-GFP/hnRNP-rtTA mice treated with dox when compared with untreated control mice (supplemental data). Since these effects were found in mice expressing both 11CGG RNA and 90CGG RNA, these changes are likely to be caused by the doxycycline treatment and not to expression of the transgene.
However, 2 other markers, cytochrome C and GPX1, only showed significant effects in the dox treated TRE-90CGG-EGFP/hnRNP-rtTA mice, but not in the dox treated TRE-11CGG-GFP/hnRNP-rtTA mice and also not in dox treated monogenic TRE-99CGG-eGFP mice. GPX1 is a glutathione oxidase, functions in the detoxification of hydrogen peroxide, and is one of the most important antioxidant enzymes. It prevents apoptosis by caspase 3 through inhibition of cytochrome C.28 We found that expression of GPX1 is significantly downregulated to 45 ± 8% (p < 0.05) in liver of mice expressing 90CGG RNA compared with untreated mice (Fig. 4C). GPX1 RNA levels in the liver of 11CGG RNA expressing mice was normal at 98 ± 16% of untreated mouse livers (p = 0.9). In addition, we found that cytochrome C, which is part of the same pathway, is significantly upregulated in the same 90CGG expressing livers to 155 ± 17% (p < 0.05) when compared with untreated mouse livers. The 11CGG expressing livers showed normal levels 81.1 ± 8% of cytochrome C (p = 0.3; Fig. 4C).
Since the pathway with GPX1 and cytochrome C can affect apoptosis through caspase 3 activation, we performed an immunostaining for cleaved caspase 3 in the liver. Indeed, we could demonstrate semi-quantitatively elevated levels of activated caspase 3 in the livers of 90CGG expressing mice, when compared with 11CGG expressing or untreated mice (Fig. 4G–I). Both control groups of TRE-11CGG-GFP/hnRNP-rtTA mice with dox and untreated mice show the same, low levels of cleaved caspase 3. Taken together, the data indicate that in vivo expression of expanded CGG RNA leads to toxicity in the liver by affecting ROS signaling and thus inducing steatosis and apoptosis.
Discussion
We have successfully generated a new inducible mouse model for FXTAS expressing CGG RNA under control of the Tet-On promoter. Our transgenic mice combined with the hnRNP-rtTA driver mouse show expression upon doxycycline administration in all tissues examined. The system shows no evidence of leaky transcription and allows for the study of the effects of expanded CGG RNA expression compared with similar levels of control size CGG RNA expression. It has been shown before that over expression of an expanded CGG repeat is toxic in Drosophila,29,30 but using this murine model we showed in vivo in a vertebrate model that toxicity depends on overexpression of an expanded CGG repeat and is not seen with overexpression of RNA with a normal sized CGG repeat. Findings in this new mouse model also show that expression of 90CGG RNA leads to mitochondrial dysfunction in the liver, ultimately resulting in liver pathology and death.
Previously, it has been shown in vitro by Hoem et al.31 that there is a threshold for CGG repeat length to induce pathology in neuroblastoma-derived SK cells that is not due simply to the amount of RNA transcribed. In the present study we confirm that pathology and toxicity is not caused by a molarity effect from overexpression of large amounts of CGG-containing RNA in the absence of a large CGG repeat expansion. Because pathology was only found with 90CGG RNA and not 11CGG RNA in vivo, there must be an in vivo threshold for CGG repeat toxicity between 11 and 90 CGG repeats, although the size of this threshold remains to be determined. These findings are also consistent with the in vitro cell culture studies by others11,12,31 that show effects of CGG repeat expansions on viability and inclusion formation that are dependent on the size of the repeat. This suggests that the pathological processes involved in FXTAS differ from other repeat associated disease such as myotonic dystrophy where overexpression of a normal-sized repeat RNA can induce pathology as well.32 The use of the Tet-On system allows us not only to control the expression of the nCGG RNA by adding dox, but it also gives us good controls to check for side-effects of transgene integration sites. Since no abnormalities were found in bigenic TRE-90CGG-eGFP/hnRNP-rtTA mice not treated with dox, we can rule out that the effects found in the mice that were exposed to dox, are caused by the transgene integration site. Since it is expected that if the site of transgene insertion would affect mitochondrial function in the liver, we would also see the effects in mice not treated with dox. Since we do not observe any pathology in mice not treated with dox, this allows us to specifically study the effects of the transgene and not its site of integration. Furthermore, since also the monogenic mice treated with dox and the bigenic TRE-11CGG-eGFP/hnRNP-rtTA control mice treated with dox, do not show an aberrant expression of gpx1, cytochrome C, or activated caspase 3 in the liver, we can also rule out the effects of dox on these markers. Therefore, we can rule out several side effects and pinpoint the effects to the expression of expanded CGG RNA, making our model very valid for studying the Fmr1 premutation and FXTAS.
The deleterious effects of expanded CGG RNA expression on the normal functioning of the liver was not expected, since human FXTAS patients are not known to display liver damage or dysfunction. Also no inclusions have been observed in the livers of FXTAS patients or mouse models.3 This observation can be explained by the fact that in this artificial system the CGG repeat RNA is overexpressed 600 times in liver when compared with endogenous Fmr1 mRNA levels. And also since CGG RNA expression levels are highest in liver compared to other tissues, this can explain the unexpected deleterious effect on this organ. In addition, the fact that the liver was selectively affected rather than other organs may be explained by the fact that the liver is the first organ to clear dox from the body and the liver is one of the organs with the highest content of gpx1, and alterations in its regulation can contribute to several pathologies related to oxidative stress.33 Hepatic mitochondria are recognized as a major source of oxidative stress, which in turn can regulate vital liver functions and disease pathogenesis. Both in alcoholic and in non-alcoholic steatosis of the liver there is a major role for the depletion of glutathion in mitochondrial dysfunction. Thus, although FXTAS patients do not display liver dysfunction, the bigenic TRE-90CGG-eGFP/hnRNP-rtTA mouse model enables in vivo studies to investigate the toxic effect of RNA containing an expanded CGG repeat outside the Fmr1 message.
Due to the liver problems bigenic TRE-90CGG-eGFP/hnRNP-rtTA mice we were not able to treat the mice long enough with dox in order to induce the formation of ubiquitin positive inclusions and pathology in the brain. Still, inclusion formation and brain pathology would be interesting to study using this inducible model (i.e., TRE-90CGG-eGFP) combined with different rtTA driver lines that specifically expresses in the brain. Currently, these studies are ongoing in our lab using PrP, pcpL7, and CamKIIα drivers. On the other hand, many inclusions have also been observed outside the CNS both in FXTAS patients and in the exCGG-KI mouse model.3 Therefore, inclusions formation in other tissues besides the brain will be interesting to study. Nevertheless, in this model the study of inclusion formation seems not to be possible since the expression of the expanded CGG RNA can never be long enough and other pathogenic triggers result in such dramatic cellular dysfunctioning that the mice die. This dysfunctioning seems to be caused by the free RNA with the expanded CGG repeat, rather than RNA that is accumulated in inclusions.
Recently, RAN translation was identified as a potential pathogenic trigger for FXTAS. In case of RAN translation. The exact mechanisms underlying RAN translation and its contribution to the pathogenesis of FXTAS are still unkown, but it would be very interesting to determine if RAN tranlation would play a role in the mitochondrial dysfunction described in this study. Unfortunately, no (commercial) antibodies are available to us yet, to perform these experiments.Evidence for mitochondrial dysfunction has been described in studies using cultured neurons from the exCGG-KI mouse,27 and also in studies in fibroblasts and post-mortem brain materials from premutation carriers with and without FXTAS.26,34 Therefore we reasoned that the steatosis we observed in the mice expressing 90CGG RNA might also be related to defective mitochondrial signaling. After testing a range of markers for mitochondrial function, we found that several were affected by dox treatment in both 11CGG RNA expressing mice as well as in the 90CGG RNA expressing mice. This was not entirely unexpected because the antibiotic dox is processed and metabolized by the liver and it is known that tetracyclines affect mitochondrial function.35 Nevertheless, we did find the mitochondrial enzymes GPX1 and cytochrome C to be differently expressed, specifically in the liver of the 90CGG expressing mice, without any effect in the 11CGG RNA expressing mice. Unfortunately, we were not able to show if this is also the case in the brain, since we are not able to treat the mice long enough with doxycycline to induce inclusion formation in the brain. Future experiments with other rtTA driver lines should shed a light on the question of whether these same mitochondrial markers are also affected in brains expressing expanded CGG RNA for a longer period, as might be expected from the present results. In fact, considerable evidence links GPX1 to neurodegenerative disease including Alzheimer, Parkinson and Huntington. GPX1 is localized in glial cells and its expression regulates vulnerability for neurotoxins. Increased vulnerability to toxins may be important because of the possibility that exposure to environmental toxins, general anesthetics and chemotherapeutic agents, may contribute to the risk for premutation carriers to develop FXTAS.36,37 This suggests that mitochondrial dysfunction, and possibly GPX1, may be a common pathological process in neurodegenerative diseases, including FXTAS. Further studies on altered GPX1 and cytochrome C expression related to CGG repeats could offer a new perspective on the development of new biomarkers for FXTAS and future therapeutic intervention studies.
Materials and Methods
Construct and mice
The pCMV-RL plasmid (Promega) was modified to express eGFP (enhanced green fluorescent protein) with an FMR1 5′ UTR under the control of the tetracycline responsive promoter element (TRE). Utilizing oligonucleotide poly-linkers the CMV promoter in pRL-CMV was replaced with the TRE promoter from the pTRE2 vector (Clontech). Immediately downstream of the TRE promoter, FMR1 5′UTRs containing 11, and 90 CGG repeats were cloned upstream of the chimeric intron in pRL-CMV. The eGFP reporter from the eGFPn-1 vector (Clontech) was cloned downstream of the chimeric intron followed by a SV40 poly A signal. The resulting plasmids are TRE-11CGG-eGFP, and TRE-90CGG-eGFP. The FMR1 5′ UTR containing 90 CGG repeats was cloned from the parent plasmid CMV-_FMR1_-FL between _Blp_I and _Nhe_I restriction sites (all restriction enzymes from New England Biolabs Inc.) The FMR1 5′ UTR containing 11 CGG repeats was amplified by PCR from mouse genomic DNA, followed by a _Blp_I-_Xho_I restriction enzyme digestion, and cloned into TRE-90CGG-eGFP. Transgenic mice were generated by injecting linearized contstruct into oocytes of Janvier .c57bl6 mice. For each construct we had several founders. We used the founders that gave optimal expression, combined with good breeding results. For the TRE-90CGG-eGFP construct we had 2 different founders, that both die when bred with hnRNP-rtTA driver mice and treated with dox.
TRE-nCGG-eGFP mice were crossed with the hnRNP-rtTA driver line24 and bigenic mice were treated with dox directly after weaning at an age of 3 weeks. In the initial experiments we treated mice with dox either in food or drinking water, we have not observed any differences between mice treated with dox water or dox food (Bio Services). Dox was stable in food pellets and had a concentration of 1 g/kg. Dox drinking water contained 2 mg/ml doxycycline hyclate (Sigma) in 5% sucrose (Sigma) and was refreshed every 2–3 d. All animal experiments were conducted with the permission of the local animal welfare committee (DEC).
Genotyping
For genotyping toe clips were incubated overnight at 55°C in TDB (50 mM KCl, 10 mM Tris-HCl pH9, 0.1% Triton X-100 and 0.15 µg proteinase K). After heat inactivation samples were spun down and 1 µl was used for a PCR. The TRE transgene was amplified using forward primer 5′-GCTTAGATCTCTCGAGTTTAC-3′ and reverse primer 5′-ATGGAGGTCAAAACAGCGTG-3′. The rtTA transgene was amplified using forward primer 5′-CAGCAGGCAGCATATCAAGGT-3′ and reverse primer 5′-GCCGTGGGCCACTTTACAC-3′.
RNA and protein isolation
Livers were homogenized in 500 µl HEPES buffer (10 mM HEPES, 300 mM KCl, 3 mMMgCl2, 0.1 mM CaCl2, 0.45% Triton, 0.05% Tween-20; pH 7.6) containing Complete protease inhibitor (Roche), 3 mM DTT and 40 units RNAse OUT (Invitrogen). RNA was isolated from 100 µl of homogenate using RNA Bee according to the manufacturer’s instructions. For protein isolation, the remaining homogenate was spun down 15000g at 4°C for 15 minutes. The supernatant was collected and the protein concentration was measured using the BCA kit (Pierce).
Quantitative RT PCR
Reverse transcriptase was performed in 1 µg of RNA using iScript cDNA synthesis kit (Biorad) according to manufacturer’s instructions. RNA was treated with DNase before cDNA synthesis. Q-PCR using SYBR Green (KAPA Biosystems) was performed on 0.1 µl RT product. Cycling conditions were an initial denaturation of 3 minutes at 95°C, followed 40 cycles of 5 seconds 95°C and 30 seconds 60°C. As a reference gene actin was used and statistical analysis was performed with a t-test. See supplemental data for primer sequences.
Western blotting
Thirty µg of total protein homogenate was loaded to Criterion XT precast gels (4-12% bis-tris) (Biorad) and run in MOPS buffer (0.05M Mops, 0.05M Tris, 3.5 mM SDS, 1mM EDTA, pH 7.7). The gel was electroblotted to a nitrocellulose membrane in TG buffer (0.192M glycine, 0.025M Tris, 20% methanol). After blocking in PBS-Tween, the membrane was incubated overnight with rabbit anti-GFP (1:100.000 Abcam) and mouse anti-GAPDH (1:200.000 Chemicon) antibodies. The secondary antibodies were goat-anti-rabbit IRDye 800cW and donkey-anti-mouse IRDye 680LT (both 1:10.000; Li-Cor). The membrane was scanned with an Odyssey Infrared Imager.
Immunohistochemistry
Tissues were fixed overnight in 4% paraformaldehyde and embedded in paraffin according to standard protocols. Sections (7 µm) were deparaffinised followed by antigen retrieval using microwave treatment in 0.01 M sodium citrate. Endogenous peroxidase activity was blocked and immunostaining was performed overnight at 4°C using mouse anti-GFP (1:2000 Roche) or rabbit anti-cleaved Caspase 3 (1:100 Cell Signaling Technology) antibodies. Antigen-antibody complexes were visualized by incubation with DAB substrate (Dako) after incubation with Brightvision poly-HRP-linker (Immunologic). Slides were counterstained with hamatoxylin and mounted with Entellan (Merck Millipore International).
Funding Statement
This work was supported by National Institutes of Health grant number NINDS NS079775 [to RFB and RW], and by NIDCR DE019583 and NIA AG032119 [to PJH], and E-Rare project number 40-42900-98-1001/113301201 from ZonMW [to RKH] and the Netherlands Brain Foundation project number F2012(1)-101 [to RW].
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors wish to acknowledge the contribution of Christina Merakou, Fatwa Adikusuma, Sotirios Zervopoulos, Laura Ashley van Dijk, and Rob Verhagen to this work.
Supplemental Materials
Supplemental data for this article can be accessed on the publisher's website.
1027469_supplementary_figures_and_tables.zip
References
- 1.Willemsen R, Levenga J, Oostra B. CGG repeat in the FMR1 gene: size matters. ClinGenet 2011; 80:214–25; PMID: 21651511; http://dx.doi.org/10.1111/j.1399-0004.2011.01723.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jacquemont S, Hagerman RJ, Leehey MA, Hall DA, Levine RA, Brunberg JA, Zhang L, Jardini T, Gane LW, Harris SW, et al. Penetrance of the fragile x-associated tremor/ataxia syndrome in a premutation carrier population. JAMA 2004; 291:460–9; PMID: 14747503; http://dx.doi.org/10.1001/jama.291.4.460 [DOI] [PubMed] [Google Scholar]
- 3.Hunsaker MR, Greco CM, Spath MA, Smits AP, Navarro CS, Tassone F, Kros JM, Severijnen LA, Berry-Kravis EM, Berman RF, et al. Widespread non-central nervous system organ pathology in fragile X premutation carriers with fragile X-associated tremor/ataxia syndrome and CGG knock-in mice. ActaNeuropathol 2011; 122:467–79; PMID: 21785977; http://dx.doi.org/10.1007/s00401-011-0860-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hagerman R, Hagerman P. Advances in clinical and molecular understanding of the FMR1 premutation and fragile X-associated tremor/ataxia syndrome. LancetNeurol 2013; 12:786–98; PMID: 23867198; http://dx.doi.org/10.1016/S1474-4422(13)70125-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tassone F, Long KP, Tong TH, Lo J, Gane LW, Berry-Kravis E, Nguyen D, Mu LY, Laffin J, Bailey DB Jr., et al. FMR1 CGG allele size and prevalence ascertained through newborn screening in the United States. GenomeMed 2012; 4:100; PMID: 23259642; http://dx.doi.org/10.1186/gm401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hantash FM, Goos DM, Crossley B, Anderson B, Zhang K, Sun W, Strom CM. FMR1 premutation carrier frequency in patients undergoing routine population-based carrier screening: Insights into the prevalence of fragile X syndrome, fragile X-associated tremor/ataxia syndrome, and fragile X-associated primary ovarian insufficiency in the United States. GenetMed 2011; 13:39–45; PMID: 21116185; http://dx.doi.org/10.1097/GIM.0b013e3181fa9fad [DOI] [PubMed] [Google Scholar]
- 7.Tassone F, Hagerman RJ, Taylor AK, Gane LW, Godfrey TE, Hagerman PJ. Elevated levels of FMR1 mRNA in carrier males: A new mechanism of involvement in the Fragile-X syndrome. Am J HumGenet 2000; 66:6–15; PMID: 10631132; http://dx.doi.org/10.1086/302720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greco CM, Hagerman RJ, Tassone F, Chudley AE, Del Bigio MR, Jacquemont S, Leehey M, Hagerman PJ. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain 2002; 125:1760–71; PMID: 12135967; http://dx.doi.org/10.1093/brain/awf184 [DOI] [PubMed] [Google Scholar]
- 9.Greco CM, Berman RF, Martin RM, Tassone F, Schwartz PH, Chang A, Trapp BD, Iwahashi C, Brunberg J, Grigsby J, et al. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS). Brain 2006; 129:243–55; PMID: 16332642; http://dx.doi.org/10.1093/brain/awh683 [DOI] [PubMed] [Google Scholar]
- 10.Greco CM, Tassone F, Garcia-Arocena D, Tartaglia N, Coffey SM, Vartanian TK, Brunberg JA, Hagerman PJ, Hagerman RJ. Clinical and neuropathologic findings in a woman with the FMR1 premutation and multiple sclerosis. ArchNeurol 2008; 65:1114–6; PMID: 18695063; http://dx.doi.org/10.1001/archneur.65.8.1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sellier C, Rau F, Liu Y, Tassone F, Hukema RK, Gattoni R, Schneider A, Richard S, Willemsen R, Elliott DJ, et al. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EmboJ 2010; 29:1248–61; PMID: 20186122; http://dx.doi.org/10.1038/emboj.2010.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sellier C, Freyermuth F, Tabet R, Tran T, He F, Ruffenach F, Alunni V, Moine H, Thibault C, Page A, et al. Sequestration of DROSHA and DGCR8 by Expanded CGG RNA Repeats Alters MicroRNA Processing in Fragile X-Associated Tremor/Ataxia Syndrome. CellRep 2013; 3:869–80; PMID: 23478018; http://dx.doi.org/10.1016/j.celrep.2013.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tassone F, Iwahashi C, Hagerman PJ. FMR1 RNA withinthe intranuclear inclusions of fragile X-associated Tremor/Ataxia syndrome (FXTAS). RNAbiology 2004; 1:103–5; PMID: 17179750; http://dx.doi.org/10.4161/rna.1.2.1035 [DOI] [PubMed] [Google Scholar]
- 14.Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M, Renoux AJ, Chen KC, Scaglione KM, Basrur V, et al. CGG Repeat-Associated Translation Mediates Neurodegeneration in Fragile X Tremor Ataxia Syndrome. Neuron 2013; 78:440–55; PMID: 23602499; http://dx.doi.org/10.1016/j.neuron.2013.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Willemsen R, Hoogeveen-Westerveld M, Reis S, Holstege J, Severijnen L, Nieuwenhuizen I, Schrier M, VanUnen L, Tassone F, Hoogeveen AT, et al. The FMR1 CGG repeat mouse displays ubiquitin-positive intranuclear neuronal inclusions; implications for the cerebellar tremor/ataxia syndrome. Hum MolGenet 2003; 12:949–59; PMID: 12700164; http://dx.doi.org/10.1093/hmg/ddg114 [DOI] [PubMed] [Google Scholar]
- 16.Entezam A, Usdin K. ATR protects the genome against CGG*CGG-repeat expansion in Fragile X premutation mice. Nucleic AcidsRes 2007; 36:1050–6; PMID: 18160412; http://dx.doi.org/10.1093/nar/gkm1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Entezam A, Biacsi R, Orrison B, Saha T, Hoffman GE, Grabczyk E, Nussbaum RL, Usdin K. Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model. Gene 2007; 395:125–34; PMID: 17442505; http://dx.doi.org/10.1016/j.gene.2007.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brouwer JR, Mientjes EJ, Bakker CE, Nieuwenhuizen IM, Severijnen LA, Van der Linde HC, Nelson DL, Oostra BA, Willemsen R. Elevated Fmr1 mRNA levels and reduced protein expression in a mouse model with an unmethylated Fragile X full mutation. Exp CellRes 2007; 313:244–53; PMID: 17150213; http://dx.doi.org/10.1016/j.yexcr.2006.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Dam D, Errijgers V, Kooy RF, Willemsen R, Mientjes E, Oostra BA, De Deyn PP. Cognitive decline, neuromotor and behavioural disturbances in a mouse model for Fragile-X-associated tremor/ataxia syndrome (FXTAS). Behavioural BrainResearch 2005; 162:233–9; PMID: 15876460; http://dx.doi.org/10.1016/j.bbr.2005.03.007 [DOI] [PubMed] [Google Scholar]
- 20.Hunsaker MR, Wenzel HJ, Willemsen R, Berman RF. Progressive spatial processing deficits in a mouse model of the fragile X premutation. BehavNeurosci 2009; 123:1315–24; PMID: 20001115; http://dx.doi.org/10.1037/a0017616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hunsaker MR, von Leden RE, Ta BT, Goodrich-Hunsaker NJ, Arque G, Kim K, Willemsen R, Berman RF. Motor Deficits on a Ladder Rung Task in Male and Female Adolescent and Adult CGG Knock-in Mice. Behav BrainRes 2011; 222:117–21; PMID: 21440572; http://dx.doi.org/10.1016/j.bbr.2011.03.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hunsaker MR, Goodrich-Hunsaker NJ, Willemsen R, Berman RF. Temporal Ordering Deficits in Female CGG KI Mice Heterozygous for the Fragile X Premutation. Behav BrainRes 2010; 213:263–8; PMID: 20478339; http://dx.doi.org/10.1016/j.bbr.2010.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hunsaker MR, Arque G, Berman RF, Willemsen R, Hukema RK. Mouse models of the fragile x premutation and the fragile X associated tremor/ataxia syndrome. Results Probl CellDiffer 2012; 54:255–69; PMID: 22009357; http://dx.doi.org/10.1007/978-3-642-21649-7_14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katsantoni EZ, Anghelescu NE, Rottier R, Moerland M, Antoniou M, de Crom R, Grosveld F, Strouboulis J. Ubiquitous expression of the rtTA2S-M2 inducible system in transgenic mice driven by the human hnRNPA2B1/CBX3 CpG island. BMC DevBiol 2007; 7:108; PMID: 17900353; http://dx.doi.org/10.1186/1471-213X-7-108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tamimi TI, Elgouhari HM, Alkhouri N, Yerian LM, Berk MP, Lopez R, Schauer PR, Zein NN, Feldstein AE. An apoptosis panel for nonalcoholic steatohepatitis diagnosis. JHepatol 2011; 54:1224–9; PMID: 21145805; http://dx.doi.org/10.1016/j.jhep.2010.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ross-Inta C, Omanska-Klusek A, Wong S, Barrow C, Garcia-Arocena D, Iwahashi C, Berry-Kravis E, Hagerman R, Hagerman PJ, Giulivi C. Evidence of mitochondrial dysfunction in fragile X-associated tremor/ataxia syndrome. BiochemJ 2010; 429:545–52; PMID: 20513237; http://dx.doi.org/10.1042/BJ20091960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaplan ES, Cao Z, Hulsizer S, Tassone F, Berman RF, Hagerman PJ, Pessah IN. Early mitochondrial abnormalities in hippocampal neurons cultured from Fmr1 premutation mouse model. JNeurochem 2012; 123(4):613–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lei XG, Cheng WH, McClung JP. Metabolic regulation and function of glutathione peroxidase-1. Annu RevNutr 2007; 27:41–61; PMID: 17465855; http://dx.doi.org/10.1146/annurev.nutr.27.061406.093716 [DOI] [PubMed] [Google Scholar]
- 29.Jin P, Duan R, Qurashi A, Qin Y, Tian D, Rosser TC, Liu H, Feng Y, Warren ST. Pur alpha Binds to rCGG Repeats and Modulates Repeat-Mediated Neurodegeneration in a Drosophila Model of Fragile X Tremor/Ataxia Syndrome. Neuron 2007; 55:556–64; PMID: 17698009; http://dx.doi.org/10.1016/j.neuron.2007.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sofola OA, Jin P, Qin Y, Duan R, Liu H, de Haro M, Nelson DL, Botas J. RNA-Binding Proteins hnRNP A2/B1 and CUGBP1 Suppress Fragile X CGG Premutation Repeat-Induced Neurodegeneration in a Drosophila Model of FXTAS. Neuron 2007; 55:565–71; PMID: 17698010; http://dx.doi.org/10.1016/j.neuron.2007.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoem G, Raske CR, Garcia-Arocena D, Tassone F, Sanchez E, Ludwig AL, Iwahashi CK, Kumar M, Yang JE, Hagerman PJ. CGG-repeat length threshold for FMR1 RNA pathogenesis in a cellular model for FXTAS. Hum MolGenet 2011; 20:2161–70; PMID: 21389081; http://dx.doi.org/10.1093/hmg/ddr101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mahadevan M, Tsilfidis C, Saborin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barcelo J, O'Hoy K, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3' untranslated region of the gene. Science 1992; 255:1253–5; PMID: 1546325; http://dx.doi.org/10.1126/science.1546325 [DOI] [PubMed] [Google Scholar]
- 33.Mari M, Morales A, Colell A, Garcia-Ruiz C, Fernandez-Checa JC. Mitochondrial glutathione, a key survival antioxidant. Antioxid RedoxSignal 2009; 11:2685–700; PMID: 19558212; http://dx.doi.org/10.1089/ars.2009.2695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Napoli E, Ross-Inta C, Wong S, Omanska-Klusek A, Barrow C, Iwahashi C, Garcia-Arocena D, Sakaguchi D, Berry-Kravis E, Hagerman R, et al. Altered Zinc Transport Disrupts Mitochondrial Protein Processing/Import in Fragile X-Associated Tremor/Ataxia Syndrome. Hum MolGenet 2011; 20:3079–92; PMID: 21558427; http://dx.doi.org/10.1093/hmg/ddr211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neustadt J, Pieczenik SR. Medication-induced mitochondrial damage and disease. Mol Nutr FoodRes 2008; 52:780–8; PMID: 18626887; http://dx.doi.org/10.1002/mnfr.200700075 [DOI] [PubMed] [Google Scholar]
- 36.Paul R, Pessah IN, Gane L, Ono M, Hagerman PJ, Brunberg JA, Tassone F, Bourgeois JA, Adams PE, Nguyen DV, et al. Early onset of neurological symptoms in fragile X premutation carriers exposed to neurotoxins. Neurotoxicology 2010; 31:399–402; PMID: 20466021; http://dx.doi.org/10.1016/j.neuro.2010.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Dwyer JP, Clabby C, Crown J, Barton DE, Hutchinson M. Fragile X-associated tremor/ataxia syndrome presenting in a woman after chemotherapy. Neurology 2005; 65:331–2; PMID: 16043816; http://dx.doi.org/10.1212/01.wnl.0000168865.36352.53 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1027469_supplementary_figures_and_tables.zip