TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis (original) (raw)
. Author manuscript; available in PMC: 2015 Nov 20.
Published in final edited form as: Leukemia. 2009 Mar 5;23(5):905–911. doi: 10.1038/leu.2009.47
Abstract
High-throughput DNA sequence analysis was used to screen for TET2 mutations in bone marrow-derived DNA from 239 patients with _BCR-ABL_-negative myeloproliferative neoplasms (MPNs). Thirty-two mutations (19 frameshift, 10 nonsense, 3 missense; mostly involving exons 4 and 12) were identified for an overall mutational frequency of ~13%. Specific diagnoses included polycythemia vera (PV; _n_=89), essential thrombocythemia (ET; _n_=57), primary myelofibrosis (PMF; _n_=60), post-PV MF (_n_=14), post-ET MF (_n_=7) and blast phase PV/ET/MF (_n_=12); the corresponding mutational frequencies were ~16, 5, 17, 14, 14 and 17% (_P_=0.50). Mutant TET2 was detected in ~17 and ~7% of _JAK2_V617F-positive and -negative cases, respectively (_P_=0.04). However, this apparent clustering of the two mutations was accounted for by an independent association between mutant TET2 and advanced age; mutational frequency was ~23% in patients ≥60 years old versus ~4% in younger patients (P<0.0001). The presence of mutant TET2 did not affect survival, leukemic transformation or thrombosis in either PV or PMF; a correlation with hemoglobin <10 g per 100 ml in PMF was noted (_P_=0.05). We conclude that TET2 mutations occur in both _JAK2_V617F-positive and -negative MPN, are more prevalent in older patients, display similar frequencies across MPN subcategories and disease stages, and hold limited prognostic relevance.
Keywords: JAK2, MPL, myeloproliferative, polycythemia, thrombocythemia, myelofibrosis
Introduction
The recent descriptions of _JAK2_V617F, JAK2 exon 12 and _MPL_515 mutations in myeloproliferative neoplasms (MPNs) underline JAK-STAT as the signaling pathway of interest in these diseases.1–9 However, despite the fact that the expression of these mutations in mouse hematopoietic stem cells induces MPN phenotype,10–14 their precise pathogenetic contribution in human disease has not been clarified. First, neither JAK2 nor MPL mutations are always found in patients with MPN and, when present, they are not necessarily specific to a particular clinicopathological entity.15–17 Second, although JAK2/MPL mutations contribute to MPN pathogenesis and phenotypic determination, there is growing evidence that suggests an additional role for mutant allele burden, host genetic factors and for the presence or absence of other coexisting mutations.11,18–22 For example, the presence of a JAK2 mutation appears to be essential for, but not specific to, the polycythemia vera (PV) phenotype, which is also characterized by a higher mutant allele burden, compared with that seen in other MPNs, including essential thrombocythemia (ET).23,24 Similarly, mutant MPL often overshadows _JAK2_V617F when the two coexist, and this scenario is usually associated with primary myelofibrosis (PMF) or ET.19,22 These observations underscore the complexity of pathogenetic mechanisms in MPN and the need for identifying additional molecular alterations, in tandem with clinical correlative studies.
In a recent communication, Delhommeau et al.25 showed that TET2 (TET oncogene family member 2), a putative tumor suppressor gene located in the minimal loss-of-heterozygosity (LOH) region of chromosome 4q24 in patients with myeloid malignancies, harbored the loss-of-function mutations that coexisted with _JAK2_V617F and sometimes predated its acquisition. The authors screened 181 _JAK2_V617F-positive patients with PV, ET or PMF, and documented an overall mutational frequency of ~14%. They also showed clonal involvement of both multipotent and committed myeloid progenitors and increased engrafting capacity, observed in nonobese diabetic–severe combined immunodeficient (NOD–SCID) mice, of _TET2_-mutated human hematopoietic stem cells from MPN patients. Following this seminal observation, we have reported an even higher TET2 mutational frequency of 29% in patients with systemic mastocytosis (SM),26 and also documented the mutation in a spectrum of myeloid neoplasms, Including myelodysplastic syndrome and chronic myelomonocytic leukemia.27 In SM, mutant TET2 segregated with _KIT_D816V, occurred in both indolent and aggressive SM variants, was significantly associated with monocytosis, and did not appear to affect survival.26 The purpose of this study was to confirm the prevalence of TET2 mutations in _JAK2_V617F-positive MPN, estimate mutational frequency in the absence of _JAK2_V617F and during different disease stages and establish clinical and laboratory correlates in the context of PV, ET or PMF.
Materials and methods
After approval by the institutional review board, the Mayo Clinic database of adult MPN patients (age ≥18 years) was reviewed to select consecutive patients with _BCR-ABL_-negative classic MPN representing different disease stages and in whom stored bone marrow (BM) cells were available for DNA extraction and mutation analysis. As such, our study population included patients with PV, ET, PMF, post-PV MF, post-ET MF and acute myeloid leukemia (AML) arising from antecedent PV, ET or MF. Diagnoses were established on the basis of the 2001 World Health Organization (WHO) criteria.28 Mutation screening for _JAK2_V617F (reverse transcriptase PCR), JAK2 exon 12 mutations and _MPL_W515L/K were performed using BM-derived cells, according to previously published methods.29,30 In patients who were alive, every attempt was made to update follow-up information by means of a questionnaire/telephone call sent to both patients and their primary doctors, and the ‘date of last contact’ reflected this time point, and not the last time they were seen at the Mayo Clinic. All BM specimens were either collected or reviewed at our institution to ensure accuracy of diagnosis. The management mostly consisted of what was considered at the time as ‘standard’ therapy and reflected the individual physician’s best clinical judgment. All parameters used for statistical analysis, except for those addressing prognosis (for example, survival, leukemic or fibrotic transformation and thrombosis), were those obtained at the time of diagnosis and before any therapeutic intervention.
High-throughput DNA sequence analysis was used to screen for TET2 mutations in BM-derived DNA. M13-appended gene-specific primers were designed to amplify and sequence all coding exons of all isoforms of TET2, including TET2 isoform A (NM_001127208–2002 amino acids) and TET2 isoform B (NM_017628–1165 amino acids). PCR primers were designed to amplify and sequence <500 bp amplicons, and overlapping PCR amplicons were designed for all exons >400 bp to ensure complete coverage. For each PCR reaction, 20 ng of genomic DNA was used for PCR amplification using a Duncan Water Bath Thermal Cycler, followed by magnetic bead purification (SPRI, Agencourt Bioscience, Beverly, MA, USA). Bidirectional sequencing was performed using ABI 3730 capillary sequencers (Agencourt Bioscience) as described previously.31 Missense and nonsense mutations were detected in bidirectional sequence traces using a Mutation Surveyor (Softgenetics Inc., State College, PA, USA). All traces were reviewed manually and with the Mutation Surveyor for the presence of frameshift mutations. Frameshift and nonsense mutations were annotated according to the predicted effects on TET2-coding sequence using TET2 isoform A as the predominant isoform.
All frameshift and nonsense mutations were scored as pathogenic TET2 mutations on the basis of the observation that we have not observed germline frameshift/nonsense mutations in >500 paired tumor and normal samples. Nonsynonymous mutations were first compared with published SNP data (dbSNP, http://www.ncbi.nlm.nih.gov/projects/SNP), such as previously annotated SNPs, were not considered pathogenic mutations. Missense mutations that were not included in the published SNP database were annotated as somatic mutations on the basis of published data showing that these are somatic mutations25 or sequence analysis of paired tumor/normal MPN/MDS samples that showed that these mutations are present in tumor and not in matched normal DNA. Samples with missense alleles that were neither in dbSNP nor could be shown to be somatic in paired samples were censored from analysis. We, therefore, classify samples as ‘TET2-wt’ if the sample was wild-type in all coding exons of TET2, and as ‘TET2-mutant’ if the sample contained a frameshift, nonsense or proven somatic missense mutation.
The statistical procedures used were conventional, and all data were analyzed by using StatView (SAS Institute, Cary, NC, USA). All _P_-values were two-tailed and statistical significance was set at the level of P<0.05. Continuous variables were summarized as medians and ranges. Categorical variables were described as count and relative frequency (%). Comparison between categorical variables was performed by Chi-squared statistics. Comparison between categorical and continuous variables was performed by either the Mann–Whitney _U_-test or the Kruskal–Wallis test. The association of variables selected from univariate analysis was explored using logistic regression models. Survival curves for patients with and without TET2 mutation were constructed by Kaplan–Meier method taking, the interval from the date of diagnosis to death or last contact, and compared using the Log-rank test.
Results
A total of 239 patients with _BCR-ABL_-negative classic MPN were included in this study: PV (_n_=89), ET (_n_=57), PMF (_n_=60), post-PV MF (_n_=14), post-ET MF (_n_=7) and blast-phase PV/ET/MF (_n_=12). Among the latter were four patients with post-PV AML, four with post-ET AML and four with post-PMF AML. The overall (_n_=239) TET2 mutational frequency was ~13% and included 32 mutations that most frequently involved exons 4 and 12: 19 frameshift, 10 nonsense and 3 missense (Table 1). None of the mutations were associated with a karyotypically apparent 4q24 rearrangement. Disease-specific mutational frequencies were ~16% in PV, ~5% in ET, ~17% in PMF, ~14% in post-PV MF, ~14% in post-ET MF and ~17% in blast-phase PV/ET/PMF (_P_=0.50). All 239 study patients were also screened for _JAK2_V617F; TET2 mutations occurred in 5 (~7%) of 75 _JAK2_V617F-negative versus 27 (~17%) of 164 _JAK2_V617F-positive cases (_P_=0.04). The corresponding figures after excluding chronic and advanced phases of PV (excluded because of their almost invariable association with a JAK2 mutation) were 8 and 15% (_P_=0.17). However, the _JAK2_V617F allele burden did not correlate with the presence of mutant TET2 (_n_=108; _P_=0.95). Information on _MPL_W515L/K status was available in 73 patients, and the five mutation-positive cases as well as two other patients with JAK2 exon 12 mutations did not carry mutant TET2.
Table 1.
TET2 mutation details in 32 patients with myeloproliferative neoplasms
| Exon | Nucleotide change | Consequence | Mutation type | Diagnosis | JAK2V617F mutational status | Age | Sex |
|---|---|---|---|---|---|---|---|
| 4 | 370_delC | Frameshift | Frameshift | PV | Pos. | 69 | F |
| 4 | 3990_4000delAAGTCGTTAT | Frameshift | Frameshift | PV | Pos. | 65 | F |
| 4 | 1726 C>G | S509X | Nonsense | PV | Pos. | 76 | M |
| 4 | 2568 C>T | Q790X | Nonsense | PV | Pos. | 66 | M |
| 4 | 3081_3082insacagcagcaaacacagcaaccccaa | Frameshift | Frameshift | PV | Pos. | 51 | M |
| 4 | 2946 C>T | Q916X | Nonsense | PV | Pos. | 52 | F |
| 8 | 266 C>A | S1291X | Nonsense | PV | Pos. | 60 | M |
| 11 | 411 C>T | R1465X | Nonsense | PV | Pos. | 61 | F |
| 12 | 699_delA | Frameshift | Frameshift | PV | Pos. | 74 | F |
| 12 | 1233_1243delCTGACATTGGG | Frameshift | Frameshift | PV | Pos. | 77 | M |
| 12 | 484_485insT | Frameshift | Frameshift | PV | Pos. | 84 | F |
| 12 | 209 C>T | R1516X | Nonsense | PV | Pos. | 72 | M |
| 12 | 537_538delAG | Frameshift | Frameshift | PV | Pos. | 81 | M |
| 12 | 722 C>T | Q1687X | Nonsense | PV | Pos. | 66 | M |
| 4 | 2036_2037insG | Frameshift | Frameshift | PMF | Pos. | 63 | M |
| 4 | 3509_3510delTT | Frameshift | Frameshift | PMF | Pos. | 58 | M |
| 4 | 1142_1145delTTCC | Frameshift | Frameshift | PMF | Pos. | 78 | M |
| 7 | 331 A>T | D1242 V | Missense | PMF | Neg. | 60 | F |
| 7 | 370_371insA | Frameshift | Frameshift | PMF | Neg. | 64 | M |
| 10 | 231 C>T | R1358C | Missense | PMF | Pos. | 69 | M |
| 11 | 301_delA | Frameshift | Frameshift | PMF | Neg. | 74 | M |
| 12 | 640 T>G | Y1659X | Nonsense | PMF | Neg. | 56 | M |
| 12 | 1423_1424inscatggcttggctctttgggaagccaaa | Frameshift | Frameshift | PMF | Pos. | 60 | F |
| 12 | 272 C>T | Q1537X | Nonsense | PMF | Pos. | 70 | M |
| 4 | 438 C>T | Q80X | Nonsense | ET | Neg. | 80 | M |
| 7 | 384_delA | Frameshift | Frameshift | ET | Pos. | 77 | M |
| 12 | 1044_1048delCTAAT | Frameshift | Frameshift | ET | Pos. | 51 | F |
| 4 | 1171_delA | Frameshift | Frameshift | Post-PV MF | Pos. | 69 | M |
| 12 | 1281 T>C | I1873T | Missense | Post-PV MF | Pos. | 74 | M |
| 4 | 1671_delC | Frameshift | Frameshift | Post-ET MF | Pos. | 66 | M |
| 4 | 1137_1140delACCT | Frameshift | Frameshift | Post-ET AML | Pos. | 68 | M |
| 4 | 2655_2656insA | Frameshift | Frameshift | Post-PV AML | Pos. | 62 | F |
Considering all 239 study patients, both mutant TET2 (P<0.0001) and _JAK2_V617F (_P_=0.008) clustered with older age. This was also true when analysis was restricted to ET, PMF or their advanced stages (_n_=132; _P_=0.001 and 0.05, respectively); as pointed out in the above paragraph, chronic and advanced phases of PV were excluded in the latter analysis because of their almost perfect association with a JAK2 mutation. The association between _JAK2_V617F and older age remained significant or near significant, when _TET2_-mutated patients were excluded from either the overall analysis (_n_=207; _P_=0.03) or the analysis restricted to ET, PMF or their advanced stages (_n_=117; _P_=0.08). However, a multivariable analysis of all 239 study patients, which included mutational status, age and sex as covariates, showed an independent association only between the presence of mutant TET2 and advanced age; mutational frequency was 23% in patients ≥60 years of age versus 4% in younger patients (P<0.0001).
Accurate clinical correlative studies of novel mutations in MPN require separate analysis of patients with ‘chronic phase’ PV, ET or PMF. Accordingly, the clinical features (Table 2) and events presented during the course of disease (Table 3) for PV, ET and PMF were reviewed in the context of their TET2 mutational status. As the number of _TET2_-mutated patients with ET was small (_n_=3), we did not attempt a formal comparison between mutated and unmutated ET cases. However, as is evident from Tables 2 and 3, it is reasonable to mention that the three _TET2_-mutated ET patients were older compared with their unmutated counterparts, and were followed up for 19 to 144 months without fibrotic or leukemic transformation. Table 4 lists the _P_-values during statistical comparison of relevant clinical and laboratory features in PV and PMF, and shows the lack of convincing evidence for specific genotype–phenotype correlates, with the one exception of a significant association between the presence of mutant TET2 and a hemoglobin level of <10 g per 100 ml in PMF. Table 5 provides a comprehensive account of pre- and post-disease transformation details in PV, ET or MF patients who underwent conversion into the fibrotic or leukemic phase of their disease. Once again, there was no overt evidence to suggest that mutant TET2 influenced disease duration before or after fibrotic or leukemic transformation. Finally, Kaplan–Meier survival curves between _TET2_-mutated and -unmutated patients with either PV (Figure 1) or PMF (Figure 2) were not significantly different.
Table 2.
Presenting clinical and laboratory features of 206 _TET2_-mutated or -unmutated patients with PV, ET or PMF
| PV(all patients) | TET2mutated | TET2unmutated | ET(all patients) | TET2mutated | TET2unmutated | PMF(all patients) | TET2mutated | TET2unmutated | |
|---|---|---|---|---|---|---|---|---|---|
| N (%) | 89 (100) | 14 (16) | 75 (84) | 57 (100) | 3 (5) | 54 (95) | 60 (100) | 10 (17) | 50 (83) |
| Males, n (%) | 43 (48) | 9 (64) | 34 (45) | 20 (35) | 2 (67) | 18 (33) | 38 (63) | 8 (80) | 30 (60) |
| Age in years, median (range) | 63 (21–93) | 68 (51–84) | 60 (21–93) | 49 (18–88) | 77 (51–80) | 47 (18–88) | 59 (35–78) | 62 (48–78) | 57 (35–76) |
| Palpable splenomegaly, n (%) | 28 (33) | 5 (36) | 23 (32) | 15 (27) | 0 | 15 (29) | 36 (71) | 5(63) | 31(72) |
| Hemoglobin (g per 100 ml),median (range) | 18.3 (15–23) | 18.6 (16.6–20.5) | 18.2 (15–23) | 14 (10.7–16.4) | 12.6 (12–16.3) | 14 (10.7–16.4) | 11.6 (6.7–14.9) | 10.5 (6.7–12) | 11.6 (8.6–14.9) |
| Leukocyte count (×109/l),median (range) | 11.5 (4.3–120) | 12.3 (4.3–24.8) | 11.3 (4.6–120) | 9.7 (3.4–29.8) | 8 (6–10.9) | 9.7 (3.4–29.8) | 8.3 (1.8–77.1) | 9.3 (8.2–20.2) | 6.7 (1.8–77.1) |
| Monocyte count (×109/l),median (range) | NA | NA | NA | NA | NA | NA | 0.4 (0–3.1) | 0.6 (0–1.6) | 0.4 (0–3.1) |
| Platelet count (×109/l), median(range) | 493 (132–2088) | 499 (160–787) | 486 (132–2088) | 1041 (623–3300) | 983 (840–1000) | 1046 (623–3300) | 246 (12–768) | 211 (81–524) | 248 (12–768) |
| Pruritus, n (%) | 27 (38) | 6 (43) | 21 (36) | NA | NA | NA | NA | NA | NA |
| Arterial thrombosis, n (%) | 7 (8) | 0 | 7 (10) | 11 (20) | 1 (33) | 10 (19) | NA | NA | NA |
| Venous thrombosis, n (%) | 12 (14) | 1 (7) | 11 (15) | 3 (5) | 0 | 3 (6) | NA | NA | NA |
| _JAK2_V617F, n (%) | 80 (90) | 14 (100) | 66 (88) | 26 (46) | 2 (67) | 24 (44) | 33 (55) | 6 (60) | 27 (54) |
Table 3.
Post-diagnosis clinical events in 206 _TET2_-mutated or -unmutated patients with PV, ET or PMF
| PV(all patients) | TET2mutated | TET2unmutated | ET(all patients) | TET2mutated | TET2unmutated | PMF(all patients) | TET2mutated | TET2unmutated | |
|---|---|---|---|---|---|---|---|---|---|
| N (%) | 89 (100) | 14 (16) | 75 (84) | 57 (100) | 3 (5) | 54 (95) | 60 (100) | 10 (17) | 50 (83) |
| Disease duration in months, median (range) | 60 (1–310) | 57 (1–191) | 60 (1–310) | 163 (0.3–398) | 89 (19–144) | 170 (0.3–398) | 48 (5–200) | 26 (6–200) | 55 (5–197) |
| Arterial thrombosis after diagnosis, n (%) | 15 (18) | 1 (7) | 14 (20) | 16 (29) | 2 (67) | 14 (27) | NA | NA | NA |
| Venous thrombosis after diagnosis, n (%) | 9 (11) | 1 (7) | 8 (11) | 7 (13) | 1 (33) | 6 (12) | NA | NA | NA |
| Leukemictransformation, n (%) | 4 (5) | 1 (7) | 3 (4) | 2 (4) | 0 | 2 (4) | 3 (5) | 1 (10) | 2 (4) |
| Fibrotic transformation, n (%) | 6 (7) | 1 (7) | 5 (7) | 3 (5) | 0 | 3 (6) | NA | NA | NA |
| Deaths, n (%) | 19 (21) | 3 (21) | 16 (21) | 13 (23) | 2 (67) | 11 (20) | 29 (48) | 7 (70) | 22 (44) |
Table 4.
_P_-values during univariate analysis correlating the presence of TET2 mutations with clinical and laboratory features at presentation and follow-up
| Polycythemia vera(total n=89; TET2mutated=14) | Primary myelofibrosis(total n=60; TET2mutated=10) | |
|---|---|---|
| Older age | 0.04 | 0.15 |
| Sex | 0.19 | 0.23 |
| _JAK2_V617F mutationalstatus | 0.17 | 0.72 |
| Hemoglobin level | 0.96 (_n_=86) | NA |
| Hemoglobin <10 g per100 ml | NA | 0.05 (_n_=45) |
| Leukocyte count | 0.83 (_n_=85) | 0.16 (_n_=45) |
| Leukocyte count <4 or>30×109/l | NA | 0.27 (_n_=45) |
| Leukocyte count>25×109/l | NA | 0.53 (_n_=45) |
| Platelet count | 0.79 (_n_=86) | 0.89 (_n_=45) |
| Platelet count<100×109/l | NA | 0.64 (_n_=45) |
| Monocyte count<1×109/l | NA | 0.64 (_n_=45) |
| Circulating blast count≥1% | NA | 0.83 (_n_=45) |
| Hypercatabolicsymptoms | NA | 0.56 (_n_=45) |
| Spleen size | 0.78 (_n_=86) | 0.86 (_n_=45) |
| Pruritus | 0.64 (_n_=72) | NA |
| Arterial thrombosis atdiagnosis | 0.23 (_n_=87) | NA |
| Arterial thrombosis atfollow-up | 0.25 (_n_=84) | NA |
| Venous thrombosis atdiagnosis | 0.43 (_n_=87) | NA |
| Venous thrombosis atfollow-up | 0.63 (_n_=84) | NA |
| Leukemic transformation | 0.61 | 0.43 |
| Fibrotic transformation | 0.79 | NA |
| Follow-up period | 0.82 | 0.08 |
| Survival | 0.86 | 0.16 |
Table 5.
Clinical features of 33 _TET2_-mutated or -unmutated patients with post-PV MF, post-ET MF or post-PV/ET/MF AML
| Post-PVMF | TET2mutated | TET2unmutated | Post-ET MF | TET2mutated | TET2unmutated | Post-PV/ET/PMF AML | TET2mutated | TET2unmutated | |
|---|---|---|---|---|---|---|---|---|---|
| N (%) | 14 (100) | 2 (14) | 12 (86) | 7 (100) | 1 (14) | 6 (86) | 12 (100) | 2 (17) | 10 (83) |
| Males, n (%) | 9 (64) | 2 (100) | 7 (58) | 6 (86) | 1 (100) | 5 (83) | 10 (83) | 1 (50) | 9 (90) |
| Age in years, median (range) | 64.5 (43–74) | 71.5 (69–74) | 59.5 (43–72) | 65 (53–73) | 68 | 64 (53–73) | 64 (53–81) | 65 (62–68) | 64 (53–81) |
| Post-PV AML, n (%) | NA | NA | NA | NA | NA | NA | 4 (33) | 1 (25) | 3 (75) |
| Post-ET AML, n (%) | NA | NA | NA | NA | NA | NA | 4 (33) | 1 (25) | 3 (75) |
| Post-PMF AML, n (%) | NA | NA | NA | NA | NA | NA | 4 (33) | 0 | 4 (100) |
| _JAK2_V617F, n (%) | 14 (100) | 2 (100) | 12 (100) | 4 (57) | 1 (100) | 3 (50) | 7 (58) | 2 (100) | 5 (50) |
| Deaths, n (%) | 9 (64) | 2 (100) | 7 (58) | 6 (86) | 1 (100) | 5 (83) | 12 (100) | 2 (100) | 10 (100) |
| Pre-transformation disease duration inmonths, median (range) | 126 (25–294) | 61 (30–92) | 138 (25–294) | 84 (54–168) | 84 | 101.1 (54–168) | 92.8 (13–217) | 103.8 (62–145) | 92.8 (13–217) |
| Post-transformation disease duration inmonths, median (range) | 41 (4–249) | 20 (4–35) | 51 (20–249) | 29 (20–156) | 29 | 46 (20–156) | 4 (1–11) | 7 (6–9) | 4 (1–11) |
Figure 1.
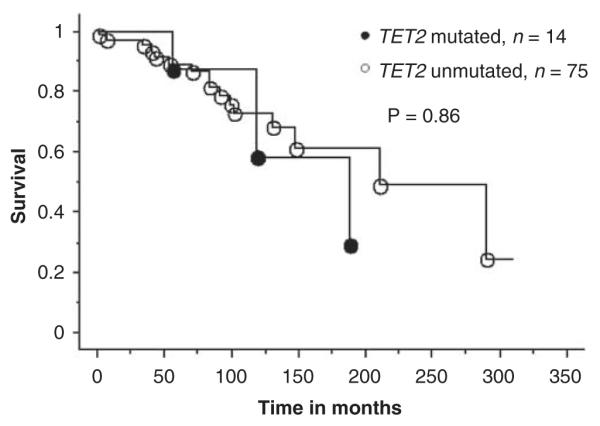
Survival curves for 89 patients with polycythemia vera stratified by their TET2 mutational status (14 mutated and 75 unmutated patients).
Figure 2.
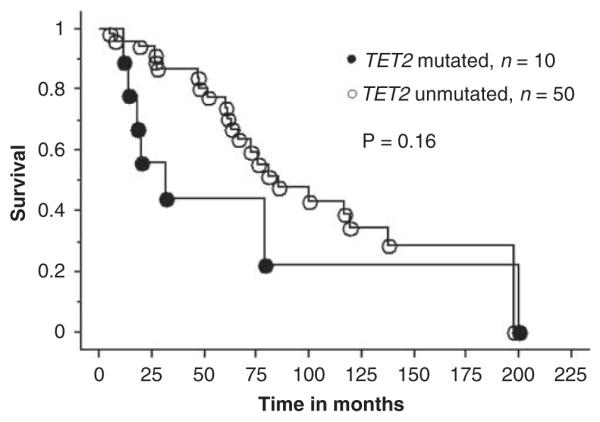
Survival curves for 60 patients with primary myelofibrosis stratified by their TET2 mutational status (10 mutated and 50 unmutated patients).
Discussion
The TET family of proteins share highly conserved regions, but their biological function is not known. TET1 is short for ‘ten–eleven translocation 1′ and was the name given to a novel gene located at chromosome 10q22, which was identified as the fusion partner of MLL during an AML-associated chromosomal translocation, t(10;11)(q22;q23).32 Subsequent genomic database exploration identified two homologous human proteins that were accordingly designated as TET2 and TET3. It is interesting that TET2 is located at chromosome 4q24, which is a breakpoint that is also involved in other AML-associated translocations: t(3;4)(q26;q24), t(4;5)(q24;p16), t(4;7)(q24;q21) and del(4)(q23q24). In 2005, Viguie et al.33 suggested that these 4q24 rearrangements occurred in multipotent myeloid–lymphoid progenitor cells, and by using BAC clones and fluorescent in situ hybridization, were able to show a 0.5-Mb commonly deleted region that accompanied the 4q24 breakpoint.33 In 2008, Delhommeau et al.25 reported acquired LOH/deletions at chromosome 4q24 in patients with MPN, and identified TET2 as the gene of interest contained within the minimally deleted/homozygosed region.
TET2 has multiple isoforms, including TET2 isoform A (NM 001127208–2002 amino acids) and TET2 isoform B (NM 017628–1165 amino acids); notably, many of the somatic mutations identified to date are observed in coding sequence specific to isoform A. TET2 isoform A spans ~133 kb, including 12 exons, and is predicted to encode a protein of 2002 amino acids. At the time of this study, the function of TET2 in normal and malignant hematopoiesis has not been fully elucidated; however, the enhanced engraftment of _TET2_-mutant MPN patient cells in NOD–SCID mice suggests that TET2 might be involved in self-renewal pathways relevant to hematopoietic transformation.25 Delhommeau et al.25 subsequently sequenced TET2 in 181 _JAK2_V617F-positive patients with PV, ET and PMF, and discovered frameshift, nonsense and missense mutations in ~14% of the patients.25
The TET2 mutational frequency observed in this study (~17%) among _JAK2_V617F-positve MPNs is remarkably similar to that reported by Delhommeau et al.25 In addition, our study shows the occurrence of the mutation in _JAK2_V617F-negative MPN, perhaps at a lesser frequency (~7%). This apparent segregation of mutant TET2 with _JAK2_V617F is similar to the association between mutant TET2 and _KIT_D816 V, which was seen in our recent report involving SM patients.26 However, the latter observation is confounded by the fact that _KIT_D816V is present in more than 90% of patients with SM and that _KIT_D816V negativity in our above-cited report probably resulted from the use of inadequately sensitive mutation screening assay (PCR sequencing), and therefore represented a low mutant allele burden rather than a true absence of the mutation.34 Similarly, in this study, multivariable analysis revealed that the association between mutant TET2 and _JAK2_V617F was fully accounted for by a strong and independent association between the former and advanced age. In this regard, it is to be recalled that both _JAK2_V617F and mutant _MPL_515 are also more prevalent in older patients with MPN.22,30,35,36 Furthermore, this study showed that the higher prevalence of _JAK2_V617F in older patients was not accounted for by the presence of mutant TET2. Collectively, these observations support the concept of ‘genetic instability’ in MPN, and underscore the importance of accounting for age during clinical correlative studies concerning MPN-associated mutations. Whether or not differences in the mutational profile of young and older patients with MPN will translate into varied responses during targeted therapy remains to be seen.
In this study, TET2 mutational frequencies were not significantly different across MPN subcategories or specific disease stages. In this regard, as discussed above, the apparently lower incidence of TET2 mutation in ET patients (~5%) may have been related to their younger age range (vide supra). Furthermore, although the numbers involved were too low to allow making statistically valid conclusions, it is reasonable to mention that the pre- and post-disease transformation time intervals among informative patients with ET, PV or PMF were not overtly different to suggest an influence from mutant TET2 (Table 5). These observations lessen the possibility that mutant TET2 emerges or disappears over time and, coupled with the absence of significant correlations between the presence of mutant TET2 and survival or leukemic transformation, at least in PV and PMF, undermine the potential value of the mutation as a prognostic tool. These data also suggest the possibility that there are additional genetic or epigenetic events that target TET2 or other genes in related pathways in TET2 wild-type patients.
There were no evident mutational hotspots detected either in this study or in our earlier report on SM.26 The TET2 mutations were spread over several exons and regions of the gene, and most resulted in either frameshift or stop codon alterations. Such changes usually result in truncated translation, and therefore inadequate production of a potentially tumor suppressor protein. Such a scenario would be consistent with the aforementioned observations from Delhommeau et al.,25 which indicated, in a subset of MPN/MDS patients, biallelic inactivation of TET2 observed because of biallelic mutations, deletion of the remaining wild-type copy or somatic isodisomy for mutant TET2. When we compared the results from our two successive TET2 mutation studies involving PV/ET/MF and SM patients, respectively, we noticed that none of the former, but 5 of the 12 _TET2_-mutated SM patients displayed more than one TET2 mutation.26 Multiple TET2 mutations in the same patient were also infrequent in our other patients with chronic myelomonocytic leukemia, myelodysplastic syndrome or acute leukemia.27 These observations need to be extended through an analysis of copy number and zygosity using SNP arrays to assess the relative frequency of homozygous TET2 inactivation because of mutation, deletion and LOH in the different myeloid malignancies. If this observation was to be validated by SNP array analysis and through an analysis of additional patient cohorts, it would suggest that biallelic inactivation of TET2 is more common in some diseases, such as SM, than in others, such as _JAK2_V617F-positive MPN.
Unlike the case with _JAK2_V617F,37 the presence of mutant TET2 did not appear to significantly influence leukocyte or platelet count in patients with PV or PMF. Hemoglobin level in PV or _JAK2_V617F allele burden in mutation-positive MPN patients was also not affected. Similarly, unlike the case with SM,26 the presence of mutant TET2 in PMF did not correlate with monocytosis. At the same time, the association between mutant TET2 and anemia in PMF did not escape our attention. However, because of the relatively small number of patients studied, we prefer to postpone additional comments regarding genotype–phenotype correlates, and instead recommend that these data be treated as preliminary. For now, we can reasonably conclude that TET2 mutations are even less specific than _JAK2_V617F in the context of myeloid malignancies, occur in the presence or absence of other MPN relevant mutations and are more likely to be detected in older patients. Larger studies including an adequate number of _TET2_-mutated patients are needed to properly assess not only the prognostic relevance of mutant TET2 by itself, but also its potential prognostic interaction with other MPN-associated molecular markers. In the latter, it is interesting to glean from Table 5 that all _TET2_-mutated patients who have undergone fibrotic or leukemic transformation also carried _JAK2_V617F.
Acknowledgements
Adriana Heguy and Igor Dolgalev from Memorial Sloan-Kettering Cancer Center, New York, NY, USA are acknowledged for their assistance with sequence analysis. This study was supported in part by grants from the Myeloproliferative Disorders Foundation, Chicago, IL, USA. DGG is an investigator of the Howard Hughes Medical Institute and is a Doris Duke Charitable Foundation Distinguished Clinical Scientist. RLL is an Early Career Award recipient of the Howard Hughes Medical Institute, a Clinical Scientist Development Award recipient of the Doris Duke Charitable Foundation and is the Geoffrey Beene Junior Chair at Memorial Sloan Kettering Cancer Center.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 2.James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 3.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 4.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 5.Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3:e270. doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356:459–468. doi: 10.1056/NEJMoa065202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108:3472–3476. doi: 10.1182/blood-2006-04-018879. [DOI] [PubMed] [Google Scholar]
- 8.Pardanani A, Lasho TL, Finke C, Hanson CA, Tefferi A. Prevalence and clinicopathologic correlates of JAK2 exon 12 mutations in JAK2V617F-negative polycythemia vera. Leukemia. 2007;21:1960–1963. doi: 10.1038/sj.leu.2404810. [DOI] [PubMed] [Google Scholar]
- 9.Pietra D, Li S, Brisci A, Passamonti F, Rumi E, Theocharides A, et al. Somatic mutations of JAK2 exon 12 in patients with JAK2 (V617F)-negative myeloproliferative disorders. Blood. 2008;111:1686–1689. doi: 10.1182/blood-2007-07-101576. [DOI] [PubMed] [Google Scholar]
- 10.Wernig G, Mercher T, Okabe R, Levine RL, Lee BH, Gilliland DG. Expression of Jak2V617F causes a polycythemia vera-like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood. 2006;107:4274–4281. doi: 10.1182/blood-2005-12-4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tiedt R, Hao-Shen H, Sobas MA, Looser R, Dirnhofer S, Schwaller J, et al. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood. 2008;111:3931–3940. doi: 10.1182/blood-2007-08-107748. [DOI] [PubMed] [Google Scholar]
- 12.Shide K, Shimoda HK, Kumano T, Karube K, Kameda T, Takenaka K, et al. Development of ET, primary myelofibrosis and PV in mice expressing JAK2 V617F. Leukemia. 2008;22:87–95. doi: 10.1038/sj.leu.2405043. [DOI] [PubMed] [Google Scholar]
- 13.Lacout C, Pisani DF, Tulliez M, Gachelin FM, Vainchenker W, Villeval JL. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood. 2006;108:1652–1660. doi: 10.1182/blood-2006-02-002030. [DOI] [PubMed] [Google Scholar]
- 14.Zaleskas VM, Krause DS, Lazarides K, Patel N, Hu Y, Li S, et al. Molecular pathogenesis and therapy of polycythemia induced in mice by JAK2 V617F. PLoS ONE. 2006;1:e18. doi: 10.1371/journal.pone.0000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steensma DP, Dewald GW, Lasho TL, Powell HL, McClure RF, Levine RL, et al. The JAK2 V617F activating tyrosine kinase mutation is an infrequent event in both ‘atypical’ myeloproliferative disorders and myelodysplastic syndromes. Blood. 2005;106:1207–1209. doi: 10.1182/blood-2005-03-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vizmanos JL, Ormazabal C, Larrayoz MJ, Cross NC, Calasanz MJ. JAK2 V617F mutation in classic chronic myeloproliferative diseases: a report on a series of 349 patients. Leukemia. 2006;20:534–535. doi: 10.1038/sj.leu.2404086. [DOI] [PubMed] [Google Scholar]
- 17.Jones AV, Kreil S, Zoi K, Waghorn K, Curtis C, Zhang L, et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood. 2005;106:2162–2168. doi: 10.1182/blood-2005-03-1320. [DOI] [PubMed] [Google Scholar]
- 18.Pardanani A, Fridley BL, Lasho TL, Gilliland DG, Tefferi A. Host genetic variation contributes to phenotypic diversity in myeloproliferative disorders. Blood. 2008;111:2785–2789. doi: 10.1182/blood-2007-06-095703. [DOI] [PubMed] [Google Scholar]
- 19.Lasho TL, Pardanani A, McClure RF, Mesa RA, Levine RL, Gary Gilliland D, et al. Concurrent MPL515 and JAK2V617F mutations in myelofibrosis: chronology of clonal emergence and changes in mutant allele burden over time. Br J Haematol. 2006;135:683–687. doi: 10.1111/j.1365-2141.2006.06348.x. [DOI] [PubMed] [Google Scholar]
- 20.Kralovics R, Teo SS, Li S, Theocharides A, Buser AS, Tichelli A, et al. Acquisition of the V617F mutation of JAK2 is a late genetic event in a subset of patients with myeloproliferative disorders. Blood. 2006;108:1377–1380. doi: 10.1182/blood-2005-11-009605. [DOI] [PubMed] [Google Scholar]
- 21.Pardanani A, Lasho TL, Finke C, Mesa RA, Hogan WJ, Ketterling RP, et al. Extending Jak2V617F and MplW515 mutation analysis to single hematopoietic colonies and B and T lymphocytes. Stem Cells. 2007;25:2358–2362. doi: 10.1634/stemcells.2007-0175. [DOI] [PubMed] [Google Scholar]
- 22.Vannucchi AM, Antonioli E, Guglielmelli P, Pancrazzi A, Guerini V, Barosi G, et al. Characteristics and clinical correlates of MPL 515W>L/K mutation in essential thrombocythemia. Blood. 2008;112:844–847. doi: 10.1182/blood-2008-01-135897. [DOI] [PubMed] [Google Scholar]
- 23.Levine RL, Belisle C, Wadleigh M, Zahrieh D, Lee S, Chagnon P, et al. X-inactivation-based clonality analysis and quantitative JAK2V617F assessment reveal a strong association between clonality and JAK2V617F in PV but not ET/MMM, and identifies a subset of JAK2V617F-negative ET and MMM patients with clonal hematopoiesis. Blood. 2006;107:4139–4141. doi: 10.1182/blood-2005-09-3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lippert E, Boissinot M, Kralovics R, Girodon F, Dobo I, Praloran V, et al. The JAK2-V617F mutation is frequently present at diagnosis in patients with essential thrombocythemia and polycythemia vera. Blood. 2006;108:1865–1867. doi: 10.1182/blood-2006-01-013540. [DOI] [PubMed] [Google Scholar]
- 25.Delhommeau F, Dupont S, James C, Masse A, le Couedic JP, Valle VD, et al. TET2 is a novel tumor suppressor gene inactivated in myeloproliferative neoplasms: identification of a pre-JAK2 V617F event. ASH Annu Meet Abstr. 2008;112:lba–lb3. Late-breaking abstract. [Google Scholar]
- 26.Tefferi A, Levine RL, Lim K-H, Abdel-Wahab O, Lasho TL, Patel J, et al. Frequent TET2 mutations in systemic mastocytosis: Clinical, KITD816V and FIP1L1-PDGFRA correlates. Leukemia. 2009 doi: 10.1038/leu.2009.37. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tefferi A, Lim K-H, Abdel-Wahab O, Lasho TL, Patel J, Patnaik MM, et al. Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia. 2009 doi: 10.1038/leu.2009.59. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaffe ES, Harris NL, Stein H, Vardiman JW. World Health Organization Classification of Tumours of Hematopoietic and Lymphoid Tissues. IARC Press; Lyon, France: 2001. pp. 1–351. [Google Scholar]
- 29.Pardanani A, Reeder TL, Kimlinger TK, Baek JY, Li CY, Butterfield JH, et al. Flt-3 and c-kit mutation studies in a spectrum of chronic myeloid disorders including systemic mast cell disease. Leuk Res. 2003;27:739–742. doi: 10.1016/s0145-2126(02)00303-x. [DOI] [PubMed] [Google Scholar]
- 30.Tefferi A, Lasho TL, Huang J, Finke C, Mesa RA, Li CY, et al. Low JAK2V617F allele burden in primary myelofibrosis, compared to either a higher allele burden or unmutated status, is associated with inferior overall and leukemia-free survival. Leukemia. 2008;22:756–761. doi: 10.1038/sj.leu.2405097. [DOI] [PubMed] [Google Scholar]
- 31.Loriaux MM, Levine RL, Tyner JW, Frohling S, Scholl C, Stoffregen EP, et al. High-throughput sequence analysis of the tyrosine kinome in acute myeloid leukemia. Blood. 2008;111:4788–4796. doi: 10.1182/blood-2007-07-101394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lorsbach RB, Moore J, Mathew S, Raimondi SC, Mukatira ST, Downing JR. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23) Leukemia. 2003;17:637–641. doi: 10.1038/sj.leu.2402834. [DOI] [PubMed] [Google Scholar]
- 33.Viguie F, Aboura A, Bouscary D, Ramond S, Delmer A, Tachdjian G, et al. Common 4q24 deletion in four cases of hematopoietic malignancy: early stem cell involvement? Leukemia. 2005;19:1411–1415. doi: 10.1038/sj.leu.2403818. [DOI] [PubMed] [Google Scholar]
- 34.Garcia-Montero AC, Jara-Acevedo M, Teodosio C, Sanchez ML, Nunez R, Prados A, et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood. 2006;108:2366–2372. doi: 10.1182/blood-2006-04-015545. [DOI] [PubMed] [Google Scholar]
- 35.Tefferi A, Lasho TL, Schwager SM, Steensma DP, Mesa RA, Li CY, et al. The JAK2 tyrosine kinase mutation in myelofibrosis with myeloid metaplasia: lineage specificity and clinical correlates. Br J Haematol. 2005;131:320–328. doi: 10.1111/j.1365-2141.2005.05776.x. [DOI] [PubMed] [Google Scholar]
- 36.Wolanskyj AP, Lasho TL, Schwager SM, McClure RF, Wadleigh M, Lee SJ, et al. JAK2 mutation in essential thrombocythaemia: clinical associations and long-term prognostic relevance. Br J Haematol. 2005;131:208–213. doi: 10.1111/j.1365-2141.2005.05764.x. [DOI] [PubMed] [Google Scholar]
- 37.Vannucchi AM, Antonioli E, Guglielmelli P, Pardanani A, Tefferi A. Clinical correlates of JAK2V617F presence or allele burden in myeloproliferative neoplasms: a critical reappraisal. Leukemia. 2008;22:1299–1307. doi: 10.1038/leu.2008.113. [DOI] [PubMed] [Google Scholar]