The Vps33a gene regulates behavior and cerebellar Purkinje cell number (original) (raw)
. Author manuscript; available in PMC: 2015 Dec 14.
Abstract
A mutation in the Vps33a gene causes Hermansky–Pudlak Syndrome (HPS)-like-symptoms in the buff (bf) mouse mutant. The encoded product, Vps33a, is a member of the Sec1 and Class C multi-protein complex that regulates vesicle trafficking to specialized lysosome-related organelles. As Sec1 signaling pathways have been implicated in pre-synaptic function, we examined brain size, cerebellar cell number and the behavioral phenotype of bf mutants. Standardized behavioral tests (SHIRPA protocols) demonstrated significant motor deficits (e.g., grip strength, righting reflex and touch escape) in bf mutants, worsening with age. Histological examination of brain revealed significant Purkinje cell loss that was confirmed with staining for calbindin, a calcium binding protein enriched in Purkinje cells. This pathologic finding was progressive, as older bf mutants (13–14 months) showed a greater attrition of neurons, with their cerebella appearing to be particularly reduced (~30%) in size relative to those of age-matched-control cohorts. These studies suggest that loss of Purkinje neurons is the most obvious neurological atrophy in the bf mutant, a structural change that generates motor coordination deficits and impaired postural phenotypes. It is conceivable therefore that death of cerebellar cells may alsobea clinical feature of HPS patients, a pathological event which has not been reported in the literature. In general, the bf mutant may be a potentially new and useful model for understanding Purkinje cell development and function.
Keywords: Vps 33a, Hermansky-Pudlak syndrome, Purkinje cell, Cerebellum, GFAP, SHIRPA
1. Introduction
Hermansky–Pudlak Syndrome (HPS) is a genetically heterogeneous disease with a wide range of physiological abnormalities, including loss of visual acuity, prolonged bleeding times and early death caused by fibrotic lung disease. The clinical signs of HPS are caused by defects in the biogenesis of specialized lysosome-related organelles including melanosomes, lysosomes and platelet dense granules (Li et al., 2004). The bf mutant models the pathophysiology of HPS, and by positional cloning of the bf gene, a missense substitution of the Vps33a allele gives rise to the bf phenotype (Suzuki et al., 2003). Vps33a is a member of the Sec1 and Class C multi-protein complex whose correct assembly could be required for the normal function of intracellular protein trafficking from the Golgi to the vacuole (Robinson et al., 1988; Wada et al., 1990). In the brain, loss of Sec1 function results in the reduction of synaptic response to light and impairs synaptic vesicle trafficking at multiple steps, ultimately affecting neurotransmitter release (Halachmi and Lev, 1996). All of these data suggest an important and direct role for Sec1 in pre-synaptic function, with an emphasis on synaptic vesicle endocytosis.
The mammalian cerebellum is primarily concerned with postural control and motor coordination. The most prominent and distinctive neuron of the cerebellar cortex is the Purkinje cell whose axons project to deep cerebellar nuclei and to several extra-cerebellar structures, particularly the vestibular nuclei (e.g., cerebellovestibular fibers). Consequently, Purkinje neurite loss is associated with ataxia (abnormal posture and gait phenotypes) (Oppenheimer, 1984; Walton, 1985). There are a number of mouse mutants with Purkinje neuropathies, ataxia and tremors (Caddy and Biscoe, 1979; Caddy and Sidman, 1981; Heckroth et al., 1989; Herrup and Trenkner, 1987; Mullen et al., 1976; Rizai, 1972; Sonmez and Herrup, 1984; Wassef et al., 1987; Yuasa et al., 1993), all of which have provided insights into Purkinje cell development and function. Vps54, a member of the Vps family mutated in the wobbler (wr/wr) mouse, severely compromises motoneuron survival (Schmitt-John et al., 2005) and causes abnormal motility on a wire grid, weight loss and low grip strength. As the Vps33a mouse models HPS, we have characterized this mutant further in terms of its neurological features and have determined whether postural control and motor coordination abnormalities are observable phenotypes in the mutant.
2. Results
2.1. General behavioral deficits in bf mice
A series of experiments were performed to test the general behavior of bf mutants compared with wild-type and heterozygous control animals. We performed 35 different tests outlined in SHIRPA protocols (Rogers et al., 1997) and 4 standard tests for examination of motor functions. As illustrated in Fig. 1A, young bf mice (8–13 weeks) exhibited significant impairments in several behavioral tests. In the touch escape test for instance, C57/BL6 controls reached 100% success rate while bf mutants reached only 50% success rate. This defect was augmented in older mice confirming a progressive impairment of motor functions with age (Fig. 1B). In the righting reflex test, the success rate was 100% for the wild-type control mice, whereas the success rate for the bf mice was only 58%. The righting reflex defect was also more severely accentuated with age as none of the older bf mutant mice reached the success rate criterion (Fig. 1B). Likewise, in the grip strength test, none of the bf mutants scored whether young or old. We also noticed other apparent deficits in the wire suspension, rotating cylinder and ladder tests in young bf mice, but these did not reach statistical significance (Fig. 1A). In sharp contrast, older bf mice showed severe defects in the aforementioned tests. Of potential interest, old heterozygous bf/+ mice (13–14 months) displayed significant deficits in some of the SHIRPA behavioral protocols indicating that possessing a single mutated allele of the Vps33a gene may predispose animals to subtle behavioral abnormalities during the aging process. However, this conclusion is tentative as aged C57/Bl6+/+ controls were not tested.
Fig. 1.
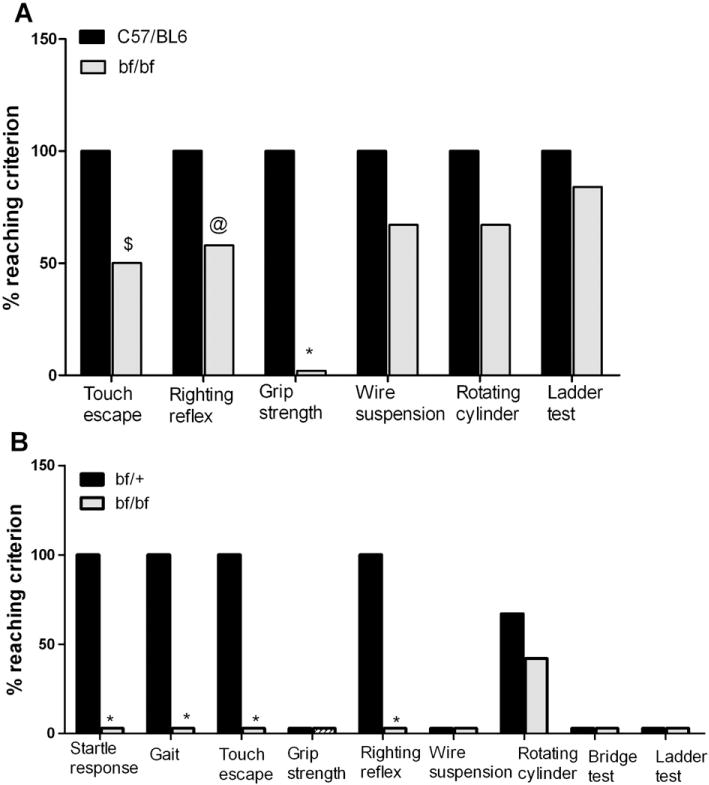
SHIRPA behavior tests. (A) Young (8–13 weeks) bf and C57/BL6 controls were tested (12 mice in each group). (B) Old (13–14 months) bf mice and their heterozygous controls were tested (12 mice each). The defect in startle response was found only in old bf mice. Results are presented only for those SHIRPA tests in which significances were noted. Twenty five other SHIRPA tests in which no significant differences were found are not presented. $_P_≤0.01; @_P_≤0.03; *_P_≤0.001.
To determine if there are defects in auditory capability, the startle response test (SHIRPA protocols) was performed. The startle response was severely deficient only in older bf mutants (Fig. 1B). Young bf mice showed a grossly normal auditory startle response (data not shown), whereas older bf mice showed a diminished or absent startle response. Likewise, there was no walking (gait) defect in the young mutant mice (data not shown), but 13–14 months bf mice were severely affected in gait (Fig. 1B). Heterozygous control mice scored 100% whereas the bf mutant mice did not score in this test. Abnormal gait was observed in all old bf mice. Their back legs were severely affected resulting in a characteristic wobbling behavior (see video Supplement 1) further confirming the motor impairments in bf mice.
2.2. Curving behavior of bf mice
Video capture of body movements and analysis of locomotor activity of young bf mice revealed a rotational asymmetry in activity and body posture (Fig. 2). In contrast, age-matched control C57/BL6 mice showed normal body movement and locomotor activity with little or no rotational asymmetry. This difference in postural symmetry suggests a subtle, but pathological motor deficit in bf mutants.
Fig. 2.
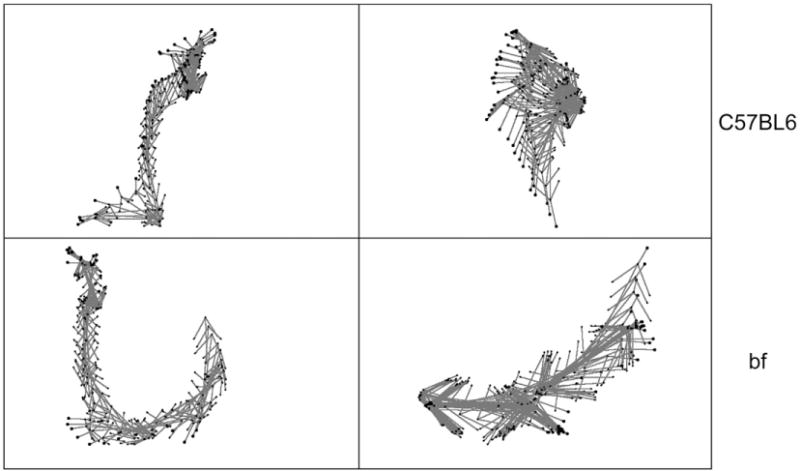
Analysis of motion and postural symmetry. Video capture of body movements reveals asymmetry (or curving) behavior of bf (lower panel) compared with movement of wild-type C57BL6 mice (top panel). Two animals were videotaped, and subtle body movements and motion analysis were processed as described in methods.
2.3. Cerebellar defects in bf mice
General examination of brain sections of young bf mice did not reveal any size difference in the cerebellum (Fig. 3A panel 1). In contrast old bf mice (calbindin stain) revealed an apparent reduction of cerebellar size compared to their age-matched heterozygous controls (Fig. 3A panels 2 and 3). Though cerebellum size is reduced in the bf mutant, foliation is not disturbed. This initial finding of decreased cerebellum size was confirmed by non-invasive MR imaging. MR imaging of the brain revealed no abnormality in size of the cerebellum relative to the total brain volume in young bf mice (Table 1). However, in aged mutant mice the size of the cerebellum was clearly reduced. There was a significant (20%) reduction of cerebellar volume at the age of 8 months in bf mice. The reduction progressed to 30% at 13–14 months (Table 1). MR imaging and 3D rendering of old bf brains clearly shows that the relative volume of cerebellum was reduced (Fig. 4). There is however no significant difference in the total brain volume in bf mice compared with their age-matched controls, and bf mice were not smaller in body size than wild-type mice. The size of total brain and cerebellum was likewise measured in sections (Fig. 3B) with Image J software National Institutes of Health (NIH) and statistical significance was determined using the GraphPad Prism 5.0 software, Inc., San Diego, CA). While a small (12.5%) nominal decrease in mutant total brain size was noted, this was not significantly different from wild-type. In contrast there was a relatively large (31.4%) decrease in size of mutant cerebella, consistent with the MR data.
Fig. 3.
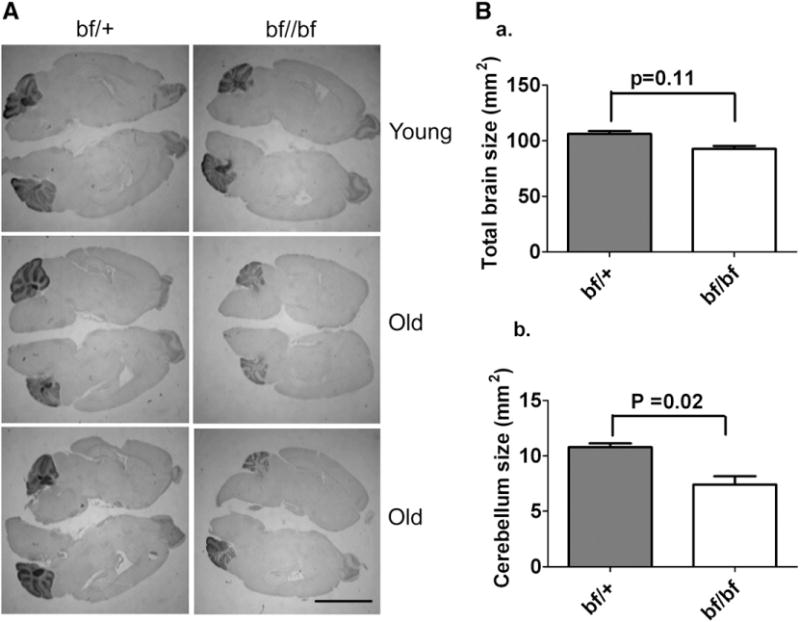
(A) Effect of the Vps33a mutation on the cerebellum. Immunocytochemical stain (calbindin) of brain sections from young and old bf mice. Specific calbindin stain highlighting cerebellar regions indicates a smaller cerebellum in old bf mutants (panels 2 and 3) compared with heterozygous controls. There is no difference in the size of cerebellum in young bf mice (panel 1). Hematoxylin and eosin was used as counter stain. The pictures are representative of three different animals. Scale bar, 5 μm. (B) Measurement of total brain and cerebellum size by Image J software: There is a nominal 12.5% reduction in the total brain size in bf mutants (92.9 mm2) but this is not significantly different (_P_=0.11) from bf/+ controls (106 mm2) (a). The cerebellum size (bf/+ 10.8 mm2, bf 7.40 mm2) in bf mutants is significantly (_P_=0.02) reduced (31.4%) compared to their controls (b). 3 wild-type and 3 mutant mice were used for determining sizes. Similar results (i.e. a selective reduction in size of mutant cerebella) were obtained when sections were stained with cresyl violet (not shown).
Table 1.
Cerebellum volume at different ages in bf mutants
| Age | (Volume mm3) | |||
|---|---|---|---|---|
| Cerebellum | Total brain | |||
| C57/BL6 (or) bf/+ | bf/bf | C57/BL6 (or) bf/+ | bf/bf | |
| 11–13 weeks | 43±2 | 39±3 | 466±3 | 469±14 |
| 8 months | 48±2 | 38±2* | 478±7 | 480±10 |
| >1 year | 45±3 | 32±3* | 470±6 | 460±5 |
Fig. 4.
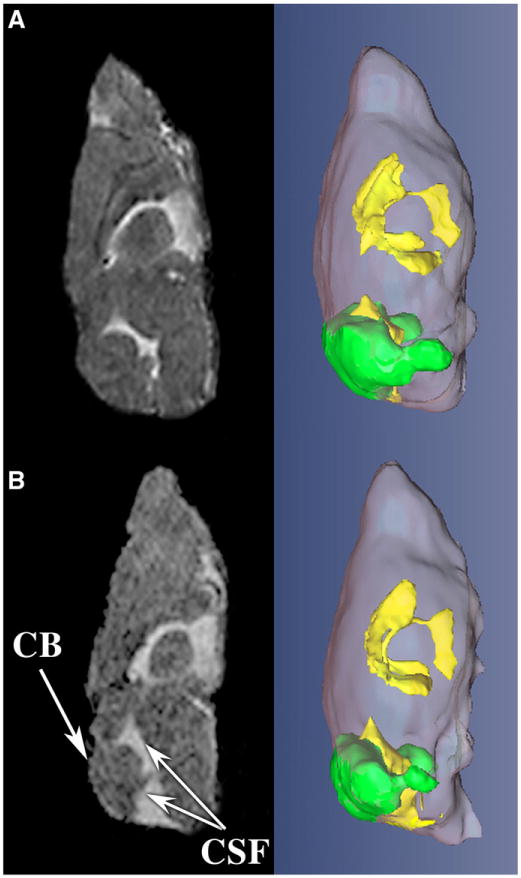
MRI analysis illustrates abnormal cerebellar volume in bf. A representative, central slice of MR imaging (left) and 3D renderings (right) show decrease in cerebellum volume (CB, green) and accumulation of more cerebral spinal fluid (CSF, yellow) in the bf mouse (panel B) as compared to control (panel A).
2.4. Immunohistochemical analysis of bf Purkinje cells
To investigate the possibility of specific cell loss, brain sections of young and old bf mice were immunostained with anti-calbindin, which highlights cerebellar Purkinje cells. In young bf cerebellum there is no significant loss of Purkinje cells (data not shown). When individual Purkinje cells (Fig. 5A) were closely enumerated, there was a 70% loss of Purkinje cells in old mutants compared to their controls (Table 2). The loss of Purkinje cells was apparent in all lobules of the cerebellum and there is no consistency in the loss of these cells in the lobes affected (Fig. 5A). The decrease in number of Purkinje cells of bf mice was also observed by Nissl and hematoxylin and eosin stains (not shown). The molecular layer (brown) and granular layer (blue) (Fig. 5B) thickness of the cerebellum of old bf mutants measured with Image J (NIH) software revealed that the thickness of both layers was reduced in the mutants. There was a significant 50% reduction in the molecular layer (bf/+ 38.3±4.9 μm; bf 18.9±3.4 μm) and 40% reduction in the granular layer (bf/+ 56.6±6.1 μm; bf 33.1±9.2 μm) compared to their age-matched controls.
Fig. 5.
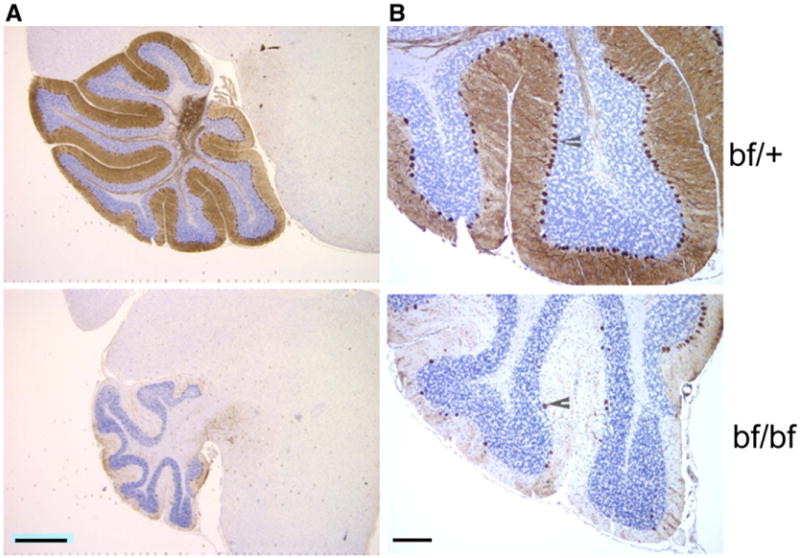
Purkinje cell loss in old bf. (A) Brain sections from old (13–14 months) bf/bf and bf/+ controls were stained with calbindin to clearly show the cerebellum. Scale bar 2 μm. (B) Higher magnification specifically highlights Purkinje cells. Arrowheads indicate Purkinje cells in the cerebellum. Three different animals were used for the study, and from each animal at least 2 different brain sections were used. Scale bar 40 μm.
Table 2.
Purkinje cells in representative brain sections stained with calbindin
| Number of Purkinje cells in cerebellum | ||
|---|---|---|
| Young | Old | |
| C57/BL6 or bf/+ | 738±35 | 875±29 |
| bf/bf | 689±31 | 270±37* |
2.5. Analysis of Glial Fibrillar Acidic Protein (GFAP) in the cerebellum
The most obvious observation in the cerebellum was loss of Purkinje cells in older bf mutant mice. In addition GFAP was slightly up-regulated in the young bf cerebellum (data not shown). This up-regulation of GFAP expression was more pronounced in bf cerebellum (Fig. 6A) at 13–14 months. Western blot analysis of cerebellar protein extracts confirmed this up-regulation in old bf mutants (Fig. 6B). This indicates that glial cells, including astrocytes in which GFAP is predominantly expressed, might be activated. GFAP was up-regulated in brain stem and deep cerebellar nuclei of bf mice where calbindin-immunopositive Purkinje cell axons terminated. The up-regulation of GFAP suggests that the cells are activated because of neuronal degeneration.
Fig. 6.
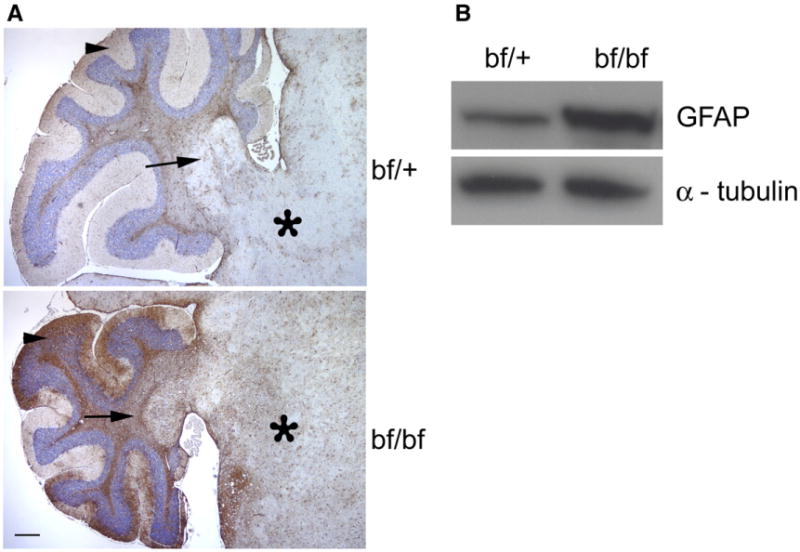
Up-regulation of GFAP in bf. (A) Brain sections of old (13–14 months) bf and controls were immunostained with GFAP antibody. GFAP over expression was observed in cerebellum in bf mice compared to their controls in the regions indicated as arrow head (molecular layer), arrow (deep nuclei) and asterisk (Brain stem). Pictures are representative of three different animals. Scale bar, 100 μm. (B) Western blot analysis of old bf brain cerebellum extracts. The up-regulation of GFAP is apparent as a more intense signal with bf mutant compared to age-matched control. α-tubulin was used as loading control.
3. Discussion
The present study was initiated to investigate whether Vps33a contributes to cerebellar functions. An abnormal behavioral phenotype was noticed in bf mutants kept for aging studies. Vps33a corresponds to the mouse bf locus and encodes a homologue of yeast vps33 (Suzuki et al., 2003). Vps33, a homologue of Sec-1p-like regulators of membrane fusion, forms the class C Vps complex by interacting with other Vps proteins (Rieder and Emr, 1997). In yeast the Vps complex is required for soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE)-mediated vesicle docking and fusion with the vacuole (Peterson and Emr, 2001; Sato et al., 2000; Wurmser et al., 2000).
Members of the Sec-1p/Munc18 (SM) family of proteins have been implicated in the vesicle docking process (Voets et al., 2001; Weimer et al., 2003). In eucaryotic cells membrane trafficking pathways require SM family proteins, a group of conserved 60–80 kDa cytosolic proteins (Toonen and Verhage, 2003). UNC-18, originally identified in C. elegans belongs to the SM protein family, and UNC-18 mutants exhibit uncoordinated locomotion (Brenner, 1974). There are seven SM proteins in mammals (Munc18-1, -2, and -c, Sly-1, Vps45, and Vps33a and b). Drosophila has four SM proteins (ROP, Sly1, Vps45, and Vps33), and yeast has a similar number (Sec-1p, Sly-1p, Vps45p, Vps33p) (Toonen and Verhage, 2003). Recently, Dulubova et al. reported that the SM proteins (Sec1/Munc18-like proteins) and neuronal SNARE proteins (synaptobrevin/VAMP on synaptic vesicles, and SNAP-25 and syntaxin-1A/1B on plasma membrane) are required for intracellular membrane fusion (Dulubova et al., 2007). Vps33a, as a member of the SM protein family, could be involved in synaptic vesicle exocytosis.
In HPS, defects in intracellular vesicle trafficking lead to the dysfunction of lysosome-related organelles. The well-known effects of HPS disease are on pigmentation, platelet function and lysosome secretion. The HPS genes control a wide range of physiological processes including immune recognition, neuronal functions, lung surfactant trafficking (Li et al., 2004) and the transport of cystine (Chintala et al., 2005).
We describe for the first time Purkinje cell abnormalities in a mouse model of human HPS disease. We did not observe a decrease in cerebellar Purkinje cells in young adult bf mice. However, bf mice older than 8 months exhibited obvious loss of Purkinje cells. Thus the onset of behavioral defects significantly precedes observable Purkinje cell degeneration. Movements of the hind legs are severely affected in the bf mutant (Supplement 1). The mutant mouse walks in a manner very different from that of the wild-type. The cerebellum is the key regulator of movement and plays an important role in locomotion and balance (Morton and Bastian, 2007). The most obvious defect due to cerebellum damage is gate ataxia or lack of coordination of movement.
The impaired response of bf mice to hand touch and startle response (Fig. 1) suggests that vps33a gene mutations may cause sensory defects in addition to the obvious cerebellar defects. Also, motor deficits may contribute to the observed defects in righting reflex, grip strength, balance and locomotion. Likewise, the odd wiggle in the hindquarters of mutant bf mice suggests possible spinal cord or neuromuscular defects in vps33a mutations. The absence of the startle response in old bf mutants suggests deafness. However, the cause for this phenotype has not been investigated.
The cerebellum is the most powerful machine in the brain containing multiple neurons involved in deciphering information (Manto, 2007). Recent reviews provide highlights from 5 decades of research on the Lurcher (LC) mouse, a neurological mutant characterized by a wobbly, lurching gait which is caused by extensive degeneration of Purkinje cellsby3months after birth (Vogel et al., 2007). The gene mutated in lurcher is the d2glutamate receptor (GluRd2; gene symbol, Grid2) (Zuo et al., 1997). In contrast, the number of Purkinje cells of the bf mutant is not significantly reduced till 1 year. This suggests that the Vps33a protein is required for maintenance rather than development of Purkinje cells. Similarly to the +/Lc mouse, bf mice exhibit behavioral deficiencies when they are young (8–13 weeks) and old (13–14 months) (Fig. 1). Significant reduction in the volume of the bf cerebellum begins at 8 months, to 30% reduction at 13–14 months (Table 1). This further indicates that Vps33a may be required for the maintenance of cerebellar functions. Consistent with this possibility, the Vps33a mRNA mutated in bf mice is ubiquitous (Huizing et al., 2001) and highly expressed in mouse brain including the cerebellum (www.brain-map.org). Interestingly there is accumulation of cerebrospinal fluid (CSF) around the cerebellum in bf mutants especially in old mutants (Fig. 4). The biological basis for the accumulation of CSF is unknown. The cerebellar size is reduced, and the resulting space may be filled with CSF. Alternatively the accumulation of CSF may reduce cerebellar size.
Another HPS mouse mutant, mocha, exhibits neurological defects (Kantheti et al., 1998; Noebels and Sidman, 1989). Behavior abnormalities have been reported in other mouse pigment mutants. The two allelic mouse mutants, yellow submarine (Ysb) and light coat and circling (Lcc) exhibit recessive circling behavior and deafness together with semi-dominant yellow coat color (Dong et al., 2002). The sut/sut (subtle gray) mouse, mutated in the cystine/glutamate exchanger (xCT) responsible for pheomelanin synthesis (Chintala et al., 2005), develops brain atrophy in early adulthood exhibiting ventricular enlargement, thinning of cortex and shrinkage of striatum (Shih et al., 2006). The dilute-lethal (dl) recessive mutation produces a dilute coat color (Searle, 1952), severe convulsive limb movements and ataxia which precedes the abnormal expression of tyrosine hydroxylase and the degeneration of Purkinje cells (Sawada et al., 1999a; Sawada et al., 1999b). A recent review (Dusart et al., 2006) summarizes Purkinje cell death in 5 additional mutant mice (nervous (nr), purkinje cell degeneration (pcd), Lurcher (Lc), toppler (top) and woozy (wz)) and also reports new data on tambaleante (tbl). These authors conclude that Purkinje cell loss in these mutants may be via apoptosis, autophagy and necrosis or a combination of these processes.
The mechanism of Purkinje cell loss has been extensively investigated in the case of the Lc mutant. The mutated Grid2 receptors in Lc are involved in autophagy through the interaction of Grid2 with Beclin via nPIST forming a signaling complex (Yue et al., 2002). nPIST is colocalized with Grid2 at postsynaptic membranes of Purkinje cell tertiary spines and has a presumptive adaptor protein function (Neudauer et al., 2001). The interaction of nPIST with Beclin1, a human ortholog of yeast autophagic gene Apg6/vps30p provides a link between autophagy and neuronal cell death. Beclin1 is a Bcl-2 interacting protein (Liang et al., 1998) and inhibits the effect of Beclin1-mediated autophagy. Thus Bcl-2 is not only antiapoptotic but also antiautophagic, and it regulates levels of autophagic processes for cell survival (Pattingre et al., 2005). Since Beclin1 is a human ortholog of the yeast autophagic gene Apg6/vps30p protein (Yue et al., 2002), we hypothesize that vps33a mutated in the bf mouse, could be involved in autophagy by interacting with Bcl-2.
An unidentified gene mutated in Tambaleante (tbl/tbl) mice causes Purkinje cell degeneration beginning at about 2 months and progressing to complete degeneration at one year (Rossi et al., 1995; Wassef etal.,1987). Degeneration is accompanied by accumulation of dense cytoplasmic material (Dusart et al., 2006) immunoreactive to antibodies against human lysosomal-associated membrane proteins LAMP-1 (Karlsson and Carlsson, 1998). The electron microscopic analysis of tbl mutant cerebella revealed the presence of numerous structures exhibiting features of autophagic vacuoles (AVs) including multilamellar bodies in degenerating Purkinje cells (Dusart et al., 2006). Purkinje cell degeneration in bf, a model for lysosome-related organelle dysfunction, may similarly be due to abnormal AV trafficking.
In young bf mutants GFAP expressed in glial cells was slightly increased in the cerebellum, and Purkinje cell number was normal. In old bf mutants Purkinje cell number was reduced to 30% of controls and GFAP expression was significantly increased in the cerebellum (Fig. 6) suggesting a relationship of GFAP expression to Purkinje cell degeneration. In agreement with these findings, GFAP positive cells are increased in the cerebellum of the toppler mutant (Duchala et al., 2004) which likewise exhibits Purkinje cell degeneration. The up-regulation of GFAP is associated with damage to neurons in CNS (Condorelli et al., 1999) and also has been widely used as a marker for neuronal damage in cerebral ischemia (Herrmann and Ehrenreich, 2003; Petzold et al., 2006; Vissers et al., 2006). The up-regulation of GFAP in bf mutants may be due to altered interaction of glial cells and the Purkinje cells since there is an obvious degeneration of Purkinje cells.
Additional studies are required to understand the mechanism of Purkinje cell death in bf mutants. These studies suggest that selected HPS patients may have neurological problems later in life. Possible abnormalities in VPS33A should be considered in these patients. It is presently not possible to directly test patients with deficiencies in the buff gene (VPS33A) for cerebellar defects since human patients with this subtype of HPS have not yet been identified. While old bf mutants are deaf, there is no report that patients with HPS have hearing defects. We suggest nevertheless, based on our observations in buff HPS mice, that HPS patients should be screened for hearing defects. There are reports of deafness in patients with Ocular Coetaneous Albinism (OCA) (Lezirovitz et al., 2006) another pigment deficiency disease.
In summary we report that the bf mouse, a model for HPS disease mutated in Vps33a, exhibits striking behavioral deficiencies and Purkinje cell degeneration. The degeneration of Purkinje cells progresses with age, reaching 70% loss in 1 year old mutant mice compared to heterozygous age-matched controls. Loss of Purkinje cells is accompanied by decrease in size of the cerebellum. Purkinje cell death is a likely cause of the observed bf behavioral defects including motor deficits and ataxia. The behavior abnormalities clearly correlate with the neurological defects. Thus the bf mouse is a model for understanding neurodegenerative disorders.
4. Experimental procedures
4.1. Mice
Mice carrying the spontaneous mutation bf on the C57BL/6J strain were obtained from The Jackson Laboratory (Bar Harbor, ME) and subsequently bred at Roswell Park Cancer Institute. All animal procedures outlined in this study were approved by The Roswell Park Cancer Institute Animal Care Use Committee.
4.2. Evaluation of behavioral impairments
Screenings of behavioral phenotypes were assessed in young (8–13 weeks) and old (13–14 months) bf mutants and their controls (C57BL/6J and bf/+) with well established behavioral procedures described in the SHIRPA protocols of the ENU Mutagenesis Program (Rogers et al., 1997). This procedure involves the use of a set of 35 tests, which profiles the spontaneous behavior of mice qualitatively. Twelve animals were used for each SHIRPA protocol category. Very briefly, the methods (described in full by Rogers et al., 1997) are as follows. The grip strength test was performed by allowing the mouse to grasp a metal net with its front legs while holding the mouse by the tail. The mouse was pulled back, and the grip strength was evaluated by hand. In the touch escape test movement of the mouse away from a side finger touch was evaluated. The righting reflex test measured the ability of the mouse to assume an upright position after being flipped gently backwards. The gait test measured observable abnormalities in gait/posture while walking a distance of 1–2 ft. The startle response test evaluated responses like a backward flick of pinnae and/or jumping in response to hand clapping and/or use of a simple clicker 30 cm above the arena.
4.3. Spontaneous, exploratory behaviors
To study the aforementioned mutants, male mice were filmed using a Sony Model TRV 900 videotape recorder equipped with a standard 35 mm Sony mini-DV tape. In brief, a given wild-type or mutant mouse was placed in a clean, cylindrical bucket (27 cm diameter×30 cm height) and was allowed to explore this novel environment for 1 min. Recorded spontaneous exploratory behaviors were then digitized for each mouse using a Peak Motus Program. This was achieved by marking several constant points on every animal recorded including the nose, occiput, left and right shoulders, left and right hips, and the base of the tail. The aforementioned coordinates were followed for multiple events of 3 s duration to generate a spatial diagram of motor activities by plotting the range of circular motion angles.
4.4. Examination of motor functions
Motor functions were evaluated by standard tests including wire suspension, rotating cylinder, bridge test and slanting ladder (Krizkova and Vozeh, 2004). Both young (_n_=12) and old (n=12) bf mutants and their age-matched homozygous (_n_=12) as well as heterozygous (bf/+) control mice (_n_=12) were tested for these motor and coordination functions. For the wire suspension test, mice were allowed to hang by their front legs on a 1 mm diameter horizontal wire which was suspended at the height of 40 cm. A foam cushion protected animals from injury upon falling. The criterion for success was the ability to hang from the wire for 1 min without falling. The rotating cylinder (15 cm diameter×20 cm length) test evaluated the ability to remain on the 1 rpm rotating cylinder for 1 min. In the bridge test (30 cm length×2 cm width×40 cm height) mice were placed on the bridge and tested for the ability to cross the bridge within a 2 min period. For the slanting ladder test (35 cm length×4 cm width×4 mm diameter) and with a 60° horizontal inclination angle, mice were placed on the middle of the ladder and tested for the ability to climb to the top or down to the floor within 1 min. All of these tests were performed in the same sterile hood and at the same time of the day.
4.5. Immunohistochemistry
Brains were dissected from age-matched and sex-matched wild-type/heterozygous control and mutant bf mice and fixed in formalin for subsequent paraffin blocks. Paraffin sections were cut at 5 μm, placed on charged slides and dried in a 60 °C oven for 1 h. Slides were deparaffinized in three changes of xylene and re-hydrated using graded alcohols. Endogenous peroxidase was quenched with aqueous 3% H2O2 for 15 min and washed with PBS/T. Antigen retrieval was then carried out with citrate buffer (pH 6) in the microwave for 10 min; slides were allowed to cool for 15 min and washed in PBS/T. The slides were then placed on a DAKO auto-stainer and immunostaining was performed as follows. Slides were washed with PBS/T followed by Casein 0.03% [in PBS/T] for a blocking step for 30 min. Primary antibodies calbindin (diluted 1/7000=1.1 μg/ml) or Glial Fibrillar Acidic Protein (GFAP) (diluted to 1.5 μg/ml) were applied to the slides and left for 1 h at room temperature. An isotype-matched control [1.1 μg/ml mIgG] was used on a duplicate slide in place of the primary antibody as a negative control. After washing with PBS/T, biotinylated secondary antibody [goat anti- mouse] was added to the solution and incubated for 30 min. A PBS/T wash was followed by the streptavidin peroxidase reagent [Zymed] for 30 min. Finally the chromogen DAB [DAKO, Carpinteria CA] was applied for 5 min to determine the color reaction product. The slides were then counterstained with hematoxylin, dehydrated, cleared and cover-slipped.
4.6. Western blot analysis
Brain samples were collected from old bf/+ and bf mice, and cerebellum was dissected carefully without any other part of the brain. Western blot analysis was performed as described (Chintala et al., 2007). Briefly, protein extract was prepared from the cerebellum in lysis buffer with protease inhibitor cocktail (Roche Molecular Biochemicals, IN). 30 μg of protein was separated on 4–12% gradient gels and transferred to polyvinylidene difluoride membrane. The membrane was blocked for 1 h with 5% milk in phosphate buffered saline (PBS-T) with 0.1% Tween-20 followed by incubation with anti-GFAP (Sigma) at 1:1000 dilution. The membrane was washed and incubated with anti-mouse horseradish peroxidase-conjugated secondary antibody for 1 h and washed with PBS-T, and the bound antibody was detected with ECL Advance (GE Life Sciences, NJ). Mouse monoclonal anti-α-tubulin (Sigma) was used a loading control.
4.7. Analysis of Purkinje cell number
To assess Purkinje cell number, brains were sectioned and stained with anti-calbindin. Purkinje cells were counted by visualizing cells under the microscopeinthe entire cerebellum. Three different mice from each group were used for the experiment, and two brain sections from each mouse were used to count the total number of Purkinje cells. Data were analyzed with unpaired _t_-tests.
4.8. Magnetic resonance imaging (MRI)
MRI scanning was performed with different age groups: (i) young (8–13 weeks) bf and their control C57/BL6, (ii) 8 months bf and their bf/+ heterozygous controls and (iii) >1 year old bf and their bf/+ heterozygous controls. Three different mice from each age group were imaged.
Magnetic resonance imaging was carried out on a 4.7 Tesla MR Scanner incorporating AVANCE™ digital electronics using a 60 mm removable gradient coil insert and a custom designed 35 mm RF transceiver coil (Bruker BioSpec platform with ParaVision® Version 3.1 Operating System, Bruker Medical, Billerica, MA). Anesthesia was induced with 4% isoflurane and maintained with 1–2% isoflurane during imaging. The respiratory rate of mice was monitored with an MR compatible monitoring and gating system (Model 1025, SA Instruments, Inc., Stony Brook, NY). A small foam pillow was taped down over the mouse heads to immobilize the heads during scan acquisitions. Several scout scans were first acquired to determine position of the brain within the RF coil. A high resolution, T2-weighted 3D scan with RARE phase encoding (Rapid Acquisition with Relaxation Enhancement) was acquired in the sagittal orientation to delineate cerebellum and total brain volume. T2-weighted imaging was chosen as it offers good contrast of cerebral spinal fluid (CSF) and arbor vitae within the cerebellum without the use of manganese as a contrast agent, which is neurotoxic in high doses. Scanning parameters of the 3D scan are as follows: effective echo time=85 ms, TRmin=3 s, field of view = 24×16×16, matrix size = 128×96×96, echo train length = 16, 2 averages). The scanner was gated to acquire data between breaths, thus actual TR time varied slightly, dependent upon respiratory rate. Following acquisition, image data were transferred to a graphic workstation for image analysis with Analyze 5.0 software (AnalyzeDirect, Overland Park, KS). An “object map” delineating the cerebellum and remaining brain regions of interest (ROI’s) was acquired by manual segmentation of the image data by an iterative process of visual inspection of all three orientations (coronal, sagittal, and axial). A 3×3×3 median filter was applied to the object map to smooth irregularities that occur via manual segmentation. Volumes of each ROI were calculated by the software, and percent volume of the cerebellum was calculated by dividing the volume of the cerebellum by the sum of the cerebellum and remaining brain.
Segmented datasets outlining cerebellum, CSF, and the remainder of the brain were imported into amira™ for 3D surface rendering (Mercury Computer Systems, Chelmsford, MA). Colors were assigned to specific regions of interest e.g. cerebellum (green), CSF (yellow), remainder of the brain (pink) and rendered as 3D surfaces. Brains were given a transparency value of 0.687 (out of 1.0) to allow the visualization of CSF within the central locations of the brain.
4.9. Statistical analysis
Fisher’s exact test was used to compare two groups that differ in portion by group with _P_≤0.05 considered as significant. We scored 100% when all the animals in the group succeeded in the test, determined the percent of animals that did not succeed in the test and compared for statistical significance. Purkinje cell number and cerebellum volume data are presented as mean±SEM. Significance was determined by using the standard _t_-test.
Supplementary Material
Video
Acknowledgments
We thank Dr. Youcef Rustum Chairman, Cancer Biology department for his help in pursuing this research. We also thank Gerald Jahreis, Madonna Reddington and Jian Tan for the technical support. We acknowledge Andrews Chris and Chunqiao Tian for the statistical analysis of behavior data. This study was supported by Grants from the National Institutes of Health (grants HL-51480, HL-31698, and EY-12104) to R.T.S. This research used core facilities supported in part by Roswell Park Cancer Institute’s National Cancer Institute-funded Cancer Center Support Grant CA-16056.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.brainres.2009.02.035.
References
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caddy KW, Biscoe TJ. Structural and quantitative studies on the normal C3H and Lurcher mutant mouse. Philos Trans R Soc Lond B Biol Sci. 1979;287:167–201. doi: 10.1098/rstb.1979.0055. [DOI] [PubMed] [Google Scholar]
- Caddy KW, Sidman RL. Purkinje cells and granule cells in the cerebellum of the Stumbler mutant mouse. Brain Res. 1981;227:221–236. doi: 10.1016/0165-3806(81)90110-3. [DOI] [PubMed] [Google Scholar]
- Chintala S, Li W, Lamoreux ML, Ito S, Wakamatsu K, Sviderskaya EV, Bennett DC, Park YM, Gahl WA, Huizing M, Spritz RA, Ben S, Novak EK, Tan J, Swank RT. Slc7a11 gene controls production of pheomelanin pigment and proliferation of cultured cells. Proc Natl Acad Sci U S A. 2005;102:10964–10969. doi: 10.1073/pnas.0502856102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chintala S, Tan J, Gautam R, Rusiniak ME, Guo X, Li W, Gahl WA, Huizing M, Spritz RA, Hutton S, Novak EK, Swank RT. The Slc35d3 gene, encoding an orphan nucleotide sugar transporter, regulates platelet-dense granules. Blood. 2007;109:1533–1540. doi: 10.1182/blood-2006-08-040196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condorelli DF, Nicoletti VG, Dell’Albani P, Barresi V, Caruso A, Conticello SG, Belluardo N, Giuffrida Stella AM. GFAPbeta mRNA expression in the normal rat brain and after neuronal injury. Neurochem Res. 1999;24:709–714. doi: 10.1023/a:1021016828704. [DOI] [PubMed] [Google Scholar]
- Dong S, Leung KK, Pelling AL, Lee PY, Tang AS, Heng HH, Tsui LC, Tease C, Fisher G, Steel KP, Cheah KS. Circling, deafness, and yellow coat displayed by yellow submarine (ysb) and light coat and circling (lcc) mice with mutations on chromosome 3. Genomics. 2002;79:777–784. doi: 10.1006/geno.2002.6783. [DOI] [PubMed] [Google Scholar]
- Duchala CS, Shick HE, Garcia J, Deweese DM, Sun X, Stewart VJ, Macklin WB. The toppler mouse: a novel mutant exhibiting loss of Purkinje cells. J Comp Neurol. 2004;476:113–129. doi: 10.1002/cne.20206. [DOI] [PubMed] [Google Scholar]
- Dulubova I, Khvotchev M, Liu S, Huryeva I, Sudhof TC, Rizo J. Munc18-1 binds directly to the neuronal SNARE complex. Proc Natl Acad Sci. 2007;104:2697–2702. doi: 10.1073/pnas.0611318104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusart I, Guenet JL, Sotelo C. Purkinje cell death: differences between developmental cell death and neurodegenerative death in mutant mice. Cerebellum. 2006;5:163–173. doi: 10.1080/14734220600699373. [DOI] [PubMed] [Google Scholar]
- Halachmi N, Lev Z. The Sec1 family: a novel family of proteins involved in synaptic transmission and general secretion. J Neurochem. 1996;66:889–897. doi: 10.1046/j.1471-4159.1996.66030889.x. [DOI] [PubMed] [Google Scholar]
- Heckroth JA, Goldowitz D, Eisenman LM. Purkinje cell reduction in the reeler mutant mouse: a quantitative immunohistochemical study. J Comp Neurol. 1989;279:546–555. doi: 10.1002/cne.902790404. [DOI] [PubMed] [Google Scholar]
- Herrmann M, Ehrenreich H. Brain derived proteins as markers of acute stroke: their relation to pathophysiology, outcome prediction and neuroprotective drug monitoring. Restor Neurol Neurosci. 2003;21:177–190. [PubMed] [Google Scholar]
- Herrup K, Trenkner E. Regional differences in cytoarchitecture of the weaver cerebellum suggest a new model for weaver gene action. Neuroscience. 1987;23:871–885. doi: 10.1016/0306-4522(87)90164-3. [DOI] [PubMed] [Google Scholar]
- Huizing M, Didier A, Walenta J, Anikster Y, Gahl WA, Kramer H. Molecular cloning and characterization of human VPS18, VPS11, VPS16, and VPS33. Gene. 2001;264:241–247. doi: 10.1016/s0378-1119(01)00333-x. [DOI] [PubMed] [Google Scholar]
- Kantheti P, Qiao X, Diaz ME, Peden AA, Meyer GE, Carskadon SL, Kapfhamer D, Sufalko D, Robinson MS, Noebels JL, Burmeister M. Mutation in AP-3 delta in the mocha mouse links endosomal transport to storage deficiency in platelets, melanosomes, and synaptic vesicles. Neuron. 1998;21:111–122. doi: 10.1016/s0896-6273(00)80519-x. [DOI] [PubMed] [Google Scholar]
- Karlsson K, Carlsson SR. Sorting of lysosomal membrane glycoproteins lamp-1 and lamp-2 into vesicles distinct from mannose 6-phosphate receptor/gamma-adaptin vesicles at the trans-Golgi network. J Biol Chem. 1998;273:18966–18973. doi: 10.1074/jbc.273.30.18966. [DOI] [PubMed] [Google Scholar]
- Krizkova A, Vozeh F. Development of early motor learning and topical motor skills in a model of cerebellar degeneration. Behav Brain Res. 2004;150:65–72. doi: 10.1016/S0166-4328(03)00247-X. [DOI] [PubMed] [Google Scholar]
- Lezirovitz K, Nicastro FS, Pardono E, Abreu-Silva RS, Batissoco AC, Neustein I, Spinelli M, Mingroni-Netto RC. Is autosomal recessive deafness associated with oculocutaneous albinism a “coincidence syndrome”? J Hum Genet. 2006;51:716–720. doi: 10.1007/s10038-006-0003-7. [DOI] [PubMed] [Google Scholar]
- Li W, Rusiniak ME, Chintala S, Gautam R, Novak EK, Swank RT. Murine Hermansky–Pudlak syndrome genes: regulators of lysosome-related organelles. Bioessays. 2004;26:616–628. doi: 10.1002/bies.20042. [DOI] [PubMed] [Google Scholar]
- Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. 1998;72:8586–8596. doi: 10.1128/jvi.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manto MABA. Cerebellum and the deciphering of motor coding. Cerebellum. 2007;6:3–6. [Google Scholar]
- Morton SM, Bastian AJ. Mechanisms of cerebellar gait ataxia. Cerebellum. 2007;6:79–86. doi: 10.1080/14734220601187741. [DOI] [PubMed] [Google Scholar]
- Mullen RJ, Eicher EM, Sidman RL. Purkinje cell degeneration, a new neurological mutation in the mouse. Proc Natl Acad Sci U S A. 1976;73:208–212. doi: 10.1073/pnas.73.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neudauer CL, Joberty G, Macara IG. PIST: a novel PDZ/coiled-coil domain binding partner for the rho-family GTPase TC10. Biochem Biophys Res Commun. 2001;280:541–547. doi: 10.1006/bbrc.2000.4160. [DOI] [PubMed] [Google Scholar]
- Noebels JL, Sidman RL. Persistent hypersynchronization of neocortical neurons in the mocha mutant of mouse. J Neurogenet. 1989;6:53–56. doi: 10.3109/01677068909107100. [DOI] [PubMed] [Google Scholar]
- Oppenheimer D, editor. Diseases of Basal Ganglia, Cerebellum and Motor Neurons. John Wiley and Sons; New York, NY: 1984. [Google Scholar]
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Peterson MR, Emr SD. The class C Vps complex functions at multiple stages of the vacuolar transport pathway. Traffic. 2001;2:476–486. doi: 10.1034/j.1600-0854.2001.20705.x. [DOI] [PubMed] [Google Scholar]
- Petzold A, Keir G, Kerr M, Kay A, Kitchen N, Smith M, Thompson EJ. Early identification of secondary brain damage in subarachnoid hemorrhage: a role for glial fibrillary acidic protein. J Neurotrauma. 2006;23:1179–1184. doi: 10.1089/neu.2006.23.1179. [DOI] [PubMed] [Google Scholar]
- Rieder SE, Emr SD. A novel RING finger protein complex essential for a late step in protein transport to the yeast vacuole. Mol Biol Cell. 1997;8:2307–2327. doi: 10.1091/mbc.8.11.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizai AYC. Abnormal rate of granule cell migration in the cerebellum of “weaver” mutant mice. Dev Biol. 1972;29:17–26. doi: 10.1016/0012-1606(72)90039-5. [DOI] [PubMed] [Google Scholar]
- Robinson JS, Klionsky DJ, Banta LM, Emr SD. Protein sorting in Saccharomyces cerevisiae: isolation of mutants defective in the delivery and processing of multiple vacuolar hydrolases. Mol Cell Biol. 1988;8:4936–4948. doi: 10.1128/mcb.8.11.4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers DC, Fisher EM, Brown SD, Peters J, Hunter AJ, Martin JE. Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm Genome. 1997;8:711–713. doi: 10.1007/s003359900551. [DOI] [PubMed] [Google Scholar]
- Rossi F, Jankovski A, Sotelo C. Target neuron controls the integrity of afferent axon phenotype: a study on the Purkinje cell-climbing fiber system in cerebellar mutant mice. J Neurosci. 1995;15:2040–2056. doi: 10.1523/JNEUROSCI.15-03-02040.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato TK, Rehling P, Peterson MR, Emr SD. Class C Vps protein complex regulates vacuolar SNARE pairing and is required for vesicle docking/fusion. Mol Cell. 2000;6:661–671. doi: 10.1016/s1097-2765(00)00064-2. [DOI] [PubMed] [Google Scholar]
- Sawada K, Komatsu S, Haga H, Oda S, Fukui Y. Abnormal expression of tyrosine hydroxylase immunoreactivity in Purkinje cells precedes the onset of ataxia in dilute-lethal mice. Brain Res. 1999a;844:188–191. doi: 10.1016/s0006-8993(99)01899-5. [DOI] [PubMed] [Google Scholar]
- Sawada K, Komatsu S, Haga H, Sun XZ, Hisano S, Fukui Y. Abnormal expression of tyrosine hydroxylase immunoreactivity in cerebellar cortex of ataxic mutant mice. Brain Res. 1999b;829:107–112. doi: 10.1016/s0006-8993(99)01347-5. [DOI] [PubMed] [Google Scholar]
- Schmitt-John T, Drepper C, Mussmann A, Hahn P, Kuhlmann M, Thiel C, Hafner M, Lengeling A, Heimann P, Jones JM, Meisler MH, Jockusch H. Mutation of Vps54 causes motor neuron disease and defective spermiogenesis in the wobbler mouse. Nat Genet. 2005;37:1213–1215. doi: 10.1038/ng1661. [DOI] [PubMed] [Google Scholar]
- Searle AG. A lethal allele of dilute in the house mouse. Heredity. 1952;6:395–401. [Google Scholar]
- Shih AY, Erb H, Sun X, Toda S, Kalivas PW, Murphy TH. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J Neurosci. 2006;26:10514–10523. doi: 10.1523/JNEUROSCI.3178-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonmez E, Herrup K. Role of staggerer gene in determining cell number in cerebellar cortex. II. Granule cell death and persistence of the external granule cell layer in young mouse chimeras. Brain Res. 1984;314:271–283. doi: 10.1016/0165-3806(84)90049-x. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Oiso N, Gautam R, Novak EK, Panthier JJ, Suprabha PG, Vida T, Swank RT, Spritz RA. The mouse organellar biogenesis mutant buff results from a mutation in Vps33a, a homologue of yeast vps33 and Drosophila carnation. Proc Natl Acad Sci U S A. 2003;100:1146–1150. doi: 10.1073/pnas.0237292100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toonen RF, Verhage M. Vesicle trafficking: pleasure and pain from SM genes. Trends Cell Biol. 2003;13:177–186. doi: 10.1016/s0962-8924(03)00031-x. [DOI] [PubMed] [Google Scholar]
- Vissers JL, Mersch ME, Rosmalen CF, van Heumen MJ, van Geel WJ, Lamers KJ, Rosmalen FM, Swinkels LM, Thomsen J, Herrmann M. Rapid immunoassay for the determination of glial fibrillary acidic protein (GFAP) in serum. Clin Chim Acta. 2006;366:336–340. doi: 10.1016/j.cca.2005.11.017. [DOI] [PubMed] [Google Scholar]
- Voets T, Toonen RF, Brian EC, de Wit H, Moser T, Rettig J, Sudhof TC, Neher E, Verhage M. Munc18–1 promotes large dense-core vesicle docking. Neuron. 2001;31:581–591. doi: 10.1016/s0896-6273(01)00391-9. [DOI] [PubMed] [Google Scholar]
- Vogel MW, Caston J, Yuzaki M, Mariani J. The Lurcher mouse: fresh insights from an old mutant. Brain Res. 2007;1140:4–18. doi: 10.1016/j.brainres.2005.11.086. [DOI] [PubMed] [Google Scholar]
- Wada Y, Kitamoto K, Kanbe T, Tanaka K, Anraku Y. The SLP1 gene of Saccharomyces cerevisiae is essential for vacuolar morphogenesis and function. Mol Cell Biol. 1990;10:2214–2223. doi: 10.1128/mcb.10.5.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton J. Brain’s Diseases of the Nervous System. Oxford university Press; Oxford: 1985. Vol. [Google Scholar]
- Wassef M, Sotelo C, Cholley B, Brehier A, Thomasset M. Cerebellar mutations affecting the postnatal survival of Purkinje cells in the mouse disclose a longitudinal pattern of differentially sensitive cells. Dev Biol. 1987;124:379–389. doi: 10.1016/0012-1606(87)90490-8. [DOI] [PubMed] [Google Scholar]
- Weimer RM, Richmond JE, Davis WS, Hadwiger G, Nonet ML, Jorgensen EM. Defects in synaptic vesicle docking in unc-18 mutants. Nat Neurosci. 2003;6:1023–1030. doi: 10.1038/nn1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurmser AE, Sato TK, Emr SD. New component of the vacuolar class C-Vps complex couples nucleotide exchange on the Ypt7 GTPase to SNARE-dependent docking and fusion. J Cell Biol. 2000;151:551–562. doi: 10.1083/jcb.151.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuasa S, Kitoh J, Oda S, Kawamura K. Obstructed migration of Purkinje cells in the developing cerebellum of the reeler mutant mouse. Anat Embryol (Berl) 1993;188:317–329. doi: 10.1007/BF00185941. [DOI] [PubMed] [Google Scholar]
- Yue Z, Horton A, Bravin M, DeJager PL, Selimi F, Heintz N. A novel protein complex linking the delta 2 glutamate receptor and autophagy: implications for neurodegeneration in lurcher mice. Neuron. 2002;35:921–933. doi: 10.1016/s0896-6273(02)00861-9. [DOI] [PubMed] [Google Scholar]
- Zuo J, De Jager PL, Takahashi KA, Jiang W, Linden DJ, Heintz N. Neurodegeneration in Lurcher mice caused by mutation in delta2 glutamate receptor gene. Nature. 1997;388:769–773. doi: 10.1038/42009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video