S-Nitrosylation of Bcl-2 Negatively Affects Autophagy in Lung Epithelial Cells (original) (raw)
. Author manuscript; available in PMC: 2017 Feb 1.
Published in final edited form as: J Cell Biochem. 2016 Feb;117(2):521–532. doi: 10.1002/jcb.25303
Abstract
Autophagy is a catabolic cellular mechanism involving lysosomal degradation of unwanted cellular components. Interaction between Beclin-1 and Bcl-2 proteins is known to play a critical role in the initiation of autophagy. We report that malignantly transformed lung epithelial cells are resistant to autophagy and express lower basal levels of autophagic proteins, Beclin-1 and LC3-II as compared to non-tumorigenic cells. Additionally, increased levels of nitric oxide (NO) and Bcl-2 were observed in transformed cells. Nitric oxide was found to negatively regulate autophagy initiation and autophagic flux by nitrosylating Bcl-2 and stabilizing its interaction with Beclin-1, resulting in inhibition of Beclin-1 activity. An increase in the apoptotic initiator caspase-9 and the apoptosis and autophagy-associated kinase p38/MAPK in both cell types indicated possible autophagy–apoptosis crosstalk. Pre-treatments with ABT-737 (Bcl-2 inhibitor) and aminoguanidine (NO inhibitor), and transfection with a non-nitrosylable Bcl-2 cysteine double-mutant plasmid resulted in increased autophagic flux (LC3-II/p62 upregulation) corresponding with decreased _S_-nitrocysteine expression, thus corroborating the regulatory role of Bcl-2 _S_-nitrosylation in autophagy. In conclusion, our study reveals a novel mechanism of autophagy resistance via post-translational modification of Bcl-2 protein by NO, which may be critical in driving cellular tumorigenesis.
Keywords: AUTOPHAGY, NITRIC OXIDE, BCL-2, BECLIN-1, _S_-NITROSYLATION
Lung cancer is one of the leading causes of cancer deaths worldwide, with over one million deaths annually [Jemal et al., 2004; Ferlay et al., 2010]. Over 200,000 new cases of lung cancer and over 150,000 related deaths were reported in the United States in 2013 alone [National Cancer Institute at the National Institutes of Health, 2014]. Most lung cancer cases are detected only at later stages of the disease when the cancer has already progressed to an aggressive stage, resulting in survival rates between 10% and 15% [Collins et al., 2007].
Dysregulation of cell death pathways such as apoptosis is considered a major barrier for curative therapy in lung cancer [Abend, 2003; Melet et al., 2008]. Autophagy is a cellular pathway that involves the removal of unwanted or damaged cellular components by lysosomal degradation. However, cancer cells can evade cell death by initiating autophagy thereby maintaining cellular energy levels until the cells can recover from the causative stress [Paglin et al., 2001; Yang et al., 2011]. One of the two major autophagic pathways involves JNK and its downstream partners Bcl-2, Beclin-1, and Vps34. Activation of JNK1 leads to phosphorylation of Bcl-2, which allows the Bcl-2–Beclin-1 complex to dissociate. The dissociated Beclin-1 forms a complex with Vps34 (Class III PI3K), leading to autophagosome formation [Mizushima and Levine, 2010]. The second major autophagy pathway is driven by IKK β, and involves downstream partners AMPK, TSC1/2, and MTORC1, which inhibits autophagosome formation upon activation [Kroemer et al., 2010]. In addition to these proteins, Atg12 and LC3 are also critical members that are required for autophagosome formation [Mizushima and Levine, 2010]. LC3 and its binding partners are responsible for autophagosome transport and maturation. Upon maturation, the autophagosome fuses with lysosomes where its internal cargo is degraded [Pattingre et al., 2008; Thorburn, 2008].
Current research indicates that long-term exposure to inhaled carcinogens is a major etiological agent for lung disease [IARC, 1990; Cogliano et al., 2011]. Hexavalent chromium [Cr(VI)] compounds have been classified as group I human carcinogens by the International Agency of Research in Cancer [Langard, 1990; De Flora, 2000; Cogliano et al., 2011]. Hexavalent chromium [Cr(VI)] is used in a wide range of processes including leather tanning, stainless steel production, wood preservation, anti-corrosion coatings, pigment in dyes, plastics and inks, among several others [Langard, 1990, 1993; De Flora, 2000]. Due to its wide usage, several epidemiological studies have confirmed that exposure to chromium compounds in various occupational settings is associated with lung disease [Simonato et al., 1991; Langard, 1993]. Cr(VI) compounds are also present in cigarette smoke, and research has indicated that there is an increased incidence of lung cancer in smokers with Cr(VI) exposure [IARC, 1990; Langard, 1990; Balansky et al., 2000]. Although the epidemiological data implicating Cr(VI) in lung carcinogenesis is strong, there has been limited success in establishing a reliable in vivo model to support the present data [Balansky et al., 2000; Bucher, 2007; Holmes et al., 2008]. To investigate the mechanisms involved in Cr(VI)-induced carcinogenesis, we developed an in vitro model by subjecting non-tumorigenic human bronchial epithelial Beas-2B cells to long-term Cr(VI) exposure, and such chronic exposure to Cr(VI) led to the malignant transformation of non-tumorigenic Beas-2B cells [Azad et al., 2010b].
Nitric oxide (NO), an important signaling molecule that functions in several cellular processes, is well known to impair autophagosome formation by inhibiting the activity of the JNK1 and IKKβ pathways through _S_-nitrosylation of these two proteins [Sarkar et al., 2011]. _S_-nitrosylation is a well-established mechanisms by which NO regulates the function of various target proteins [Stamler et al., 1992]. This post-translational modification of proteins can positively or negatively regulate various signaling pathways, proteins, and metabolic processes [Hess et al., 2005]. However, it is not known whether regulation of autophagic mediators through NO plays a role in Cr(VI)-induced malignant transformation.
In this study, we demonstrate that malignantly transformed lung epithelial cells are inherently resistant to autophagy and apoptosis in comparison to non-tumorigenic lung cells. NO-mediated _S-_nitrosylation of Bcl-2 was observed to be an important mechanism of these cellular processes due to its effect on the Bcl-2–Beclin-1 complex, and subsequent effects on the autophagic process. This is significant to the autophagy resistance we observe in malignantly transformed cells because these cells express a higher basal level of Bcl-2 in comparison to non-tumorigenic lung cells. Overall, this study reveals a novel mechanism by which NO can affect proteins downstream of the JNK1 pathway in autophagy, and is the first to demonstrate a role for Bcl-2 _S_-nitrosylation in resistance to autophagy initiation.
MATERIALS AND METHODS
CHEMICALS AND REAGENTS
Rapamycin, sodium dichromate (Na2Cr2O7·H2O) [Cr(VI)], 2,3-diaminonaphthalene (DAN), mercury(II) chloride (HgCl2), sodium hydroxide (NaOH), NO inhibitor aminoguanidine (AG), p38 inhibitor SB203580, NO donor diethylamine NONOate sodium salt, Bafilomycin A1, Chloroquine diphosphate salt, and _S_-nitrosocysteine were obtained from Sigma–Aldrich (St. Louis, MO). The fluorogenic caspase-9 substrate, LEHD–amino-4-methylcoumarin (AMC), was from Alexis Biochemicals (San Diego, CA). Diaminofluorescein (DAF)-diacetate (DA) was purchased from Molecular Probes (Eugene, OR). Antibodies for Rabbit IgG, Bcl-2, p62, Beclin-1, Atg5, Atg12, P-p38/p38, β-actin, and peroxidase-labeled secondary antibodies were purchased from Cell Signaling Technology (Danvers, MA). Lipofectamine 2000 was purchased from Life Technologies (Carlsbad, CA). The Bcl-2 inhibitor ABT-737 and a Bcl-2 antibody for immunoprecipitation were obtained from Santa Cruz Biotechnologies (Dallas, TX). pAb anti-LC3 antibody (HRP) was from Novus Biologicals (Littleton, CO).
CELL CULTURE
Nontumorigenic human bronchial epithelial Beas-2B cells were obtained from American Type Culture Collection (Manassas, VA). Beas-2B cells were malignantly transformed as previously described [Azad et al., 2010b]. Briefly Beas-2B cells were continuously exposed to 5 μM Cr(VI) in culture for 24 weeks. Cells were passaged weekly and parallel cultures grown in Cr(VI)-free medium provided passage-matched controls. The tumorigenic potential of the malignantly transformed cells was determined by various experiments. Cr(VI)-exposed Beas-2B cells are designated as B-Cr cells so as to distinguish them from the parental Beas-2B cells. Both passage-matched Beas-2B and B-Cr cells were cultured in Dulbecco’s modified Eagle medium (Thermo Scientific) supplemented with 5% FBS, 2 mM L-glutamine, 100 U/ml penicillin, and 100 mg/ml streptomycin in a 5% CO2 environment at 37°C.
PLASMIDS AND TRANSFECTION
Beas-2B and B-Cr cells were seeded in 60 mm dishes until they reached 80% confluence. The cells were then transfected with myc-tagged wild-type (WT) Bcl-2 (WT Bcl-2), double-mutant (C158A/C229A) Bcl-2 (DM Bcl-2), _si_-Bcl-2, or control (pcDNA3) plasmid using Lipofectamine 2000 transfecting agent according to the manufacturer’s protocol. Plasmid DNA (5 μg) was diluted in Opti-MEM Medium (Gibco, Life Technologies) before being added to an Opti-MEM Medium/Lipofectamine Reagent (15 μl) solution in a 1:1 ratio. This DNA–lipid complex was incubated for 5 min before being added to the cells. Cells were incubated at 37°C for 36 h, then treated with 20 μM Cr(VI) for 6 h before being lysed for Western blot analysis.
CASPASE ASSAY
Caspase activity was determined by fluorometric assay with the enzyme substrate LEHD-AMC, which is specifically cleaved by caspase-9 at the Asp residue to release the fluorescent group, AMC. Cell extracts containing 20 μg of protein were incubated with 100 mM HEPES containing 10% sucrose, 10 mM dithiothreitol, 0.1% 3-[(3-cholamidiopropyl)-dimethylammonio]-1-propane sulfonate, and 50 mM caspase substrate in a total reaction volume of 0.25 ml. The reaction mixture was incubated for 60 min at 37°C, and quantified fluorometrically at the excitation and emission wavelengths of 380 and 460 nm, respectively, with a Gen 5 2.0 All-In-One Microplate Reader (BioTek Instruments, Inc.)
APOPTOSIS ASSAY
Apoptosis was determined by Hoechst 33342 DNA fragmentation assay. Briefly, cells were incubated with 10 μg/ml Hoechst 33342 nuclear stain (Life Technologies) for 30 min at 37°C and scoring the percentage of cells having intensely condensed chromatin and/or fragmented nuclei by fluorescence microscopy (EVOS All-in-one digital inverted fluorescence microscope) with software. From random fields 1,000 nuclei were analyzed for each sample. The apoptotic index was calculated as apoptotic nuclei/total nuclei × 100 (%) using ImageJ software (Java image processing, NIH).
AUTOPHAGIC VACUOLE DETECTION
Autophagic vacuoles were detected using the Cyto-ID Autophagy Detection Kit (Enzo Scientific). After cells were seeded in 6-well plates and treated accordingly, 0.25 μl of Cyto-ID Green Detection reagent was added per ml of complete growth medium per well. Plates were incubated at 37°C for 30 min before imaging by fluorescence microscopy (EVOS All-in-one digital inverted fluorescence microscope) with software. Graphs represent percentage of stained cells in each sample well over total number of cells using ImageJ software (Java image processing, NIH).
NO DETECTION
Intracellular NO production was determined by spectrofluorometry using DAF-DA fluorescent probe. After specific treatments, cells were incubated with 10 μM DAF-DA for 30 min at 37°C. Cells were analyzed for DAF fluorescence at the excitation and emission wavelengths of 488 and 538 nm, respectively, with a Gen 5 2.0 All-In-One Microplate Reader (BioTek Instruments, Inc.).
WESTERN BLOTTING
After specific treatments, cells were incubated in lysis buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, and 1 μg/ml leupeptin (Cell Signaling), 100 mM PMSF, and a commercial protease inhibitor (Sigma–Aldrich) and phosphatase inhibitor (Pierce Biotechnology) mixture for 20 min on ice. After insoluble debris was precipitated by centrifugation at 10,000 RPM for 10 min at 4°C, the supernatant were collected and assayed for protein content with bicinchoninic acid assay kit (Thermo Scientific). Equal amount of proteins per sample (20 μg) were resolved on 10% SDS–PAGE and transferred onto 0.45 mm nitrocellulose membranes. The transferred membranes were incubated overnight at 4°C with appropriate primary antibodies diluted in 5% nonfat dry milk in Tris-buffered saline and Tween (25 mM Tris–HCl [pH 7.4], 125 mM NaCl, 0.05% Tween-20), followed by horseradish peroxidase-conjugated isotype-specific secondary antibodies for 1 h at room temperature. The immune complexes were detected by chemiluminescence (Supersignal West Pico; Pierce Biotechnology) using a MyECL Imager (Thermo Scientific), and quantified by imaging densitometry using ImageJ (NIH, Image analysis using Java) digitizing software. Mean densitometry data from independent experiments were normalized to the control.
IMMUNOPRECIPITATION
Immunoprecipitation was performed using the Pierce Classic Magnetic IP/Co-IP Kit (Thermo Scientific). Briefly, after treatments, cells were washed with PBS and lysed in IP lysis/wash buffer on ice for 5 min with periodic mixing. After centrifugation at 14,000_g_ for 10 min at 4°C, the supernatants were collected and the protein content determined by BCA protein assay. Cleared lysates were normalized and 200 μg proteins were incubated with 2.5 μg of anti-Bcl-2 antibody (Santa Cruz Biotechnology) or Rabbit IgG antibody overnight at 4°C. Pierce Protein A/G Magnetic Beads (25 μl) were repeatedly washed and collected with the magnetic stand before being incubated with the antigen sample/antibody mixture for 1 h with mixing. The immune complexes were eventually eluted with 100 μl of elution buffer, and separated by 10% SDS–PAGE and analyzed by Western blotting as described above. Whole cell lysates (10%) were used as a positive control for the protein of interest.
MEASUREMENT OF Bcl-2 _S_-NITROSYLATION
Spectrofluorometry
_S_-nitrosylation of Bcl-2 was measured as previously described [Azad et al., 2006]. In brief, cells were treated, harvested, lysed, and subjected to immunoprecipitation as described earlier. Equal volumes of immune complexes were resuspended in 500 μl of PBS and incubated with HgCl2 (200 μM) and DAN (200 μM) for 0.5 h in dark at room temperature followed by the addition of 1 M NaOH. Samples were then quantified using a Gen 5 2.0 All-In-One Microplate Reader (BioTek Instruments, Inc.) at the excitation and emission wavelengths of 375 and 450 nm, respectively.
_S_-NITROSYLATION (TMT) WESTERN BLOTTING
_S_-nitrosylation of Bcl-2 was also measured using the S_-nitrosylation Western Blot kit (Pierce, Thermo Scientific). Briefly, treated cells were lysed using HENS buffer and centrifuged at 10,000_g for 10 min before performing BCA assay to determine protein concentration. 2 μl of 1 mM MMTS was then added to each 100 μl sample, vortexed, and incubated for 30 min at room temperature to block free cysteine thiols. The protein was then precipitated with six volumes of pre-chilled acetone. Samples were then centrifuged before incubation with 1 μl labeling reagent and 2 μl of 1 M sodium ascorbate for 1–2 h at room temperature. Samples were then separated by 10% SDS–PAGE and analyzed by Western blotting as described above using the anti-TMT antibody provided with the kit.
STATISTICAL ANALYSIS
The data represent means (±SEM) from three or more independent experiments. Statistical analysis was performed by Student’s _t-_test at a significance level of P < 0.05.
RESULTS
DIFFERENTIAL AUTOPHAGIC RESPONSE IN NON-TUMORIGENIC VERSUS MALIGNANTLY TRANSFORMED LUNG CELLS
We characterized the autophagic response in both non-tumorigenic Beas-2B cells and malignantly transformed B-Cr cells (see Materials and Methods section) using fluorescence microscopy. While analysis of the fluorescence images indicated a dose and time-dependent increase in autophagy in both cell lines (Fig. 1A), the effect was more pronounced in Beas-2B cells as compared to B-Cr cells. Interestingly, autophagy is ablated and returns to control levels at the 24 h time-point in Beas-2B cells. We have previously shown that Beas-2B cells exposed to Cr(VI) for 24 h become apoptotic (evidenced by Hoechst dye staining), undergoing rapid cell death [Azad et al., 2010b]. Representative micrographs (Fig. 1B, middle panel) show that a 20 μM Cr(VI) dose triggered significant amount of autophagic vacuole formation in Beas-2B cells as compared to B-Cr cells and control. Rapamycin (autophagy inducer) was used as a positive control to indicate the effectiveness of the Cyto-ID dye to stain autophagic cells. This was further confirmed by Western blot analysis for specific autophagic proteins including Beclin-1, LC3, and Atg12. Significantly higher levels of Beclin-1, LC3-II, and Atg12 proteins were detected in Beas-2B cells as compared to B-Cr cells (Fig. 1C) in response to Cr(VI) treatment.
Fig. 1.

The initial autophagic response in Beas-2B and B-Cr cells. (A) Subconfluent (80%) monolayers of Beas-2B and B-Cr cells were treated with varying concentrations of Cr (VI) (0–50 μM) for different time points (0, 3, 6, 12, and 24 h) and analyzed for autophagic vacuole formation by fluorescence microscopy. Graphs represent the percentage of stained autophagic vacuoles normalized to untreated control. (B) Representative fluorescence micrographs of Beas-2B and B-Cr cells treated with Cr(VI) (20 μM) for 6 h and stained with Cyto-ID dye. Rapamycin (2 μM) was used as a positive control. (C) Beas-2B and B-Cr cells were treated with Cr(VI) (20, 50 μM) for 6 h, and cell lysates were analyzed for Beclin-1, LC3, and Atg12 protein expression. Ntx represents untreated controls. Data are mean ± SEM (n = 4). *P<0.05 versus Beas-2B or B-Cr untreated control.
To further confirm the autophagy-inducing effect of Cr(VI), we characterized autophagic flux by pre-treating Beas-2B and B-Cr cells with Bafilomycin A1 and Chloroquine, two specific autophagosome-lysosome late-stage autophagy inhibitors, followed by Cr (VI) treatment for three different time-points. We then performed Western blotting to analyze LC3-II and p62 (LC3-II-autophagosome binding partner); two late stage autophagy protein markers. At the 6h time-point, an increase in LC3-II expression was observed in Cr (VI)-treated cells over control (Fig. 2A and B) with Beas-2B cells showing a more pronounced increase possibly due to the fact that these cells are undergoing autophagy in response to Cr(VI) treatment. The increase in protein expression clearly dissipated in a time-dependent manner from 6 to 20 h. However, when the autophagy inhibitors are introduced, both LC3-II and p62 expression levels are increased as autophagy is arrested, and both proteins accumulate in the autophagosome, unable to be degraded due to the inhibitors. The inhibitors are not causing any accumulation of the Beclin-1 protein at any of the time points where autophagy is observed, further confirming that the lysomotropic inhibitors only affect late stage autophagy. Overall, Figure 2 shows clearly by way of autophagic flux that Cr(VI) treatment is indeed inducing autophagy in these lung cells.
Fig. 2.

Autophagic flux using bafilomycin A1 and chloroquine. (A) Beas-2B and (B) B-Cr cells were pre-treated with bafilomycin A1 (BAF, 10 nM) or chloroquine (CHL, 20 μM) for 1 h followed by Cr(VI) (20 μM) treatment for three different time points (6, 12, and 20 h). Untreated cells and cells treated with Cr(VI) (20 μM) only were utilized as negative and positive controls. Cell lysates were collected and analyzed for Beclin-1, p62, and LC3 protein expression. Ntx represents untreated controls. Representative blots from three or more experiments.
EFFECT OF THE APOPTOSIS REGULATORS Bcl-2 AND p38 ON Cr(VI)-INDUCED AUTOPHAGY
We have previously shown that apoptosis is the primary mode of cell death in response to Cr(VI) treatment, and that the mitochondrial pathway is the major apoptotic pathway involved [Azad et al., 2006, 2008]. To investigate the potential crosstalk between autophagy and apoptosis, Bcl-2 and p38 protein levels were assessed in Beas-2B and B-Cr cells treated with Cr(VI). While basal expression of phosphorylated p38 (P-p38) was higher in Beas-2B cells; basal expression of Bcl-2 was significantly higher in B-Cr cells (Fig. 3A and B). Cr(VI) treatment increased Bcl-2 and P-p38 expression in a dose-dependent manner in both Beas-2B and B-Cr cells at 6 h. We evaluated the role of Bcl-2 and p38 in mediating the autophagic process in response to Cr(VI). Beas-2B and B-Cr cells were pre-treated with either ABT-737 (Bcl-2 inhibitor) or SB203580 (p38 inhibitor), and the effect on autophagic-vacuole formation was analyzed. Both Bcl-2 and p38 inhibitors led to an increase in autophagic vacuole formation in Beas-2B and B-Cr cells, suggesting an important role for these proteins in autophagy (Fig. 3C and D). This result was validated by evaluating Beclin-1 and LC3 protein levels by Western blotting after cells were treated with ABT-737 and SB203580, respectively. Results indicated upregulation in both Beclin-1 and LC3 levels in the presence of inhibitors for Bcl-2 and p-38 in response to Cr(VI) treatment (Fig. 3E and F).
Fig. 3.

The role of Bcl-2 and p38 in autophagy. (A) Beas-2B and B-Cr cells were treated with Cr(VI) (20, 50 μM) for 6 h, and cell lysates were analyzed for Bcl-2. (B) Beas-2B and B-Cr cells were treated with Cr(VI) (20, 50 μM) for 6 h, and cell lysates were analyzed for p-p38. Blots were reprobed for total p38 levels. (C) Beas-2B and B-Cr cells were pre-treated with ABT-737 (1 μM) for 1 h followed by Cr(VI) (20 μM) treatment for 6 h and analyzed for autophagic vacuole formation by fluorescence microscopy. Graphs represent the percentage of stained autophagic vacuole normalized to untreated control. (D) Beas-2B and B-Cr cells were pre-treated with SB203580 (10 μM) for 1 h followed by Cr(VI) (20 μM) treatment for 6 h and analyzed for autophagic vacuole formation by fluorescence microscopy. Graphs represent the percentage of stained autophagic vacuole normalized to untreated control. (E) Beas-2B and B-Cr cells were pre-treated with ABT-737 (1 μM) or SB203580 (10 μM) for 1 h followed by Cr(VI) (20 μM) treatment for 6 h, and cell lysates (30 μg protein) were prepared and analyzed for Beclin-1, and (F) LC3. Ntx represents untreated controls. Densitometry was performed to determine the relative protein levels after reprobing the membrane with β-actin antibody. Data are mean ± SEM (n = 3). *P<0.05 versus Beas-2B or B-Cr untreated control. #P<0.05 B-Cr versus Beas-2B cells.
NO INHIBITS THE AUTOPHAGIC RESPONSE IN Beas-2B AND B-Cr CELLS
To determine the role of nitrosative stress in autophagy, cellular NO levels were assessed using the DAF-DA fluorescent probe (Fig. 4A). Cr(VI) induced significantly higher NO levels in B-Cr cells in comparison to Beas-2B cells. Cells were then pre-treated with either NO inhibitor Aminoguanidine (AG) or NO donor (DPTA NONOate) and analyzed for autophagic vacuole formation. NO inhibition resulted in increased autophagic vacuole formation in both Beas-2B and B-Cr cells, while pre-treatment with NO donor DPTA NONOate showed an opposite effect, which was more pronounced in Beas-2B cells (Fig. 4B). Furthermore, pre-treatment with AG increased Beclin-1 and LC3-II protein levels in both Beas-2B and B-Cr cells, while DPTA NONOate pre-treatment led to a decrease in Beclin-1 and LC3-II protein levels (Fig. 4C and D), suggesting a clear inhibitory role for NO in mediating Cr(VI)-induced autophagy.
Fig. 4.

NO inhibits the autophagic response. (A) Beas-2B and B-Cr cells were treated with Cr(VI) (20 and 50 μM) for 3 h before NO levels were analyzed by spectrofluorometric measurement of DAF fluorescence. Graphs represent relative fluorescence intensity over untreated control. (B) Beas-2B and B-Cr cells were pre-treated with AG (300 μM) or DPTA NONOate (400 μM) for 1 h followed by Cr(VI) (20 μM) treatment for 6 h and analyzed for autophagic vacuole formation by fluorescence microscopy. Graphs represent the percentage of stained autophagic vacuole normalized to untreated control. (C) Beas-2B and B-Cr cells were pre-treated with AG (300 μM) or DPTA NONOate (400 μM) for 1 h followed by Cr(VI) (20 μM) treatment for 6 h, and cell lysates (30 μg protein) were prepared and analyzed for Beclin-1 and (D) LC3 protein expression by Western blotting. Ntx represents untreated controls. Densitometry was performed to determine the relative protein levels after reprobing the membrane with β-actin antibody. Data are mean ± S.E.M (n = 3). *P<0.05 versus Beas-2B or B-Cr untreated control. #P<0.05 B-Cr versus Beas-2B cells.
EFFECT OF NO MODULATORS ON Bcl-2 EXPRESSION
NO is known to independently inhibit autophagic pathways by nitrosylating and thereby rendering key autophagic marker proteins inactive [Stamler et al., 1992; Sarkar et al., 2011]. Since Bcl-2 was observed to be a key player in autophagy and apoptosis in lung cells, and we know from previous studies that _S_-nitrosylation of Bcl-2 can alter its function [Azad et al., 2006], we evaluated whether NO-mediated Bcl-2 _S_-nitrosylation could alter Cr(VI)-induced autophagy. Beas-2B and B-Cr cells were treated with Cr(VI) and NO modulators, and cell lysates were immunoprecipitated with anti-Bcl-2 antibody followed by nitrosylation analysis using the anti-TMT antibody provided with the _S_-nitrosylation Western blot kit. This allowed for analysis of nitrosylation specific to Bcl-2 protein in our samples. Treatment with Cr(VI) had a negligible effect on Bcl-2 _S_-nitrosylation compared to control, however Bcl-2 _S_-nitrosylation was significantly enhanced by NO donor DPTA NONOate (Fig. 5A). In contrast, NO inhibitor AG had a negative effect on Bcl-2 _S_-nitrosylation. It is well-established that Bcl-2 interacts and complexes with Beclin-1 prior to autophagy [Wei et al., 2008]. This interaction occurs through the BH3 domain of Beclin-1 and prevents Beclin-1 from assembling the pre-autophagosomal proteins needed for autophagy initiation. To determine the effect of Bcl-2 nitrosylation on Bcl-2–Beclin-1 interaction, Beas-2B and B-Cr cells were treated with AG and DPTA NONOate along with Cr(VI) and immunoprecipitated using anti-Bcl-2 antibody followed by Immunoblotting for Beclin-1 protein. Interestingly, an increase in Beclin-1 protein levels with DPTA NONOate pre-treatment and a decrease with AG pre-treatment was observed, especially in Beas-2B cells (Fig. 5B). This indicated that NO-mediated Bcl-2 nitrosylation may have a direct effect on the Bcl-2–Beclin-1 binding complex, regulating the amount of Beclin-1 protein that is tethered to Bcl-2. Figure 5B also clearly shows that treatment of Beas-2B cells with Cr (VI) results in a decrease in the amount of Beclin-1 protein bound to Bcl-2 as compared to control, indicating the detachment of Beclin-1 from the Bcl-2 complex to enter into the autophagy initiation process.
Fig. 5.

Effect of NO modulators on Bcl-2 expression. (A) Beas-2B and B-Cr cells were pre-treated with AG (300 μM) or NONOate (400 μM) followed by Cr(VI) (20 μM) treatment for 6 h and cell lysates (100 μg protein) were prepared and immunoprecipitated with anti-Bcl-2 or control anti-Rabbit IgG antibody. The elutes and the input (10% of the nuclear extracts) were analyzed for Bcl-2 _S_-nitrosylation by immunoblotting with the anti-TMT antibody provided with the _S_-nitrosylation Western Blot kit. Densitometry was performed to determine the relative TMT levels after stripping and reprobing the membrane with anti-Bcl-2 antibody. (B) Beas-2B and B-Cr cells were pre-treated with AG (300 μM) or NONOate (400 μM) for 1 h followed by Cr(VI) (20 μM) treatment for 6 h and cell lysates were immunoprecipitated with anti-Bcl-2 or control anti-Rabbit IgG antibody. The elutes and the input (10% of the nuclear extracts) were analyzed for Beclin-1 expression by immunoblotting. Ntx represents untreated controls. Densitometry was performed to determine the relative Beclin-1 levels after reprobing the membrane with anti-Bcl-2 antibody. Data are mean ± SEM (n = 4). *P<0.05 versus untreated control.
_S_-NITROSYLATION OF Bcl-2 INCREASES RESISTANCE TO AUTOPHAGY
To further elucidate the effect of Bcl-2 _S_-nitrosylation on autophagy, we analyzed the effect of Bcl-2 inhibitor ABT-737 on Beclin-1 levels. Beas-2B and B-Cr cells were pre-treated with ABT-737, immunoprecipitated for Bcl-2 and analyzed for Beclin-1 and _S_-nitrosocysteine (SNO-Cys) levels by Western blotting. While ABT-737 treatment decreased Beclin-1 levels bound to Bcl-2, ABT-737 treatment also resulted in a significant decrease in _S_-nitrosocysteine expression in both Beas-2B and B-Cr cells (Fig. 6A). Once again significantly more nitrosylation is observed in B-Cr cells as compared to Beas-2B cells. This was confirmed using spectrofluorometry, which showed a decrease in Bcl-2 _S_-nitrosylation fluorescence in both Beas-2B and B-Cr cells with ABT-737 pre-treatment (Fig. 6B). To confirm that NO-mediated _S_-nitrosylation of Bcl-2 may be an important mechanism in the Cr(VI)-induced autophagic response, Beas-2B and B-Cr cells were transfected with either wild-type (WT) Bcl-2, non-nitrosylable (C158A/C229A) double-mutant (DM) Bcl-2, or pcDNA3 (control) plasmid, and their effect on the autophagic response was assessed. Expression levels of Beclin-1 and LC3-II were increased in both Beas-2B and B-Cr cells transfected with DM Bcl-2, while _S_-nitroscysteine expression was decreased (Fig. 6C). Spectrofluorometry data also demonstrated a sharp decrease in _S_-nitrosylation in cells transfected with the Bcl-2 DM plasmid in comparison to pcDNA3 and Bcl-2 WT transfected cells (Fig. 6D).
Fig. 6.

_S_-nitrosylation of Bcl-2 in autophagy. (A) Beas-2B and B-Cr cells were pre-treated with ABT-737 (1 μM) for 1 h followed by Cr(VI) (20, 50 μM) treatment for 6 h. Cell lysates were immunoprecipitated with anti-Bcl-2 or control anti-Rabbit IgG antibody. The elutes and the input (10% of the nuclear extracts) were analyzed for Beclin-1 and _S_-nitrosocysteine (SNO-Cys) expression by immunoblotting. Membranes were stripped and reprobed with anti-Bcl-2 antibody for control. (B) Bcl-2 immunoprecipitates from above were analyzed for _S_-nitrosylation by Spectrofluorometry. Graphs represent relative fluorescence intensity over untreated control. (C) Beas-2B and B-Cr cells were transfected with pcDNA3, WT, and DM Bcl-2 plasmids respectively for 36 h followed by Cr(VI) (20 μM) treatment for 6 h and cell lysates were analyzed for Beclin-1, LC3 I/II, and _S_-nitrosocysteine (SNO-Cys) using Western blotting. (D) Beas-2B and B-Cr cells were transfected with pcDNA3, WT, and DM Bcl-2 plasmids respectively for 36 h followed by Cr (VI) (20 μM) treatment for 6 h and cell lysates were analyzed for _S_-nitrosylation by Spectrofluorometry. Ntx represents untreated controls. Graphs represent relative fluorescence intensity over untreated control. Data are mean ± SEM (n = 3). *P<0.05 versus untreated control. #P<0.05 Beas-2B/B-Cr cells (treated with ABT) versus Beas-2B/B-Cr cells (no ABT treatment).
FUNCTIONAL ROLE OF Bcl-2 IN AUTOPHAGY
To further establish the novel role of NO-mediated _S_-nitrosylation of Bcl-2 in autophagy, cells were transfected with either _si_-Bcl-2, Bcl-2 DM, Bcl-2 WT, or pcDNA plasmids. Western blot data demonstrates that Beas-2B and B-Cr cells transfected with the _si_-Bcl-2 or DM-Bcl-2 plasmids showed higher basal levels of Beclin-1 and LC3-II proteins (Fig. 7A and B). Furthermore, Bcl-2 knockdown negated the effect of both NO donor (DPTA NONOate) and NO inhibitor (AG) on the expression of Beclin-1 and LC3-II protein levels. The absence of significant _S_-nitroscysteine expression in _si_-Bcl-2 or DM-Bcl-2 plasmid transfected cells further indicates that nitrosylation of Bcl-2 may be an important regulatory mechanism of autophagy, which may also regulate Beclin-1 and LC3-II protein expression levels.
Fig. 7.

Functional role of Bcl-2 in autophagy. (A) Beas-2B and (B) B-Cr cells were transfected with pcDNA3, WT, DM and _si_-Bcl-2 plasmids respectively for 36 h followed by pre-treatment with AG (300 μM) or NONOate (400 μM) for 1 h and Cr(VI) (20 μM) treatment for 6 h. Cell lysates were collected and analyzed for Beclin-1, LC3 I/II, Bcl-2, and _S_-nitro-Cysteine using Western blotting. Ntx represents untreated controls. Densitometry was performed to determine the relative protein levels after reprobing the membrane with β-actin antibody. Representative blots from three or more experiments.
DISCUSSION
Autophagy is a tightly regulated lysosomal degradation mechanism utilized by cells for maintaining homeostasis and particularly as a response to stress. The two main autophagic pathways are spearheaded by JNK1 and IKKβ. In this study, our focus is on the JNK1 pathway that involves the Bcl-2 protein. It is reported that phosphorylation of Bcl-2 leads to the dissociation of the Bcl-2-Beclin-1 complex, allowing Beclin-1 to complex with Vps34 triggering the initiation of autophagosome formation [Thorburn, 2008; Mizushima and Levine, 2010]; a process that incorporates LC3-II and several Atg-related proteins. We demonstrate that while the prototypical autophagy proteins including LC3, Beclin-1, and the Atg protein family are integral members of the autophagic pathway; p38, Bcl-2, and NO are also critical players in the process. We observed an increase in P-p38 in both Beas-2B and B-Cr cells, while Bcl-2 expression increased in B-Cr cells (Fig. 3). Since Bcl-2 is also a major anti-apoptotic protein that regulates apoptotic cell death [Cleary et al., 1986], while also being a critical member of the autophagy-apoptosis crosstalk within cells [Thorburn, 2008; Sarkar et al., 2011; Zhou et al., 2011], this result indicates an interesting link between autophagy and apoptosis.
Bcl-2 is considered an oncogene because overexpression of Bcl-2 within the cell, simultaneously with the downregulation of proapoptotic genes, results in unchecked cell proliferation—a hallmark of cancer progression. P38 is a mitogen-activated protein kinase that is activated by cell stress signals, and is involved in apoptosis and autophagy by phosphorylating key pathway members and also transcription factors. There is published data indicating p38 activation in astrocytes leads to the direct inhibition of mTOR and subsequently autophagy induction [Tang et al., 2008], while others have indicated that p38 activation in hepatocytes can lead to the suppression of autophagy-related proteolysis [Prick et al., 2006]. Resveratrol, a known anti-cancer agent, has also been shown to induce apoptosis and autophagy in T-cells by p38 MAPK activation [Ge et al., 2013]. Apoptosis analysis in our model showed an increase in apoptosis in Cr(VI)-treated Beas-2B and B-Cr cells. This data was supported by a similar trend seen in Caspase-9 activity in both Beas-2B and B-Cr cells (data not shown). Our data indicates that autophagy can possibly lead to programmed cell death as the cell stress continues to build. Most importantly, while the B-Cr cells are resistant to autophagy, the cells eventually also enter into apoptosis with prolonged Cr(VI)-induced carcinogenesis (Fig. 1A). This was also evident at the 24 h time point in Beas-2B cells where autophagy was replaced by apoptosis in response to Cr(VI) exposure. Analysis of both Beas-2B and B-Cr cells demonstrates that the malignant cells express higher endogenous levels of Bcl-2 but lower levels of total and phosphorylated form of p38 (Fig. 3). This indicates a clear functional role for p38 in our model due to the high level of autophagy induced in B-Cr cells after pre-treatment with SB203580 (p38 inhibitor). It is possible that p38 is functioning to inhibit autophagy initiation in malignantly transformed cells, which may be occurring simultaneously with the induction of an apoptotic pathway. Furthermore pretreatment with specific inhibitors of both Bcl-2 and p38 proteins resulted in a significant increase in LC3-II-autophagic vacuole formation in both Beas-2B and B-Cr cells (Fig. 3C and D). This fluorescence data was corroborated with Western blots showing an increase in Beclin-1 and LC3-II expression in both Beas-2B and B-Cr cells (Fig. 3E and F). Therefore, it is possible that without p38 activation and Bcl-2 to push these stressed cells into apoptosis, they directly initiate the autophagic process as the preferred stress response mechanism.
To establish that Cr(VI) is in fact inducing autophagy in Beas-2B and B-Cr cells, we evaluated autophagic flux. We utilized two well-defined and well-known lysosomotropic reagents that inhibit autophagy by blocking lysosomal acidification and autophagosome-lysosome fusion—Bafilomycin and Chloroquine [Brest et al., 2011; Herzog et al., 2012] in pre-treatments for Beas-2B and B-Cr cells before monitoring LC3-II and p62 turnover. Figure 2 shows that at the 6h time point, Cr(VI) treatment results in a significant increase in expression of LC3-II, which reverts to baseline levels at 20h indicating completion of the autophagy process which culminates in the degradation of LC3-II and p62 in the autophagolysosome. The degradation of LC3-II and its binding partner p62 over time is also a clear indicator of autophagic flux [Mizushima et al., 2010]. Furthermore, inhibition of LC3-II and p62 degradation by both autophagic inhibitors causes accumulation of both proteins at various time points, confirming the autophagic response observed in Figure 1.
The effect of NO modulators on autophagy and related proteins was determined by co-treating cells with autophagic inhibitors and a similar time-course analysis of LC3-II and p62 expression was performed (data not shown). Both Beas-2B and B-Cr cells pre-treated with AG and autophagy inhibitors showed accumulation of p62 and LC3-II for all three time points, thereby establishing autophagic flux similar to that seen in Figure 2. In contrast, pre-treatment with the NO donor NONOate has minimal effect on LC3-II and p62 levels as expected, even with the addition of the autophagy inhibitors. As shown in Figure 4, AG and NONOate induce and downregulate autophagy respectively, without accumulation or turnover of LC3-II and p62 when the autophagy inhibitors are added.
NO is an important cellular signaling molecule involved in many physiological and pathological processes. While NO-mediated _S_-nitrosylation of JNK1 and IKKβ is well documented as a known autophagy inhibitor, _S_-nitrosylation of Bcl-2 has only been linked to its role as an anti-apoptotic protein [Azad et al., 2008, 2010b]. The resistance of B-Cr cells to autophagy may in part be due to their elevated levels of Bcl-2 and NO expression, which can in turn lead to the _S_-nitrosylation of Bcl-2 [Azad et al., 2010a]; a process that we demonstrate is required to stabilize the Beclin-Bcl-2 complex, thereby preventing Beclin-1 release which is required for autophagy initiation. This is the first known study to demonstrate that NO-mediated _S_-nitrosylation of Bcl-2 directly affects the interaction between Beclin-1 and Bcl-2 that occurs prior to autophagy initiation (Fig. 5B). NO levels were shown to be significantly higher in B-Cr cells (Fig. 4A). Treatment with NO inhibitor AG led to a positive effect on autophagic vacuole formation, while the NO donor DPTA NONOate inhibited autophagy specifically in Beas-2B cells (Fig. 4A and B). The same result was observed at the protein level; both Beclin-1 and LC3-II expression levels increased with AG treatment and decreased with NONOate treatment (Fig. 4C and D). These results clearly indicate that NO functions within our model in an anti-autophagic role.
To determine if Bcl-2 _S_-nitrosylation is responsible for the anti-autophagic response of NO, cells were treated with AG and DPTA NONOate prior to Cr(VI) and assayed for _S_-nitrosylation specific to Bcl-2 by immunoprecipitating cell lysates with anti-Bcl-2 antibody. Figure 5 demonstrates an increase in Bcl-2 _S_-nitrosylation in both Beas-2B and B-Cr cells when pre-treated with DPTA NONOate. There was also a significant increase in Beclin-1 levels in cell lysates immunoprecipitated using Bcl-2 antibody, while Cr(VI) treatment was able to reduce Beclin-1 levels possibly by initiating some level of autophagic activation. Furthermore, inhibition of Bcl-2 decreased Beclin-1 levels in both Beas-2B and B-Cr cells, and had a negative effect on the level of _S_-nitrosylation observed supporting the data that the nitrosylation observed in Beas-2B and B-Cr cells is specific to Bcl-2 protein (Fig. 6). Furthermore, we show that the _S_-nitrosylation observed using the _S_-nitrocysteine antibody is specific to Bcl-2 by transfecting cells with a Bcl-2 mutant plasmid that had mutations of cysteines at both residues 158 and 229, rendering it incapable of _S_-nitrosylation [Azad et al., 2006, 2010b]. Transfection with Bcl-2 DM plasmid led to the ablation (Beas-2B cells) or a significant decrease (B-Cr cells) in _S_-nitrocysteine expression, which was corroborated by Spectrofluorometric analysis (Fig. 6C and D). We also utilized a Bcl-2 knockdown plasmid, _si_-Bcl-2. Figure 7 demonstrates that in the absence of Bcl-2, Beclin-1 and LC3 expression is increased in both Beas-2B and B-Cr cells and the effects previously seen with NO donor and inhibitor pre-treatment were nullified. _S_-nitrocysteine expression was significantly inhibited with _si_-Bcl-2, indicating that the nitrosylation is specific for Bcl-2 protein in our system, similar to the observation in Figure 6C. While the role of Bcl-2 _S_-nitrosylation in apoptosis regulation is well understood [Azad et al., 2006; Iyer et al., 2008], we show here that Bcl-2 _S_-nitrosylation is critical in autophagy as well. Given the involvement of p-38 MAPK activity in our system, NO-mediated nitrosylation of Bcl-2 may be linked with the JNK-1 autophagy pathway, revealing a novel mechanism of autophagy regulation downstream of JNK1.
In summary, our data provides evidence that Bcl-2 is a key player in Cr(VI)-induced autophagy in lung epithelial cells. Additionally, NO through its ability to nitrosylate Bcl-2, plays an important role as a negative regulator of autophagy by increasing Bcl-2 stability and its interaction with Beclin-1 (Fig. 8). In demonstrating the importance of _S_-nitrosylation of Bcl-2 in the regulation of autophagic protein expression, we document a novel layer of autophagy regulation, which may represent an important mechanism in cancer development and progression, and may have important implications in carcinogenesis and its prevention. If cancer cells are able to utilize autophagy as a cell survival mechanism, it would be interesting to determine the role of Bcl-2 and NO along with p38 MAPK signaling in this process. Targeting these proteins in conjunction with more traditional therapies may pave the way for novel preventive and therapeutic strategies in lung cancer.
Fig. 8.
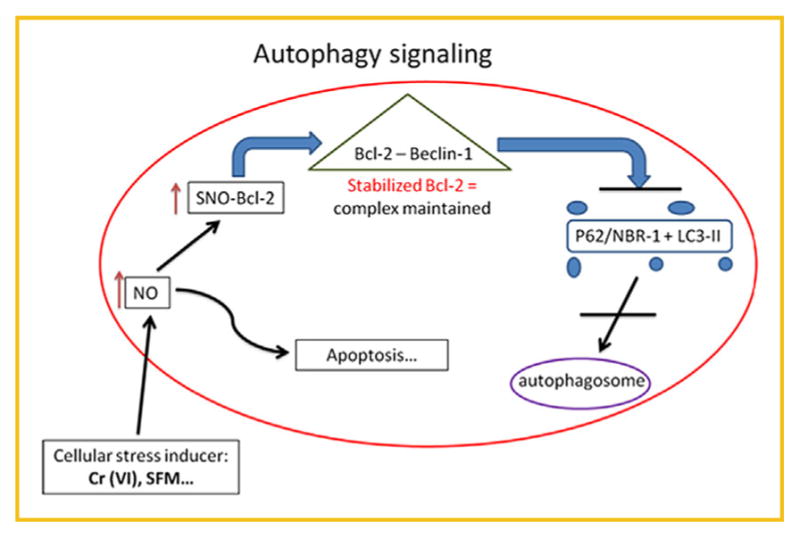
Autophagy signaling overview. The illustration identifies the proposed autophagy mechanism that occurs in Beas-2B and B-Cr cells after Cr(VI) treatment. An increase in NO levels can trigger a Bcl-2-mediated autophagy or apoptosis pathway. High levels of NO expression can lead to _S_-nitrosylation of Bcl-2; a process that stabilizes the Bcl-2-Beclin-1 complex thereby preventing the release of the Beclin-1 protein which is needed for autophagy initiation. This in turn will prevent the fusion of the LC3-II-containing autophagosome and the lysosome; a terminal step in the autophagic process.
Acknowledgments
Grant sponsor: National Institutes of Health; Grant numbers: HL112630, CA173069.
Footnotes
Conflict of interest: None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Chromium, nickel and welding. IARC Monogr Eval Carcinog Risks Hum. 49:1–648. [PMC free article] [PubMed] [Google Scholar]
- 2.Lung Cancereditor’editors. National Cancer Institute at the National Institutes of Health; http://www.cancer.gov. [Google Scholar]
- 3.Abend M. Reasons to reconsider the significance of apoptosis for cancer therapy. Int J Radiat Biol. 2003;79:927–941. doi: 10.1080/09553000310001632958. [DOI] [PubMed] [Google Scholar]
- 4.Azad N, Vallyathan V, Wang L, Tantishaiyakul V, Stehlik C, Leonard SS, Rojanasakul Y. S-nitrosylation of Bcl-2 inhibits its ubiquitin-proteasomal degradation. A novel antiapoptotic mechanism that suppresses apoptosis. J Biol Chem. 2006;281:34124–34134. doi: 10.1074/jbc.M602551200. [DOI] [PubMed] [Google Scholar]
- 5.Azad N, Iyer AK, Manosroi A, Wang L, Rojanasakul Y. Superoxide-mediated proteasomal degradation of Bcl-2 determines cell susceptibility to Cr(VI)-induced apoptosis. Carcinogenesis. 2008;29:1538–1545. doi: 10.1093/carcin/bgn137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Azad N, Iyer A, Vallyathan V, Wang L, Castranova V, Stehlik C, Rojanasakul Y. Role of oxidative/nitrosative stress-mediated Bcl-2 regulation in apoptosis and malignant transformation. Ann N Y Acad Sci. 2010a;1203:1–6. doi: 10.1111/j.1749-6632.2010.05608.x. [DOI] [PubMed] [Google Scholar]
- 7.Azad N, Iyer AK, Wang L, Lu Y, Medan D, Castranova V, Rojanasakul Y. Nitric oxide-mediated bcl-2 stabilization potentiates malignant transformation of human lung epithelial cells. Am J Respir Cell Mol Biol. 2010b;42:578–585. doi: 10.1165/rcmb.2009-0094OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balansky RM, D’Agostini F, Izzotti A, De Flora S. Less than additive interaction between cigarette smoke and chromium(VI) in inducing clastogenic damage in rodents. Carcinogenesis. 2000;21:1677–1682. doi: 10.1093/carcin/21.9.1677. [DOI] [PubMed] [Google Scholar]
- 9.Brest P, Lapaquette P, Souidi M, Lebrigand K, Cesaro A, Vouret-Craviari V, Mari B, Barbry P, Mosnier JF, Hebuterne X, Harel-Bellan A, Mograbi B, Darfeuille-Michaud A, Hofman P. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat Genet. 2011;43:242–245. doi: 10.1038/ng.762. [DOI] [PubMed] [Google Scholar]
- 10.Bucher JR. NTP toxicity studies of sodium dichromate dihydrate (CAS No. 7789-12-0) administered in drinking water to male and female F344/N rats and B6C3F1 mice and male BALB/c and am3-C57BL/6 mice. Toxic Rep Ser. 2007:1–G4. [PubMed] [Google Scholar]
- 11.Cleary ML, Smith SD, Sklar J. Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t (14;18) translocation. Cell. 1986;47:19–28. doi: 10.1016/0092-8674(86)90362-4. [DOI] [PubMed] [Google Scholar]
- 12.Cogliano VJ, Baan R, Straif K, Grosse Y, Lauby-Secretan B, El Ghissani F, Bouvard V, Benbrahim-Tallaa V, Guha L, Freeman N, Galichet C, Wild L. Preventable exposures associated with human cancers. J Natl Cancer Inst. 2011;103:1827–1839. doi: 10.1093/jnci/djr483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collins LG, Haines C, Perkel R, Enck RE. Lung cancer: Diagnosis and management. Am Fam Physician. 2007;75:56–63. [PubMed] [Google Scholar]
- 14.De Flora S. Threshold mechanisms and site specificity in chromium(VI) carcinogenesis. Carcinogenesis. 2000;21:533–541. doi: 10.1093/carcin/21.4.533. [DOI] [PubMed] [Google Scholar]
- 15.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 16.Ge J, Liu Y, Li Q, Guo X, Gu L, Ma ZG, Zhu YP. Resveratrol induces apoptosis and autophagy in T-cell acute lymphoblastic leukemia cells by inhibiting Akt/mTOR and activating p38-MAPK. Biomed Environ Sci. 2013;26:902–911. doi: 10.3967/bes2013.019. [DOI] [PubMed] [Google Scholar]
- 17.Herzog C, Yang C, Holmes A, Kaushal GP. ZVAD-fmk prevents cisplatin-induced cleavage of autophagy proteins but impairs autophagic flux and worsens renal function. Am J Physiol Renal Physiol. 2012;303:F1239–F1250. doi: 10.1152/ajprenal.00659.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 19.Holmes AL, Wise SS, Wise JP., Sr Carcinogenicity of hexavalent chromium. Indian J Med Res. 2008;128:353–372. [PubMed] [Google Scholar]
- 20.Iyer AK, Azad N, Wang L, Rojanasakul Y. Role of S-nitrosylation in apoptosis resistance and carcinogenesis. Nitric Oxide. 2008;19:146–151. doi: 10.1016/j.niox.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jemal A, Clegg LX, Ward E, Ries LA, Wu X, Jamison PM, Wingo PA, Howe HL, Anderson RN, Edwards BK. Annual report to the nation on the status of cancer, 1975–2001, with a special feature regarding survival. Cancer. 2004;101:3–27. doi: 10.1002/cncr.20288. [DOI] [PubMed] [Google Scholar]
- 22.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langard S. One hundred years of chromium and cancer: A review of epidemiological evidence and selected case reports. Am J Ind Med. 1990;17:189–215. doi: 10.1002/ajim.4700170205. [DOI] [PubMed] [Google Scholar]
- 24.Langard S. Role of chemical species and exposure characteristics in cancer among persons occupationally exposed to chromium compounds. Scand J Work Environ Health. 1993;19(Suppl 1):81–89. [PubMed] [Google Scholar]
- 25.Melet A, Song K, Bucur O, Jagani Z, Grassian AR, Khosravi-Far R. Apoptotic pathways in tumor progression and therapy. Adv Exp Med Biol. 2008;615:47–79. doi: 10.1007/978-1-4020-6554-5_4. [DOI] [PubMed] [Google Scholar]
- 26.Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12:823–830. doi: 10.1038/ncb0910-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, Domingo D, Yahalom J. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001;61:439–444. [PubMed] [Google Scholar]
- 29.Pattingre S, Espert L, Biard-Piechaczyk M, Codogno P. Regulation of macroautophagy by mTOR and Beclin 1 complexes. Biochimie. 2008;90:313–323. doi: 10.1016/j.biochi.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 30.Prick T, Thumm M, Haussinger D, Vom Dahl S. Deletion of HOG1 leads to Osmosensitivity in starvation-induced, but not rapamycin-dependent Atg8 degradation and proteolysis: further evidence for different regulatory mechanisms in yeast autophagy. Autophagy. 2006;2:241–243. doi: 10.4161/auto.2743. [DOI] [PubMed] [Google Scholar]
- 31.Sarkar S, Korolchuk VI, Renna M, Imarisio S, Fleming A, Williams A, Garcia-Arencibia M, Rose C, Luo S, Underwood BR, Kroemer G, O’Kane CJ, Rubinsztein DC. Complex inhibitory effects of nitric oxide on autophagy. Mol Cell. 2011;43:19–32. doi: 10.1016/j.molcel.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simonato L, Fletcher AC, Andersen A, Anderson K, Becker N, Chang-Claude J, Ferro G, Gerin M, Gray CN, Hansen KS. A historical prospective study of European stainless steel, mild steel, and shipyard welders. Br J Ind Med. 1991;48:145–154. doi: 10.1136/oem.48.3.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stamler JS, Simon DI, Osborne JA, Mullins ME, Jaraki O, Michel T, Singel DJ, Loscalzo J. S-nitrosylation of proteins with nitric oxide: Synthesis and characterization of biologically active compounds. Proc Natl Acad Sci USA. 1992;89:444–448. doi: 10.1073/pnas.89.1.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang G, Yue Z, Talloczy Z, Hagemann T, Cho W, Messing A, Sulzer DL, Goldman JE. Autophagy induced by Alexander disease-mutant GFAP accumulation is regulated by p38/MAPK and mTOR signaling pathways. Hum Mol Genet. 2008;17:1540–1555. doi: 10.1093/hmg/ddn042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thorburn A. Apoptosis and autophagy: Regulatory connections between two supposedly different processes. Apoptosis. 2008;13:1–9. doi: 10.1007/s10495-007-0154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30:678–688. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: Therapeutic implications. Mol Cancer Ther. 2011;10:1533–1541. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou F, Yang Y, Xing D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS J. 2011;278:403–413. doi: 10.1111/j.1742-4658.2010.07965.x. [DOI] [PubMed] [Google Scholar]