A computational and experimental study of O-glycosylation. Catalysis by human UDP-GalNAc polypeptide:GalNAc transferase-T2 (original) (raw)
. Author manuscript; available in PMC: 2016 Feb 6.
Published in final edited form as: Org Biomol Chem. 2014 May 7;12(17):2645–2655. doi: 10.1039/c3ob42569j
Abstract
It is estimated that >50% of proteins are glycosylated with sugar tags that can modulate protein activity through what has been called the sugar code. Here we present the first QM/MM calculations of human GalNAc-T2, a retaining glycosyltransferase, which initiates the biosynthesis of mucin-type O-glycans. Importantly, we have characterized a hydrogen bond between the β-phosphate of UDP and the backbone amide group from the Thr7 of the sugar acceptor (EA2 peptide) that promotes catalysis and that we propose could be a general catalytic strategy used in peptide O-glycosylation by retaining glycosyltransferases. Additional important substrate–substrate interactions have been identified, for example, between the β-phosphate of UDP with the attacking hydroxyl group from the acceptor substrate and with the substituent at the C2′ position of the transferred sugar. Our results support a front-side attack mechanism for this enzyme, with a barrier height of ~20 kcal mol−1 at the QM(M05-2X/TZVP//BP86/SVP)/CHARMM22 level, in reasonable agreement with the experimental kinetic data. Experimental and in silico mutations show that transferase activity is very sensitive to changes in residues Glu334, Asn335 and Arg362. Additionally, our calculations for different donor substrates suggest that human GalNAc-T2 would be inactive if 2′-deoxy-Gal or 2′-oxymethyl-Gal were used, while UDP-Gal is confirmed as a valid sugar donor. Finally, the analysis herein presented highlights that both the substrate–substrate and the enzyme–substrate interactions are mainly concentrated on stabilizing the negative charge developing at the UDP leaving group as the transition state is approached, identifying this as a key aspect of retaining glycosyltransferases catalysis.
A. Introduction
O-glycans are responsible for a number of unique structural features in mucin glycoproteins and numerous membrane receptors,1–3 and also impart resistance to thermal change and proteolytic attack in a number of diverse proteins.4,5 Moreover, O-linked carbohydrate side chains function as ligands for receptors (e.g. in host–microbial interactions,6 lymphocyte and leukocyte homing7,8) and as signals for protein sorting along the endocytic and biosynthetic pathway.9–13 Importantly, it has been estimated that >50% of proteins are glycosylated, being glycosylation the most abundant post-translational modification of proteins.14
The enzymes UDP-_N_-acetylgalactosamine polypeptide: _N_-acetylgalactosaminyl-transferases (GalNAcTs, EC 2.4.1.41) catalyze the transfer of GalNAc from the sugar donor UDP-GalNAc to serine and threonine residues, in what was the first committed step in mucin biosynthesis to form the Tn antigen (GalNAcα1-O-Ser/Thr).15 Subsequent extension of O-glycan formation proceeds step-wise.16 Several experiments indicate that there is a hierarchical addition of core GalNAc residues to apomucins, implying that the complete glycosylation of certain substrates is dependent on the coordinated action of multiple GalNAcTs.15 Up to 20 members have been identified in humans for this large and evolutionarily conserved family (family 27 in the CAZy17 database). Thus, understanding the catalytic mechanism of GalNAcTs would have important practical implications and would shed light on the O-glycosylation process. More generally, the mechanism for retaining glycosyl transfer stereospecificity has been a matter of debate in glycobiology for the last few decades18 and is hampering the rational design of specific drugs/inhibitors for this class of enzymes.
Two main mechanisms have been proposed in the literature for retaining glycosyltransferases. Initially, and by analogy with retaining glycosidases19,20 (despite the lack of evolutionary relatedness21), a double-displacement mechanism was proposed, with formation and subsequent cleavage of a covalent glycosyl–enzyme (CGE) intermediate involving a nucleophilic residue of the enzyme (Scheme 1A). Although some experiments might support the existence of a CGE,22,23 so far there is no conclusive evidence for it. In a recent computational study we showed that, for bovine α-1,3-galactosyltransferase (α3GalT, GT6 family), the formation of a CGE is consistent with the experimental kinetic data, even though its formation was calculated to be quite endoergic, the minima was not very stable and it may require the presence of the acceptor substrate to be formed at kinetically relevant rates.24 Others have subsequently confirmed that, computationally, this CGE can be characterized for this enzyme.25 However, as new crystal structures of retaining glycosyltransferases have been solved, it has become apparent that most retaining GTs do not present a well-positioned residue in the active site to act as the nucleo-phile.18 In fact, only the GT6 glycosyltransferase family have one. Therefore, the double-displacement mechanism might not be a universal mechanism for retaining glycosyltransferases, and alternative mechanisms have been advocated. The most favoured mechanism is a front-side attack of the acceptor nucleophile that is located on the same side as the leaving group, resulting in the formation of oxocarbenium species that could correspond to a single transition state (Scheme 1B) or to an oxocarbenium-phosphate short-lived ion pair intermediate, with the two corresponding transition states (Scheme 1C). The latest theoretical and experimental studies on retaining GTs24,26–32 lend support to this front-side attack mechanism for those retaining GTs where no good nucleophile is suitably positioned to form the CGE. In fact, even in the case of α3GalT we were able to describe a front-side attack mechanism with a potential energy barrier comparable to the one calculated for the double-displacement.24
Scheme 1.
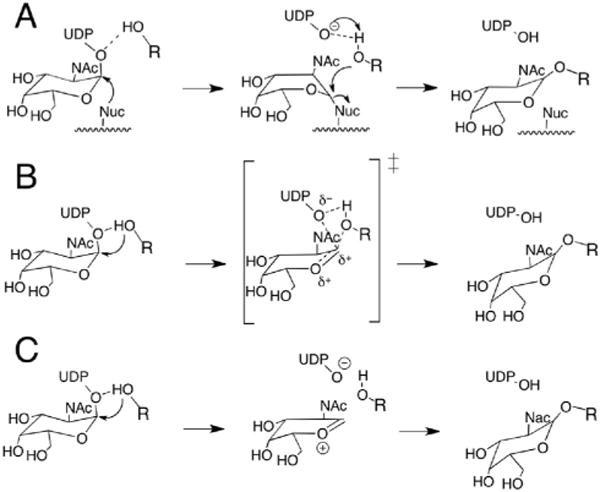
Proposed mechanisms for the retaining GTs. (A) Double-displacement mechanism with formation of a covalently bound glycosylenzyme intermediate. Front-side attack with (B) an oxocarbenium ion-like transition state or (C) a short-lived oxocarbenium–phosphate ion pair intermediate.
In the case of GalNAc-T2 (GT-A fold for the catalytic domain), the available crystallographic structures show that the nearest acidic residues that might function as nucleophiles in a double-displacement mechanism (i.e. Asp224 of the DXH motif binding Mn2+ and Glu334) are ~7 Å away from the β-phosphate oxygen. Consequently, a double displacement seems unlikely or would require a large conformational change. The latter is not observed on the timescale of the MD simulations performed on the Michaelis complex of human GalNAc-T2 by Milac et al.33 On the other hand, their results are more consistent with a front-side mechanism, since the distance between the glycosidic oxygen and the nucleophilic hydroxyl group is about 3 Å and is maintained nearly constant during the simulation, which would be at least structurally consistent with a nucleophilic role of the acceptor.33 These results, together with the available X-ray structures and site-directed mutagenesis data, point to a single-displacement mechanism as the most likely one.
In our previous work on retaining glycosyltransferases,24,30,31 we have emphasized the importance of intra- and inter-substrate interactions in catalyzing this reaction. In most cases, these interactions involve the β-phosphate group of UDP with the O2′ hydroxyl of the transferred monosaccharide, with hydroxyl groups of the acceptor molecule and, most importantly, a hydrogen bond with the hydrogen of the attacking hydroxyl. We have also noted that the particular interactions used by each enzyme:substrate system depend on the chemical identity of the substrates and on their relative binding orientation in the active site (which in turn depends on the specificity of the new glycosidic linkage). Thus, the present study is the first one to analyze how these substrate–substrate interactions act in the case of transferring GalNAc to a peptide acceptor.
We present here a combined computational and experimental work on the catalytic mechanism of human GalNAc-T2 and take it as a model of O-glycosylation by retaining glycosyltransferases. Hybrid quantum mechanical/molecular mechanical (QM/MM) calculations on the full enzyme have been used (Fig. 1). Key factors supporting catalysis have been identified and, when possible, experiments have been performed to test the computational findings.
Fig. 1.
Model system of GalNAc-T2:EA2 used in the QM/MM calculations.
B. Computational methods
An initial fully solvated ternary complex modelled by Milac et al.33 was used as a starting point in the reactivity study. This ternary complex had been built by taking the coordinates of the catalytic domain of GalNAc-T2 and of the acceptor peptide (EA2; sequence PTTDSTTPAPTTK) from the PDB Code 2FFU,34 and modelling the donor substrate (UDP-GalNAc) in the active site using as a template the human GalNAcT-10 (PDB Code 2D7I35), which contains hydrolyzed UDP-GalNAc. In the present study, all water atoms in this solvated ternary complex more than 30 Å away from the anomeric centre (C1′GalNAc) were deleted. This procedure resulted in a system with ~12 630 atoms, including ~2170 TIP3P water molecules (see Fig. 1). The Mn2+ ion present in the original X-ray structure was modelled by the computationally more convenient Mg2+, an approach that has been validated in previous studies of related systems.30,36 Moreover, some experiments have shown that GalNAc-Ts can also be active with Mg2+ and other divalent cations.37 The system was then divided into a QM and an MM zone (Scheme 2). The charge of the QM region was −1 and included 80 atoms: those from the GalNAc ring, the side chain of Thr7 from the acceptor substrate (peptide EA2), Mg2+ and its first coordination sphere (phosphate groups from UDP and the side chains of residues Asp224, His226, His359 and one crystallographic water). Five hydrogen link atoms were added to treat the QM/MM boundary with the charge shift model.38,39 An electronic embedding scheme40 was adopted in the QM/MM calculations, and no cutoffs were introduced for the nonbonding MM and QM/MM interactions. Note that only residues and water molecules within 15 Å of the anomeric centre (~2080 atoms) were allowed to move during the QM/MM calculations.
Scheme 2.
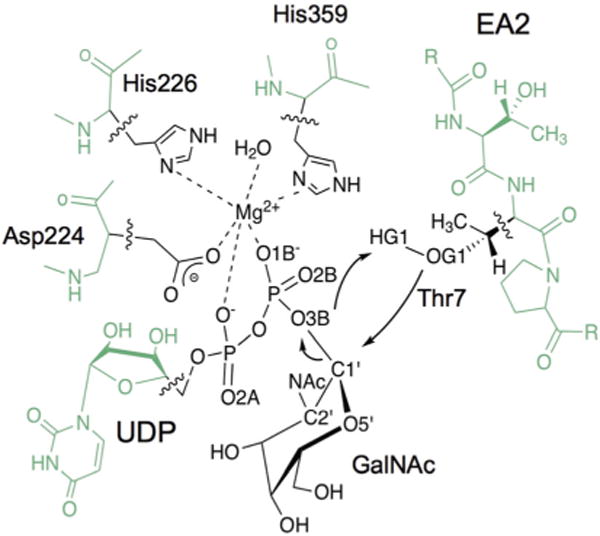
QM/MM partition considered in the present work. QM (MM) atoms are depicted in black (green). Wavy lines indicate the boundary between the QM and MM regions. The arrows indicate the distances considered in the reaction coordinates and the atoms involved are labeled.
This model of the Michaelis complex was then subjected to a NVT QM(SCC-DFTB41,42)/MM(CHARMM2243,44) molecular dynamics simulation using the dynamics module within ChemShell.45 The SHAKE procedure46 was applied at every step for the O–H bonds of the water molecules. A 10 ps MD equilibration run was followed by 80 ps of production MD. Two randomly selected snapshots from this simulation were used in QM/MM geometry optimizations with QM = (BP8647–50/SVP51), a method that we have successfully applied before to study other glycosyltransferases.24,30
Starting at these optimized reactant structures, reaction paths were scanned by performing constrained QM(BP86/SVP)/ CHARMM optimizations along suitably defined reaction coordinates in steps of 0.2 Å. This provided us with starting structures for subsequent full optimization of transition states and products. Frequency calculations were performed for the QM region to confirm that the optimized TS structures are indeed characterized by one imaginary frequency and a suitable transition vector. Additional single-point energy calculations were carried out at the M05-2X52/TZVP53 level which has proven to properly describe retaining GT systems.30,36,54 For the purpose of comparison, additional single-point energies were calculated at the BP86/TZVP, B3LYP47,48,55–57/SVP and B3LYP/TZVP levels of theory.
The electrostatic stabilization provided by different residues to the QM(M05-2X/TZVP)/CHARMM energy was examined by setting their point charges to zero in additional single-point energy calculations. A Natural Bond Orbital (NBO) analysis58–61 was also performed for some of the stationary points using the NBO program v3.162 included in Gaussian09.63
All QM/MM calculations were performed with the modular program package ChemShell using TURBOMOLE64 or Gaussian09 at the DFT level (BP86, B3LYP and M05-2X functionals) or MNDO65 at the SCC-DFTB level. MM energies and gradients were retrieved from DL_POLY,66 using the CHARMM force field. Energy minimizations were done with the low-memory Broyden–Fletcher–Goldfarb–Shanno (L-BFGS) algorithm,67,68 and the TS searches were performed with the microiterative TS optimizer that combines L-BFGS and the partitioned rational function optimizer (P-RFO).69,70 Both L-BFGS and P-RFO algorithms are implemented in the HDLCopt71 module of ChemShell.
C. Results and discussion
Our aim here is to determine if retaining O-linked glycosylation can be achieved via the controversial front-side attack mechanism, as there is no strong nucleophile in the vicinity of the anomeric center in GalNAc-T2, and to reveal the factors that allow for it.
a. Catalytic mechanism
The reaction mechanism was modeled by using both a double (RC = [d(O3BUDP−C1′α-GalNAc)−d(OG1T7−C1′α-GalNAc)]) or a triple (RC = [d(O3BUDP−C1′α-GalNAc)−d(OG1T7−C1′α-GalNAc)−d(HG1T7−O3BUDP)]) reaction coordinate, which are the ones we have used in our studies of LgtC30 and α3GalT,24,31 respectively. In both cases, the calculated potential energy profiles were very similar. Similar results were also obtained for the two frames from the molecular dynamics simulations considered in the QM/MM reactivity studies. For simplicity, the results presented in the main text will refer to frame 1 (see ESI,† for the results of frame 2).
The calculated potential energy profile is depicted in Fig. 2, together with the evolution of key distances along this front-side attack mechanism.
Fig. 2.
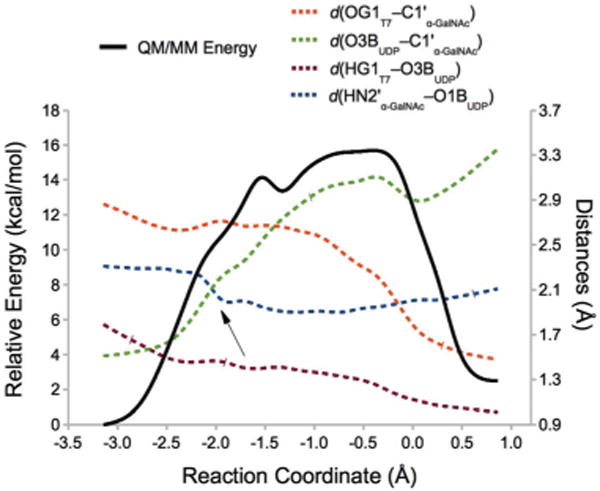
QM(BP86/SVP)/MM(CHARMM22) energy profile for the front-side attack mechanism in GalNAc-T2. Reaction coordinate (RC) = [d(O3BUDP−C1′α-GalNAc) −d(OG1T7−C1′α-GalNAc) −d(HG1T7−O3BUDP)]. The variation of several interatomic distances involved in the reaction is also depicted. HN2′ is the amine hydrogen of the NAc group. The arrow indicates the moment when the NAc group from the α-GalNAc gets properly oriented to favour the O3BUDP−C1′α-GalNAc bond-breaking process (see the main text).
As can be seen in the figure, a potential energy path reproducing the front-side attack mechanism has been obtained and with a reasonable energy maximum at ~16 kcal mol−1 (a phenomenological free energy barrier of 17.3 kcal mol−1 can be derived from the experimental _k_cat value of 3.7 s−1 at 310 K34). The distances depicted show that the reaction starts with the breakage of the UDP-GalNAc glycosidic bond. In fact, the energy required to break this linkage accounts for nearly all the potential energy barrier associated with the whole process (see ESI, Fig. S1,† for the WT enzyme with different substrates and for some mutant enzymes, see below). The potential energy surface is quite planar in the region corresponding to the potential energy maximum for the transferase reaction (with 2.8 Å < _d_(O3BUDP−C1′α-GalNAc) < 3.2 Å and 2.6 Å > d(OG1T7−C1′α-GalNAc) > 2.2 Å), which could seem to indicate a SNi-like mechanism (see Scheme 1). However, no ion-pair intermediate (IP) could be characterized so that the SNi term may be more appropriate. Note, however, that the differences between these two alternatives of the front-side attack mechanism can be very subtle in this kind of potential energy surfaces. In fact, the topology of this surface conditioned that we were also unable to find the corresponding transition state. In what follows, for the QM(BP86/SVP)/MM(CHARMM22) level we will be considering the TS guess (i.e. ?TSi; the structure corresponding to the maximum potential energy value along the RC reaction coordinate) as the effective TS for analysis.
At the QM(SCC-DFTB)/MM(CHARMM22) level a TS was easily identified with an imaginary frequency consistent with the reaction under study (see ESI Tables S1–S2 and Fig. S2–S3† for the structural and energetic results). An estimation of the free energy profile was also done by umbrella sampling calculations at the QM(SCC-DFTB)MM(CHARMM22) level of theory (ESI, Fig. S3†). A qualitative comparison with the potential energy barrier suggests that entropic effects might be relatively small for the present system, as was the case in our previous study of LgtC.30
The evolution of distances along the reaction depicted in Fig. 2 shows that the reaction starts readily with the breakage of the O3BUDP−C1′α-GalNAc bond, as the HOG1T7−O3BUDP hydrogen bond (which we have shown before to be essential in assisting the leaving group departure24) is already present in the reactants.
The QM/MM energy barriers and reaction energies calculated at different levels of theory for frame 1 are shown in Table 1. The potential energy barrier at the reference level (QM = M05-2X/TZVP) is 19.8 kcal mol−1, again in qualitative agreement with the experimentally derived one, suggesting that the front-side attack is actually feasible. The reaction turns out to be almost isoergic (0.3 kcal mol−1) at this level of theory, but slightly exoergic at others; we are not aware of any experimental data on reaction energies to compare with for GalNAc-Ts.
Table 1.
QM/MM potential energy barriers and reaction energies (in kcal mol−1)a at different levels of theory for the front-side attack mechanism for frame 1
| BP86 | B3LYP | M05-2X | ||||
|---|---|---|---|---|---|---|
| SVP | TZVP | SVP | TZVP | SVP | TZVP | |
| R | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| ?TSi | 15.7 | 10.7 | 19.3 | 14.0 | 26.3 | 19.8 |
| P | −2.3 | −0.5 | 1.8 | −1.1 | 3.6 | 0.3 |
The structures of the stationary points are depicted in Fig. 3 (key distances and atomic charges are listed in ESI, Table S4†). Some common trends are observed when comparing to the results obtained in our previous work on retaining glycosyl-transferases.24,30,31 Notably, the ?TSi is highly dissociative (d(O3BUDP−C1′α-GalNAc) = 3.10 Å), which explains why a nucleophilic substitution by the same side is possible, and proton transfer from the attacking nucleophile (OG1T7) to the leaving group oxygen (O3BUDP) takes place very late in the reaction allowing the final formation of the new glycosidic bond. The dissociative character of the TS is also reflected in the positive charge development at the C1′ and O5′ atoms (Δ_q_(C1′ + O5′) = 0.25 a.u.) in going from the R to the ?TSi. The closest residue on the β-face of the donor sugar substrate that could stabilize this positive charge is Ala307, whose carbonyl group is 4.09 Å away from the anomeric centre at the reactants and, mainly as a result of the change in puckering of the ring, gets closer at the ?TSi (3.12 Å). On the other side, the Δ_q_(O3BUDP) = −0.25 a.u. and different interactions are observed that could favour this increment of the negative charge between R and ?TSi. All this will be analysed in the following sections (only for frame 1).
Fig. 3.
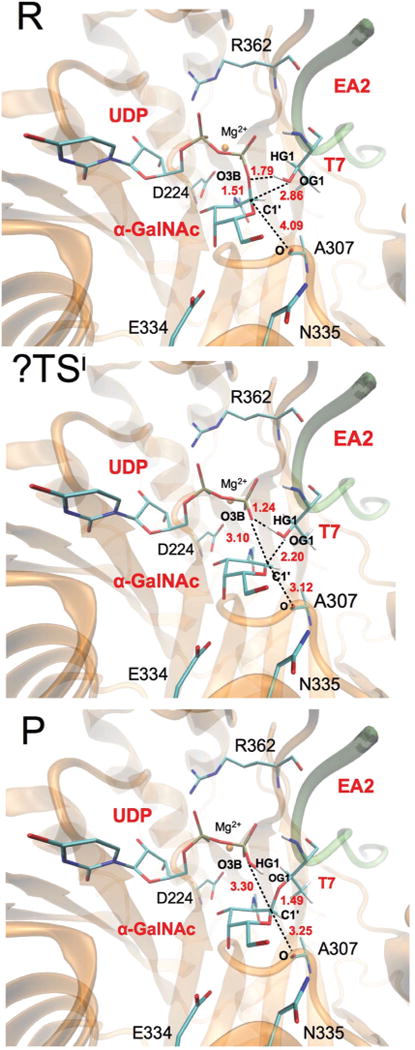
QM(BP86/SVP)/MM(CHARMM22) optimized reactants (R), transition state guess (?TSi) and products (P) for the front-side attack mechanism. The donor and part of the acceptor substrate, together with some relevant residues in the active site, are represented as sticks. Selected distances (in Å) are also indicated.
b. Inter- and intra-substrate interactions
In our previous work on other retaining glycosyltransferases (i.e. α1,3-GalT24 and LgtC30) we have characterized a series of inter- and intra-substrate interactions that seem to be vital to explain the catalytic efficiency of this family of enzymes. These interactions were mainly the hydrogen bonds of the β-phosphate of UDP with the C2′ hydroxyl group of α-Gal, or with hydroxyl groups of the acceptor substrate. In order to identify equivalent interactions in the case of human GalNAc-T2 we have performed an NBO analysis by considering the reactants and the transition state guess. Note that for the reaction catalysed by GalNAc-T2, the transferred sugar has an _N_-acetyl group (NAc) at position C2′ instead of a hydroxyl group, and the acceptor substrate is a peptide instead of another sugar as was the case in our previous studies.
As described for LgtC and α1,3-GalT, the interactions between molecular orbitals involving the leaving group oxygen O3BUDP and the attacking nucleophilic group from the acceptor substrate (here (HG1 – OG1)T7) are the most significant ones (ESI, Table S6,† and Fig. 4A,B). Secondarily, the interactions between the orbitals of OG1T7 and an antibonding molecular orbital of (C1′–O5′)GalNAc are also contributing to the stabilization of the transition state. Interestingly, a new inter-substrate interaction involving the leaving group occurs in this system: a hydrogen bond with the backbone amide of the attacking residue (i.e. Thr7 from peptide EA2, Fig. 4C and D). This interaction could be a general strategy used by retaining GTs transferring the monosaccharide to a peptide acceptor to help stabilize the transition state. For sugar acceptors, an analogous interaction involving the hydroxyl group neighbouring the attacking oxygen was also described in LgtC.30 Note from Fig. 4A and C that the two inter-substrate interactions described for GalNAc-T2 were already present in the Michaelis complex so that the two substrates are bound in the active site optimally oriented for the specificity of the reaction to be catalysed by the enzyme. Finally, another distinctive trait of GalNAc-T2 is the interaction between the 2′-_N_-acetyl group from the donor substrate and the UDP leaving group, which is only present at the transition state (Fig. 4E and F). In the reactants, NAc is interacting with Asp224, one of the residues coordinating the metallic cofactor (i.e. Mg2+), an interaction that is maintained throughout the 40 ns of MD simulation performed by Milac et al. on the Michaelis complex.33 Along the reaction, and due to the change in the sugar ring puckering (i.e. from a distorted 4C1 (puckering parameters ϕ = 137.3°, θ = 15.0°) to a 4E-like ring conformation (ϕ = 242.9°, θ = 31.3°) in R and ?TSi, respectively), NAc gets reoriented and forms a stabilizing interaction with the UDP leaving group. The distance d(HN2′α-GalNAc−O1BUDP) gets shorter in the process, from 2.31 Å in the Michaelis complex to 1.91 Å in the ?TSi and, as a result, N2′α-GalNAc gets hydrogen-bonded to O1BUDP (see Fig. 2).
Fig. 4.
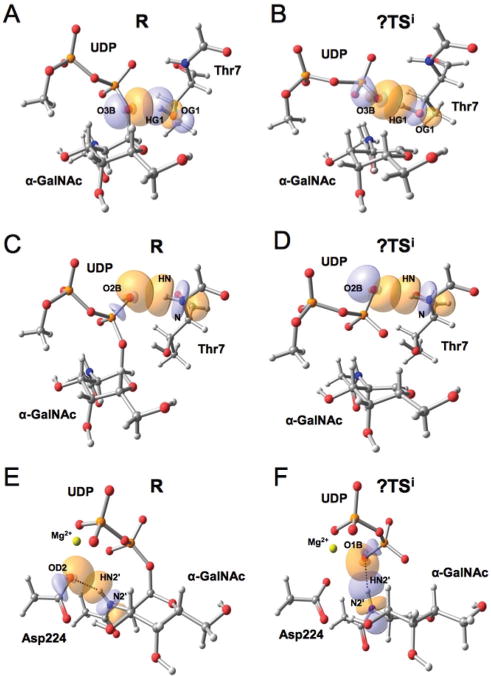
Relevant molecular orbital interactions between the substrates in GalNAc transfer by GalNAc-T2 according to a NBO analysis. These interactions involve: (A) O3BUDP and the incoming OG1T7 in the reactants, R, and (B) in the front-side attack transition state guess, ?TSi; (C) the backbone amide group of Thr7 in R and (D) in the ?TSi; (E) the NAc group of the donor substrate with Asp224 in R and (F) with UDP in the ?TSi. For clarity, just a fraction of the QM atoms is shown. The corresponding second order interaction energies are given in ESI Table S6.†
It is known that some hydrolases like OGA (a glycosidase involved in O-GlcNAcylation cycling) employ the NAc group from GlcNAc itself as a nucleophile to cleave the monosaccharide from serine/threonine.72 However, in the case of glycosyltransferases a catalytic role played by a NAc group from the donor substrate has only very recently been described for an inverting glycosyltransferase. In that work, Tvaroška et al.73 performed a QM/MM study on O-GlcNAc transferase (i.e. uridine diphospho-_N_-acetylglucosamine:poly-peptide β-_N_-acetyl-aminyltransferase, OGT) and found a substrate-assisted mechanism by the NAc group. More specifically, they described a rotation of the C2′–N2′ bond that approaches the HN2′ proton to the oxygen of the breaking glycosidic linkage, thus stabilizing the leaving group negative charge and assisting its departure. The authors hypothesize that OGT could require a mechanism like this to account for the lack of stabilization provided by an absent metal cofactor. This does not appear to be the case for GalNAc-T2, but still the NAc group appears important in catalysis.
To shed more light on the relevance of the NAc group in catalysis, alternative donor substrates were considered in silico.
We substituted this NAc group by OH, H or OCH3 in the original Michaelis complex, which corresponds to considering UDP-Gal, 2′-deoxy-Gal and 2′-oxymethyl-Gal as donor substrates. We assumed that no significant changes in the binding occur and we focused solely on the catalytic process itself. The effects of this functional group substitution on the energy and reaction barriers are summarized in Table 2.
Table 2.
QM(M05-2X/TZVP//BP86/SVP)/MM(CHARMM22) potential energy barriers (Δ_V_‡)a and reaction energies (Δ_V_R), in kcal mol−1, GalNAc-T2 with different donor substrates
| UDP-GalNAc | UDP-Gal | UDP-2′-deoxy-Gal | UDP-2′-oxymethyl-Gal | |
|---|---|---|---|---|
| Δ_V_‡ | 19.8 | 20.9 | 26.5 | 28.5 |
| Δ_V_R | 0.3 | −0.3 | 8.6 | −2.2 |
Using UDP-Gal as a donor substrate results in a potential energy barrier very similar to the one obtained with the original UDP-GalNAc substrate. Inspection of the structures shows that in the Michaelis complex the 2′-hydroxyl group of Gal is also predominantly interacting with Asp224, and that it gets reoriented along the reaction, thus behaving similarly to the NAc group in UDP-GalNAc (ESI, Fig. S4†). Interestingly, in the case of LgtC and α1,3-GalT, which use UDP-Gal as a donor substrate, the interaction of the 2′-hydroxyl group with UDP was already present in the reactants (ESI, Fig. S5†). The NAc-Asp224 interaction in the reactants for GalNAc-T2 prevailed during a 100 ps QM(SCC-DFTB)/MM(CHARMM22) MD simulation of the Michaelis complex. Altogether, our results suggest that human GalNAc-T2 may be able to transfer Gal to the peptide EA2, which is consistent with the experimental results obtained for another acceptor peptide (i.e. Muc2; sequence PTTTPISTTTMVTPTPTPTC).74 In that work, the _V_max values corresponding to the transfer of Gal-NAc and Gal were estimated in 46.1 and 79.9 pmol min−1, respectively, which implies a difference of less than 1 kcal mol−1 between the two donor substrates. Moreover, the authors concluded that giving the relatively small difference between the _K_M values for UDP-GalNAc and UDP-Gal (10 and 27 μM, respectively), UDP-Gal might actually be a naturally relevant substrate of GalNAc-T2. In that sense, note that even if our results predict a slightly higher difference (energy barrier is ~1 kcal mol−1 higher for UDP-Gal), it falls within the order of error that could be expected for the methods used.
A bigger effect is observed for the other two alternative donor substrates. As can be seen in Table 2, the energy barrier increases by ~7 and ~9 kcal mol−1 for 2′-deoxy-Gal and 2′-oxymethyl-Gal, respectively, and also the reaction energies are more affected. These results support the idea that suppressing the interaction between the 2′-NAc (OH) and UDP in UDP-GalNAc (UDP-Gal) would significantly disrupt catalysis in GalNAc-T2. According to our theoretical results, negligible or no detectable residual activity should be expected when 2′-deoxy-Gal or 2′-oxymethyl-Gal is used as the donor substrate. Unfortunately, no experimental data can be provided to test this hypothesis as these compounds are not available.
c. Enzyme–substrate interactions; key enzyme residues
Since the intermediates of the reactions catalyzed by retaining glycosyltransferases are significantly charged, the electrostatic stabilization of the transition state provided by the enzyme residues is expected to be significant. To assess these contributions in the present system, we carried out an analysis for the residues in the active space by switching off the charge of the residue and recalculating the QM/MM interaction energy. The analysis identifies four residues displaying a significant effect: Arg362, Glu334, Ala307 and Trp331, for which electrostatic stabilization energies of 18.6, 11.5, 2.8 and 2.3 kcal mol−1 were estimated, respectively, for the ?TSi as compared to the reactants (QM = M05-2X/TZVP). The most stabilizing residue (i.e. Arg362) interacts with UDP (Fig. 3), as we described in our previous studies of LgtC and α1,3-GalT, and participates in the conformational change that accompanies the binding of the donor substrate. This confirms the key role that the stabilization of the negative charge on the leaving group has in the catalytic efficiency of retaining glycosyltransferases. Arg362 is also involved in acceptor binding via a hydrogen bond that gets slightly elongated along the reaction between its carbonyl backbone and the side chain of Thr6. On the other hand, Glu334 (which is located on the β-face of the sugar ring and is negatively charged) is hydrogen-bonded to the donor substrate and is also involved in substrate binding (see Fig. 3). The carbonyl group of Ala307 is suitably positioned to stabilize the positive charge developing in C1′α-GalNAc as reflected in the 2.83 kcal mol−1 of stabilization that it provides. Tyr331 is another residue that interacts with the leaving group, in this case through a hydrogen bond with O3BUDP.
Interestingly, our analysis suggests that Asn335, a residue lying on the β-face of the donor sugar substrate that could be suggested to be the putative nucleophile in a double-displacement mechanism in GalNAc-T2, does not have a very significant effect on the stabilization of the oxocarbenium species. This is not surprising as it is located at a distance of d(OD1N335−C1′α-GalNAc) = 7.05 Å in the optimized Michaelis complex (see Fig. 3) and of 6.96 ± 0.43 Å during the simulations of this complex performed by Milac et al.33 Moreover, the carbonyl side chain of Asn335 is pointing away from the anomeric carbon, whereas the amide nitrogen is hydrogen bonded to the Ala307 backbone carbonyl.
d. Experimental and in silico mutants
Once the most important residues of the enzyme (from the catalytic point of view) were identified, several mutant forms of GalNAc-T2 were tested in silico and/or experimentally. The position 335 was also considered, given its potential relevance in catalysis upon mutation. Conservative mutations, which are presumed to preserve the structural role of the residue while targeting the chemistry in question, were applied in most cases.
Secreted forms of wild-type GalNAc-T2 or GalNAc-T2, mutated at positions E334Q, N335A, N335D, N335H, N335S and R362K, were expressed (see the Experimental section, Table S7 and Fig. S8 in ESI†) in COS-7 cells. The ability of these mutant forms of GalNAc-T2 to transfer GalNAc to EA2 peptide was tested in vitro (Fig. 5) and the results were compared to in silico determinations.
Fig. 5.
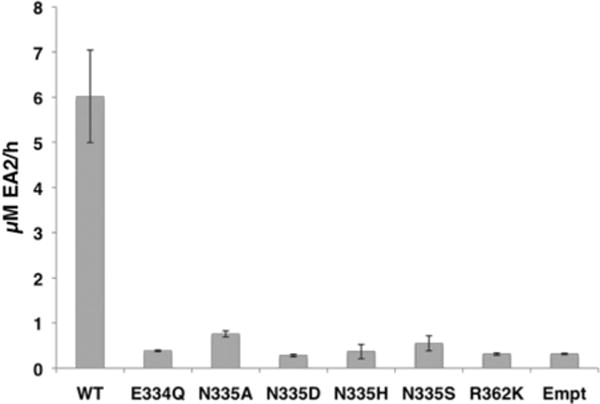
UDP-GalNAc transfer activity by wild-type and mutant human GalNAc-T2 onto the EA2 peptide. Values are the average of two experiments run by triplicate. WT: wild-type GalNAc-T2 and Empt: pIMFK4 empty vector.
Models of R362K, E334Q, N335A and N335D were also built in silico and the energy profile for the front-side attack mechanism was calculated to assess the effect of such mutations on catalysis. Again it is important to highlight that our models of the mutants were built by just replacing the side chain of the original residue. We did this with the purpose of evaluating the effect of the mutations on the catalytic mechanism itself, assuming no significant structural perturbations of the enzyme and a negligible effect on the binding of the substrates. These may not be very good assumptions for some of the mutants (even if conservative mutations have been done), since we are considering residues that are directly implicated in the binding and/or could also have a structural role in the active site. However, our aim was to estimate whether the catalytic performance of the mutants can be explained by only considering the role of the specific residues in the reaction. If important inconsistencies with the experimental kinetic results are found, it may indicate that the overall structure of the active site and/or the binding of the substrates is also affected.
The potential energy barrier and reaction energies associated with these mutants are given in Table 3. The corresponding potential energy profiles were equally planar at their maximum (ESI, Fig. S6†) and, therefore, we did not perform a proper TS search but used the maximum of the potential energy profiles as a TS guess for the analysis.
Table 3.
QM(M05-2X/TZVP//BP86/SVP)/MM(CHARMM22) potential energy barriers (Δ_V_‡)a and reaction energies (Δ_V_R), in kcal mol−1, for wild-type (WT) GalNAc-T2 and considered mutants with UDP-GalNAc and EA2 as substrates
| WT | R362K | E334Q | N335A | N335D | |
|---|---|---|---|---|---|
| Δ_V_‡ | 19.8 | 23.1 | 27.1 | 20.6 | 12.1 |
| Δ_V_R | 0.3 | 0.3 | 3.7 | −0.3 | −4.6 |
As can be seen in Table 3, for the mutant R362K we calculated an increase of ~3 kcal mol−1 in the potential energy barrier, which is quite significant taking into account that a lysine in such position would still stabilize the developing negative charge on the leaving group, although to a lesser extent (ESI, Fig. S7C†). The closer distance between Arg362 and the UDP leaving group can explain the differences found (i.e. d(NH2R362−O2AUDP)/(d(NZK362−O2AUDP) = 3.23/4.97 Å and 2.72/4.62 Å in R and ?TSi, respectively). An increment of ~3 kcal mol−1 in the energy barrier would imply less than 0.04% of residual activity. This is in agreement with the experimental result we have obtained for this mutant, for which no significant transferase activity has been measured. A similar mutation (R365K) was reported for bovine α3GalT, resulting in a small variation of _K_M and a bigger effect on _k_cat but maintaining enzyme activity.75 Therefore, GalNAc-T2 seems to be more sensitive to changes at this position.
An even higher effect is obtained for the less conservative mutation E334Q (energy barrier increase by ~7 kcal mol−1). In the reactants, the carboxylic group of Glu334, located on the β-face of α-GalNAc, is 6 Å away from the anomeric carbon; yet mutating Glu to Asn significantly reduces the electrostatic stabilization role in catalysis of position 334 (Fig. 3 and ESI, Fig. S7A†). Moreover, this is a key residue in the binding of the donor substrate via a hydrogen bond to the OH4 of UDP-GalNAc, a recurrent interaction in retaining GTs. Therefore, mutation of this residue will probably affect both the _K_M for the donor substrate and the _k_cat values. Moreover, since retaining GTs bind their substrates in an ordered and interdependent way, which have also been certified in the case of GalNAcTs,76 an increase in the _K_M value for the acceptor substrate could also be expected. In fact, the experimental E334Q mutant in GalNAc-T2 renders the enzyme inactive for the transfer reaction (Fig. 5). In the case of murine GalNAc-T1, E319Q (Glu319 being the equivalent of Glu334 in human GalNAc-T2) exhibits a residual activity of 0.04%,77 and this is also consistent with our findings.
For the in silico mutant N335A, the energy barrier remains practically unaffected (~1 kcal mol−1 higher, within the order of error of the methods, Table 3), which is consistent with our expectations since Asn335 was not found to be an important residue in terms of electrostatic stabilization (ESI, Fig. S7B†). Mutations to alanine in the equivalent asparagine residue in the murine isoform (i.e. GalNAcT-1) just had a little effect on catalysis,77 consistent with our predictions. However, the experimental data obtained here for the N335A mutation in GalNAc-T2 show no significant transferase activity for the mutant. In fact, all the recombinant mutants tested at this position (N335A, N335D, N335H and N335S) are enzymatically inactive.
The other mutation at position 335 that was tested in silico was N335D. In that case, a drop in the energy barrier of ~7 kcal mol−1 was obtained. This suggests that having a negatively charged residue on the β-face of the donor sugar substrate would turn 335 into a key position, even if it is too far away from the anomeric center to participate in a double-displacement mechanism (d(OD1N335−C1′α-GalNAc) = 7.05 Å in reactants). According to our in silico model, the presence of a strong nucleophile like an Asp at this position would facilitate the leaving group departure, basically because of a better stabilization of the positive charge on the α-GalNAc ring (ESI, Fig. S7B†) and, more importantly, would also delay the nucleophilic attack of the incoming hydroxyl group (ESI, Fig. S6A†). The latter could lead to an increase in the probability of hydrolysis, thus competing with the transferase activity. As mentioned, however, experimentally the N335D mutant does not present transferase activity. The disagreement between our in silico results for position 335 and the experimental ones most likely indicates that the mutation provokes significant changes in the structure or the mechanism that are not captured by our present model. The modification of similar residues in the β face of the sugar ring has also led to unexpected results for other retaining GTs like LgtC, where formation of a glycosyl–enzyme complex with a neighboring Asp residue has been reported.22 A deeper understanding of the effect of such mutations would require much more extensive computational work and, probably, also more experimental data; but this is outside the scope of the present study. In any case, it is clear from the experimental results that GalNAc-T2 transferase activity is very sensitive to any change at position 335 and, more generally, to any mutation of the residues highlighted by our analysis.
D. Conclusions
The presence of O-linked carbohydrates on the surface of many proteins has an important biological role and serves to modulate the physico-chemical properties of glycosylated molecules. The enzymes responsible for forming this O-linkage are glycosyltransferases. In particular, mucin-type O-glycosylation is initiated by GalNAc-Ts. Here, we present a combined computational and experimental approach to investigate the catalytic activity of the human retaining glycosyltransferase GalNAc-T2. Hybrid QM/MM calculations on the full enzyme have been used for the first time to identify the key factors supporting catalysis in protein O-glycosylation by retaining glycosyltransferases, and experiments have been carried out to validate the findings.
A front-side attack mechanism has been described, with an estimated potential energy barrier of ~20 kcal mol−1 (QM = M05-2X/TZVP), which is in reasonable agreement with the experimental kinetic data. The analysis of factors contributing to catalysis highlighted two key amino acids in the active site of the enzyme: Arg362 and Glu334. Tyr331 and the backbone of Ala307 were also found to stabilize the transition state, but to a lesser extent. Experimental and in silico mutation of residues in positions 334 and 362, and of Asn335 (which is situated on the β-face of the GalNAc ring) confirm that transferase activity is very sensitive to mutation at these positions.
Substrate–substrate interactions that contribute to catalysis by stabilizing the developing negative (positive) charge in UDP (α-GalNAc) have also been identified. Taken together, the interactions that predominate are those that stabilize the negative charge developing at the UDP as the transition state is approached, showing that this is a key aspect of retaining glycosyltransferases catalysis. Very interestingly, a new interaction that promotes catalysis has been characterised, that is, a hydrogen bond between the UDP and the amide group from the accepting Thr7 within the EA2 peptide. We propose that this can be a general feature of peptide O-glycosylation by retaining glycosyltransferases. Finally, an intra-substrate interaction involving the 2′ NAc group of α-GalNAc has also been described to stabilize the transition state. Complementary studies in silico with alternative donor substrates suggest that human GalNAc-T2 would be inactive if 2′-deoxy-Gal or 2′-oxymethyl-Gal were used, while UDP-Gal is confirmed as a valid substrate.
Supplementary Material
Supplemental
Acknowledgments
The authors wish to acknowledge Dr A. Milac for providing the coordinates of the starting model for the calculations and also Dr T. Fritz and Dr Jaya Raman for helpful discussions. We acknowledge financial support from the Spanish “Ministerio de Economía y Competitividad” through project CTQ2011-24292 and the “Ramon y Cajal” program (L.M.), and from the “Generalitat de Catalunya”, project 2009SGR409, and the intramural program of NIDCR, NIH.
Footnotes
†
Electronic supplementary information (ESI) available: Details on the experimental methods as well as supplementary results. See DOI: 10.1039/c3ob42569j
Contributor Information
Lawrence A. Tabak, Email: Lawrence.Tabak@nih.gov.
Laura Masgrau, Email: Laura.Masgrau@uab.cat.
Notes and references
- 1.Jentoft N. Trends Biochem Sci. 1990;15:291. doi: 10.1016/0968-0004(90)90014-3. [DOI] [PubMed] [Google Scholar]
- 2.Moody AM, Chui D, Reche PA, Priatel JJ, Marth JD, Reinherz EL. Cell. 2001;107:501. doi: 10.1016/s0092-8674(01)00577-3. [DOI] [PubMed] [Google Scholar]
- 3.Xu Z, Weiss A. Nat Immunol. 2002;3:764. doi: 10.1038/ni822. [DOI] [PubMed] [Google Scholar]
- 4.Sauer J, Sigurskjold BW, Christensen U, Frandsen TP, Mirgorodskaya E, Harrison M, Roepstorff P, Svensson B. Biochim Biophys Acta. 2000;1543:275. doi: 10.1016/s0167-4838(00)00232-6. [DOI] [PubMed] [Google Scholar]
- 5.Garner B, Merry AH, Royle L, Harvey DJ, Rudd PM, Thillet J. J Biol Chem. 2001;276:22200. doi: 10.1074/jbc.M102150200. [DOI] [PubMed] [Google Scholar]
- 6.Hooper LV, Gordon JI. Glycobiology. 2001;11:1R. doi: 10.1093/glycob/11.2.1r. [DOI] [PubMed] [Google Scholar]
- 7.Yeh JC, Hiraoka N, Petryniak B, Nakayama J, Ellies LG, Rabuka D, Hindsgaul O, Marth JD, Lowe JB, Fukuda M. Cell. 2001;105:957. doi: 10.1016/s0092-8674(01)00394-4. [DOI] [PubMed] [Google Scholar]
- 8.Somers WS, Tang J, Shaw GD, Camphausen RT. Cell. 2000;103:467. doi: 10.1016/s0092-8674(00)00138-0. [DOI] [PubMed] [Google Scholar]
- 9.Alfalah M, Jacob R, Preuss U, Zimmer KP, Naim H, Naim HY. Curr Biol. 1999;9:593. doi: 10.1016/s0960-9822(99)80263-2. [DOI] [PubMed] [Google Scholar]
- 10.Altschuler Y, Kinlough CL, Poland PA, Bruns JB, Apodaca G, Weisz OA, Hughey RP. Mol Biol Cell. 2000;11:819. doi: 10.1091/mbc.11.3.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Breuza L, Garcia M, Delgrossi MHH, Le Bivic A. Exp Cell Res. 2002;273:178. doi: 10.1006/excr.2001.5442. [DOI] [PubMed] [Google Scholar]
- 12.Naim HY, Joberty G, Alfalah M, Jacob R. J Biol Chem. 1999;25:17961. doi: 10.1074/jbc.274.25.17961. [DOI] [PubMed] [Google Scholar]
- 13.Zheng X, Sadler JE. J Biol Chem. 2002;277:6858. doi: 10.1074/jbc.m109857200. [DOI] [PubMed] [Google Scholar]
- 14.Schjoldager KTBG, Clausen H. Biochim Biophys Acta. 2012;1820:2079. doi: 10.1016/j.bbagen.2012.09.014. [DOI] [PubMed] [Google Scholar]
- 15.Ten Hagen KG, Fritz TA, Tabak LA. Glycobiology. 2003;13:1R. doi: 10.1093/glycob/cwg007. [DOI] [PubMed] [Google Scholar]
- 16.Strous GJ. Proc Natl Acad Sci U S A. 1979;76:2694. doi: 10.1073/pnas.76.6.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. Nucleic Acids Res. 2009;37:D233. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lairson LL, Henrissat B, Davies GJ, Withers SG. Annu Rev Biochem. 2008;77:521. doi: 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- 19.Bottoni A, Miscione GP, De Vivo M. Proteins: Struct, Funct, Bioinf. 2005;59:118. doi: 10.1002/prot.20396. [DOI] [PubMed] [Google Scholar]
- 20.Bowman AL, Grant IM, Mulholland AJ. Chem Commun. 2008:4425. doi: 10.1039/b810099c. [DOI] [PubMed] [Google Scholar]
- 21.Davies GJ, Withers SG. In: Comprehensive Biological Catalysis. Sinnott ML, editor. London: 1998. pp. 119–208. [Google Scholar]
- 22.Lairson LL, Chiu CP, Ly HD, He S, Wakarchuk WW, Strynadka NC, Withers SG. J Biol Chem. 2004;279:28339. doi: 10.1074/jbc.M400451200. [DOI] [PubMed] [Google Scholar]
- 23.Soya N, Fang Y, Palcic MM, Klassen JS. Glycobiology. 2011;21:547. doi: 10.1093/glycob/cwq190. [DOI] [PubMed] [Google Scholar]
- 24.Gómez H, Lluch JM, Masgrau L. J Am Chem Soc. 2013;135:7053. doi: 10.1021/ja4024447. [DOI] [PubMed] [Google Scholar]
- 25.Rojas-Cervellera V, Ardèvol A, Boero M, Planas A, Rovira C. Chemistry. 2013;19:14018. doi: 10.1002/chem.201302898. [DOI] [PubMed] [Google Scholar]
- 26.Tvaroška I. Carbohydr Res. 2004;339:1007. doi: 10.1016/j.carres.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 27.Errey JC, Lee SS, Gibson RP, Martinez-Fleites C, Barry CS, Jung PM, O’Sullivan AC, Davis BG, Davies GJ. Angew Chem, Int Ed. 2010;49:1234. doi: 10.1002/anie.200905096. [DOI] [PubMed] [Google Scholar]
- 28.Lee SS, Hong SY, Errey JC, Izumi A, Davies GJ, Davis BG. Nat Chem Biol. 2011;7:631. doi: 10.1038/nchembio.628. [DOI] [PubMed] [Google Scholar]
- 29.Ardèvol A, Rovira C. Angew Chem, Int Ed. 2011;50:10897. doi: 10.1002/anie.201104623. [DOI] [PubMed] [Google Scholar]
- 30.Gómez H, Polyak I, Thiel W, Lluch JM, Masgrau L. J Am Chem Soc. 2012;134:4743. doi: 10.1021/ja210490f. [DOI] [PubMed] [Google Scholar]
- 31.Gómez H, Lluch JM, Masgrau L. Carbohydr Res. 2012;356:204. doi: 10.1016/j.carres.2012.03.027. [DOI] [PubMed] [Google Scholar]
- 32.Schuman B, Evans SV, Fyles TM. PLoS One. 2013;8:e71077. doi: 10.1371/journal.pone.0071077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Milac AL, Buchete NV, Fritz TA, Hummer G, Tabak LA. J Mol Biol. 2007;373:439. doi: 10.1016/j.jmb.2007.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fritz TA, Raman J, Tabak LA. J Biol Chem. 2006;281:8613. doi: 10.1074/jbc.M513590200. [DOI] [PubMed] [Google Scholar]
- 35.Kubota T, Shiba T, Sugioka S, Furukawa S, Sawaki H, Kato R, Wakatsuki S, Narimatsu H. J Mol Biol. 2006;359:708. doi: 10.1016/j.jmb.2006.03.061. [DOI] [PubMed] [Google Scholar]
- 36.Kóňa J, Tvaroška I. Chem Pap. 2009;63:598. [Google Scholar]
- 37.Elhammer A, Kornfeld S. J Biol Chem. 1986;261:5249. [PubMed] [Google Scholar]
- 38.Sherwood P, de Vries AH, Collins SJ, Greatbanks SP, Burton NA, Vincent MA, Hillier IH. Faraday Discuss. 1997;106:79. [Google Scholar]
- 39.de Vries AH, Sherwood P, Collins SJ, Rigby AM, Rigutto M, Kramer GJ. J Phys Chem B. 1999;103:6133. [Google Scholar]
- 40.Bakowies D, Thiel W. J Phys Chem. 1996;100:10580. [Google Scholar]
- 41.Elstner M, Porezag D, Jungnickel G, Elsner J, Haugk M, Frauenheim T, Suhai S, Seifert G. Phys Rev B: Condens Matter. 1998;58:7260. [Google Scholar]
- 42.Frauenheim T, Seifert G, Elstner M, Niehaus T, Köhler C, Amkreutz M, Sternberg M, Hajnal Z, Carlo AD, Suhai S. J Phys: Condens Matter. 2002;14:3015. [Google Scholar]
- 43.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiórkiewicz-Kuczera J, Yin D, Karplus M. J Phys Chem B. 1998;102:3586. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 44.MacKerell AD, Feig M, Brooks CL. J Am Chem Soc. 2004;126:698. doi: 10.1021/ja036959e. [DOI] [PubMed] [Google Scholar]
- 45.Sherwood P, de Vries AH, Guest MF, Schreckenbach G, Catlow CRA, French SA, Sokol AA, Bromley ST, Thiel W, Turner AJ, Billeter S, Terstegen F, Thiel S, Kendrick J, Rogers SC, Casci J, Watson M, King F, Karlsen E, Sjovoll M, Fahmi A, Schäfer A, Lennartz C. J Mol Struct (THEOCHEM) 2003;632:1. [Google Scholar]
- 46.Ryckaert JP, Ciccotti G, Berendsen HJC. J Comput Phys. 1977;23:327. [Google Scholar]
- 47.Slater JC. Phys Rev. 1951;81:385. [Google Scholar]
- 48.Becke AD. Phys Rev A. 1988;38:3098. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 49.Vosko SH, Wilk L, Nusair M. Can J Phys. 1980;58:1200. [Google Scholar]
- 50.Perdew JP. Phys Rev B: Condens Matter. 1986;33:8822. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
- 51.Schäfer A, Horn H, Ahlrichs R. J Chem Phys. 1992;97:2571. [Google Scholar]
- 52.Zhao Y, Schultz NE, Truhlar DG. J Chem Theory Comput. 2006;2:364. doi: 10.1021/ct0502763. [DOI] [PubMed] [Google Scholar]
- 53.Schäfer A, Huber C, Ahlrichs R. J Chem Phys. 1994;100:5829. [Google Scholar]
- 54.Gábor IC, Alfred DF, Glenn PJ, Carlos AS. J Chem Theory Comput. 2009;5:679. [Google Scholar]
- 55.Becke AD. J Chem Phys. 1993;98:1372. [Google Scholar]
- 56.Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. J Phys Chem. 1994;98:11623. [Google Scholar]
- 57.Lee CT, Yang WT, Parr RG. Phys Rev B: Condens Matter. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 58.Foster JP, Weinhold F. J Am Chem Soc. 1980;102:7211. [Google Scholar]
- 59.Reed AE, Weinhold F. J Chem Phys. 1983;78:4066. [Google Scholar]
- 60.Reed AE, Weinstock RB, Weinhold F. J Chem Phys. 1985;83:735. [Google Scholar]
- 61.Reed AE, Curtiss LA, Weinhold F. Chem Rev. 1988;88:899. [Google Scholar]
- 62.Glendening ED, Reed AE, Carpenter JE, Weinhold F. Program NBO Version 3.1 [Google Scholar]
- 63.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian09, revision A.1. Gaussian, Inc; Wallingford CT: 2009. [Google Scholar]
- 64.Ahlrichs R, Bär M, Häser M, Horn H, Kölmel C. Chem Phys Lett. 1989;162:165. [Google Scholar]
- 65.Thiel W. Program MNDO2009, Version 7.0. Max-Planck-Institut für Kohlenforschung; Mülheim: 2005. [Google Scholar]
- 66.Smith W, Forester TR. J Mol Graphics. 1996;14:136. doi: 10.1016/s0263-7855(96)00043-4. [DOI] [PubMed] [Google Scholar]
- 67.Nocedal J. Math Comp. 1980;35:773. [Google Scholar]
- 68.Liu DC, Nocedal J. Math Program. 1989;45:503. [Google Scholar]
- 69.Banerjee A, Adams N, Simons J, Shepard R. J Phys Chem. 1985;89:52. [Google Scholar]
- 70.Baker J. J Comput Chem. 1986;7:385. [Google Scholar]
- 71.Billeter SR, Turner AJ, Thiel W. Phys Chem Chem Phys. 2000;2:2177. [Google Scholar]
- 72.Vocadlo DJ, Whiters SG. Biochemistry. 2005;44:12809. doi: 10.1021/bi051121k. [DOI] [PubMed] [Google Scholar]
- 73.Tvaroška I, Kozmon S, Wimmerová M, Koča J. J Am Chem Soc. 2012;134:15563. doi: 10.1021/ja307040m. [DOI] [PubMed] [Google Scholar]
- 74.Wandall HH, Hassan H, Mirgorodskaya E, Kristensen AK, Roepstorff P, Bennett EP, Nielsen PA, Hollingsworth MA, Burchell J, Taylor-Papadimitriou J, Clausen H. J Biol Chem. 1997;272:23503. doi: 10.1074/jbc.272.38.23503. [DOI] [PubMed] [Google Scholar]
- 75.Jamaluddin H, Tumbale P, Withers SG, Acharya KR, Brew K. J Mol Biol. 2007;369:1270. doi: 10.1016/j.jmb.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 76.Wragg S, Hagen FK, Tabak LA. J Biol Chem. 1995;270:16947. doi: 10.1074/jbc.270.28.16947. [DOI] [PubMed] [Google Scholar]
- 77.Hagen FK, Hazes B, Raffo R, deSa D, Tabak LA. J Biol Chem. 1999;274:6797. doi: 10.1074/jbc.274.10.6797. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental