Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo (original) (raw)
. Author manuscript; available in PMC: 2016 May 18.
Published in final edited form as: Leukemia. 2015 Sep 15;30(2):492–500. doi: 10.1038/leu.2015.247
Abstract
Adoptive T-cell therapy with gene-modified T-cells expressing a tumor-reactive T-cell receptor (TCR) or chimeric antigen receptor (CAR) is a rapidly growing field of translational medicine and has shown success in the treatment of B-cell malignancies and solid tumors. In all reported trials, patients have received T-cell products comprised of random compositions of CD4+ and CD8+ naïve and memory T-cells, meaning that each patient received a different therapeutic agent. This variation might have influenced the efficacy of T-cell therapy, and complicates comparison of outcomes between different patients and across trials. We analyzed CD19 CAR-expressing effector T-cells derived from different subsets (CD4+/CD8+ naïve, central memory, effector memory). T-cells derived from each of the subsets were efficiently transduced and expanded, but showed clear differences in effector function and proliferation in vitro and in vivo. Combining the most potent CD4+ and CD8+ CAR-expressing subsets resulted in synergistic antitumor effects in vivo. We show that CAR-T-cell products generated from defined T-cell subsets can provide uniform potency compared with products derived from unselected T-cells that vary in phenotypic composition. These findings have important implications for the formulation of T-cell products for adoptive therapies.
Introduction
Immunotherapy with gene-modified T-cells expressing a tumor-reactive chimeric antigen receptor (CAR) is a rapidly evolving research field1,2. Impressive responses have been achieved in some patients with refractory acute lymphoblastic leukemia (ALL), chronic lymphocytic leukemia (CLL), and non-Hodgkin’s lymphoma after infusing autologous T-cells expressing a CAR specific for the B-lineage molecule CD193–8. Tumor regression appears to correlate with the level and duration of CAR-T-cell engraftment, and the subset of patients in whom CD19 CAR-T-cells proliferate and persist in the blood have continuous on-target depletion of normal CD19+ B-cells and are more likely to remain in remission3–10.
Designing optimized CARs with enhanced signaling to sustain T-cell proliferation and survival may improve the efficacy of CAR-T-cells11–16. Generating cell products derived from subsets of CD8+ and CD4+ T-cells with superior intrinsic abilities for proliferation and survival after transfer might also enhance efficacy. CD8+ and CD4+ T-cells exist as naïve (TN), effector (TE), and memory (TM) subpopulations delineated by changes in surface phenotype after antigen exposure. TM are further divisible into central (TCM) and effector memory (TEM) subsets that differ in phenotype, transcriptional profile, and self-renewal capacity17–19. Mouse models have defined lineage relationships of these CD8+ T-cell subsets. Fate mapping of the differentiation of TN in response to antigen supports a model in which TN differentiate in a linear fashion to long-lived TCM that serve as stem cells for antigen-specific immune responses, and to shorter-lived TEM and TE cells18,20–22. CD4+ T-cells also express TN, TCM, and TEM surface markers and provide help for cytolytic T-cells and antibody producing B-cells23. Clinical trials in cancer have not considered the derivation of CAR-T-cells from defined subsets despite evidence for synergy between CD8 and CD4 cells in an HIV CAR trial that might be further enhanced by subset selection24,25; rather CD3+ T-cells are selected and non-specifically activated from PBMC with anti-CD3 mAb before transduction and expansion. This strategy simplifies manufacturing of cell products but the frequency of CD8+ and CD4+ T-cell subsets in the blood can differ markedly in individuals due to age, pathogen exposure, and the lymphocytotoxic effects of chemotherapy26,27. As a consequence, CAR-T-cell products prepared from PBMC contain divergent proportions of CD8+ and CD4+ T-cell subsets, and this heterogeneity could contribute to the differences in efficacy and toxicity observed in clinical trials3,5,6,10.
Here, we purified individual CD8+ and CD4+ T-cell subsets from normal donors and patients with B-cell malignancies before their genetic modification with a lentiviral vector encoding a CAR, enabling analysis of the functional activity of subsets and subset combinations in vitro and in vivo. Our data show that the composition of CAR-T-cell products profoundly influences function and therapeutic efficacy, and reveals synergy between CD4+ and CD8+ CAR-T-cells in mediating antitumor responses in vivo.
Materials and Methods
Cells
293T cells (ATCC_CRL-11268) were cultured in DMEM/10% FCS and 100 U/ml penicillin/streptomycin and K562 (ATCC_CCL-243), K562/CD1928, K562/ROR113, Raji (ATCC_CCL-86), Raji/ffluc29, and JeKo-1 (ATCC_CRL-3006) in RPMI-1640/5% FCS and 100 U/ml penicillin/streptomycin. Cell lines were tested monthly for the absence of mycoplasma. T-cells were cultured in RPMI-1640/10% human serum, 100 U/ml penicillin/streptomycin, 4 mM L-glutamine, 50 μM β-mercaptoethanol, and 50 U/ml IL-2. PBMC were isolated over Ficoll-Hypaque (Sigma), CD4+ or CD8+ T-cells were isolated by negative magnetic selection (Miltenyi), and then labeled for CD4/CD8/CD45RO/CD62L. TN (CD45RO−/CD62L+), TCM (CD45RO+/CD62L+), and TEM (CD45RO+/CD62L−) were sorted to >99% purity on a FACSAriaII. PBMC from CLL patients were used as autologous CLL samples.
Vector construction, preparation of lentivirus, generation of CAR-T-cells, and in vitro functional assays
The CD19 CAR included an scFv of the CD19-specific mAb FMC6330 with a 218 linker31 between the VL and the VH domains, the hinge region of IgG4 with a serine to proline substitution at position 10, the CD28 transmembrane region, a 4-1BB costimulatory domain, and intracellular CD3ζ. The CD19 CAR gene was linked to a truncated epidermal growth factor receptor (EGFRt) by T2A and cloned into the lentiviral vector epHIV732. The ROR1-specific CAR was described previously33. Lentivirus was generated by transient transfection of 293T cells34 using psPAX2 and pMD2G packaging plasmids. T-cell were activated with anti-CD3/CD28-beads (Life Technologies), and transduced one day after activation by centrifugation at 800xg for 90 minutes at 32°C with lentiviral supernatant supplemented with 1 μg/mL polybrene (Millipore). T-cells were enriched for EGFRt+ cells by FACS and expanded with an irradiated B-lymphoblastoid cell line (LCL)28. Functional in vitro assays were performed as described; cytotoxicity was analyzed by standard chromium release assays, cytokine levels by ELISA or Luminex assays; and proliferation of CAR-T-cells by carboxyfluorescein succinimidyl ester (CFSE) dilution assays13.
Immunophenotyping
T-cells were stained with combinations of the following conjugated mAbs: CD4, CD8, CD27, CD28, CD45, CD45RA, CD45RO, CD62L, and matched isotype controls (BD Biosciences). Transduced T-cells were stained with biotin-conjugated anti-EGFR antibody (ImClone Systems Incorporated) and streptavidin-PE (BD Biosciences). Flow cytometry was performed on a FACSCantoII and data analyzed with FlowJo (Treestar).
Transfer of T-cells in NOD/SCID/γc−/− (NSG) mice
Six to eight week old female NSG mice were obtained from the Jackson Laboratory or bred in-house, engrafted via tail vein with 5×105 Raji/ffluc cells, and one week later, injected i.v. with CAR- or control T-cells that had been expanded with LCL for eight or nine days. Bioluminescence imaging was performed as described13. Peripheral blood was obtained by retro-orbital bleeding and erythrocytes were removed using ammonium-chloride-potassium lysis buffer. At least five mice per experimental group were used for data analysis to provide 81% power to detect an effect size of 1.75, based on a t-test with a 1-sided 0.05 level of significance. The investigator was blinded to group allocation.
Statistics
A two-sided student’s t-test was performed using Prism Software (GraphPad). Survival was compared by a Log-rank (Mantl-Cox) test. Results with a p-value of <0.05 were considered significant.
Study approval
Blood samples were obtained from healthy donors and from patients enrolled on a clinical trial (NCT01865617) of CD19 CAR-T-cell therapy. Normal donors and patients provided written informed consent for research protocols approved by the Institutional Review Board of the FHCRC. The FHCRC Institutional Animal Care and Use Committee approved all mouse experiments.
Results
CD4+ and CD8+ T-cell subsets in normal donors and patients with B-cell malignancies
We analyzed the frequency of CD4+ and CD8+ T-cells and the fraction of TN (CD45RA+/RO−/CD62L+), TCM (CD45RA−/RO+/CD62L+), and TEM (CD45RA+/−/CD62L−) subsets in PBMC from 25 normal donors and 43 patients that were being screened for a clinical trial of CD19 CAR-T-cells. It is known that chemotherapy alters T-cell numbers and subset distribution, but variation in subsets has not been systematically analyzed for heavily pre-treated adult patients with B-cell malignancies who are candidates for CAR-T-cells. We observed substantial differences in the frequency of CD4+ and CD8+ T-cells between patients and normal donors, with most patients having a higher proportion of CD8+ and less CD4+ T-cells (Fig.1A). There was also heterogeneity in the proportions of TN, TCM, and TEM subsets in individual normal donors and patients, with patient samples containing a significantly higher mean frequency of TEM and lower mean frequency of TN cells in both CD4+ and CD8+ subsets than normal donors (Fig.1B). These data are consistent with the large variation in the frequency of CD4 and CD8 T-cells in CAR-T-cell products reported in studies in which unselected peripheral T-cells were transduced to generate T-cells for adoptive therapy10,35.
Figure 1. T-cell subset composition in PBMC from patients and healthy donors.
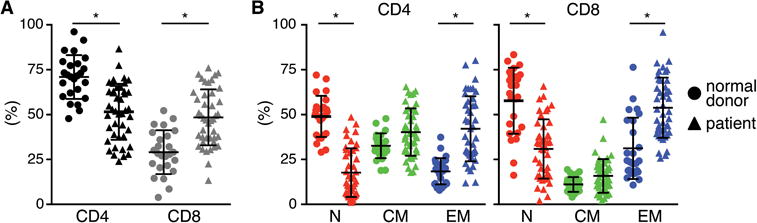
Proportions of (A) CD4+ and CD8+ T-cells, and (B) TN, TCM, and TEM cell subsets in blood of patients with B-cell malignancies and normal donors. Asterisk indicates significant differences between normal donors and patients.
CD4+ and CD8+ CAR-T-cells are transduced efficiently and differ in effector function
Strategies for purifying T-cell subsets have been developed, making it possible to prepare products derived from defined populations36. To provide insight into how heterogeneity in T-cell composition might affect therapy, we evaluated functions of purified CD4+ and CD8+ T-cells from normal donors after transduction with a CD19-specific 4-1BB/CD3ζ CAR. The CAR vector contained the marker gene EGFRt for analysis of transduction efficiency and to enable purification of CAR-T-cells32. CD4+ and CD8+ T-cells from normal donors were transduced with similar efficiency (Fig.2A). Transgene-positive CD4+ and CD8+ T-cells were enriched for EGFRt expression, expanded, and effector function was analyzed. CD8+ CAR-T-cells exhibited higher specific lytic activity than CD4+ CAR-T-cells against CD19+ Raji lymphoma and K562 cells transduced to express CD19 (K562/CD19) (Fig.2B). CD4+ CAR-T-cells produced more Th1 cytokines (IFN-γ/TNF-α/IL-2) and proliferated more vigorously after tumor cell recognition compared to CD8+ CAR-T-cells (Fig.2C,D). We observed the same results with T-cells expressing a ROR1-specific CAR with a CD28 costimulatory domain demonstrating that these differences in functions of CD4+ and CD8+ CAR-T-cells were independent of the specificity of the CAR and the costimulatory domain in the vector (Fig.S1A–C).
Figure 2. CD4+ and CD8+ CAR-T-cells differ in effector function.
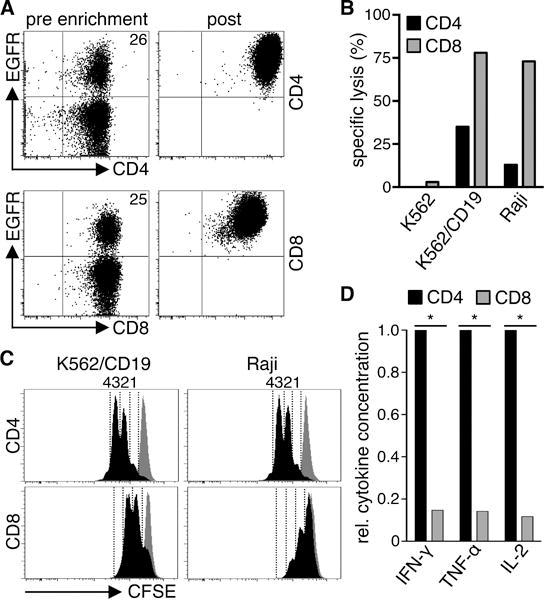
(A) Expression of EGFRt on CD4+ and CD8+ T-cells transduced with a CD19 CAR-EGFRt construct before enrichment (pre) and after enrichment and expansion (post). (B) Cytolytic activity of CD19 CAR-T-cells against 51Cr-labeled CD19+ (K562/CD19, Raji) and control (K562) target cells at an E:T ratio of 30:1 analyzed by a standard 4 h chromium release assay. (C) Proliferation of CFSE-labeled CD19 CAR-T-cells after stimulation with K562/CD19 and Raji cells for 72 h measured by CFSE dye dilution. Stimulation of CFSE-labeled CD19 CAR-T-cells with CD19− K562 cells is shown as a comparison in each histogram (gray graph). Numbers above each histogram indicate the number of cell divisions the proliferating subset underwent. Data in A–C are representative of experiments with CAR-T-cells derived from three different donors. (D) Relative cytokine production by CD19 CAR-T-cells after co-culture with K562/CD19 and Raji cells for 24 h. Data from three independent experiments with T-cells prepared from different donors were combined. Cytokine production of CD4+ cells was set as 1 and relative cytokine production of CD8+ T-cells was calculated. Asterisk indicates significant differences between CD4+ and CD8+ cells.
Analysis of CAR function in naïve and memory CD4+ T-cells
We next sort-purified CD4+ TN, TCM, and TEM from PBMC (Fig.3A), and transduced each subset with the CD19 CAR. Transduction of all three subsets was similar and not significantly different from unselected CD4+ T-cells (Fig.S2A). CAR-T-cells at the end of in vitro culture showed uniform expression of CD45RO, including CAR-T-cells derived from the CD45RO− TN subset, due to their activation and differentiation in culture. CD62L was retained on a higher fraction of CAR-T-cells derived from TN and TCM, CD28 expression was similar on CAR-T-cells from each subset, and CD27 was expressed by a higher fraction of CAR-T-cells derived from CD4+ TN (TN>TCM>TEM) (Fig.3B). CD4+ CAR-T-cells derived from TN, TCM, and TEM mediated weak lytic activity against CD19+ tumor cells consistent with data on unselected CD4+ T-cells (Fig.3C). CD4+ CAR-T-cells derived from the TN subset produced the highest levels of cytokines after stimulation with CD19+ tumor cells (Fig.3D). Greater cytokine production of CAR-T-cells from CD4+ TN cells was also observed with a ROR1-specific CD28/CD3ζ CAR (data not shown), suggesting the differences in function of CAR-T-cells derived from TN, TCM, and TEM subsets reflected cell intrinsic properties.
Figure 3. In vitro and in vivo analysis of CD19 CAR-expressing naïve and memory CD4+ T-cell subsets.
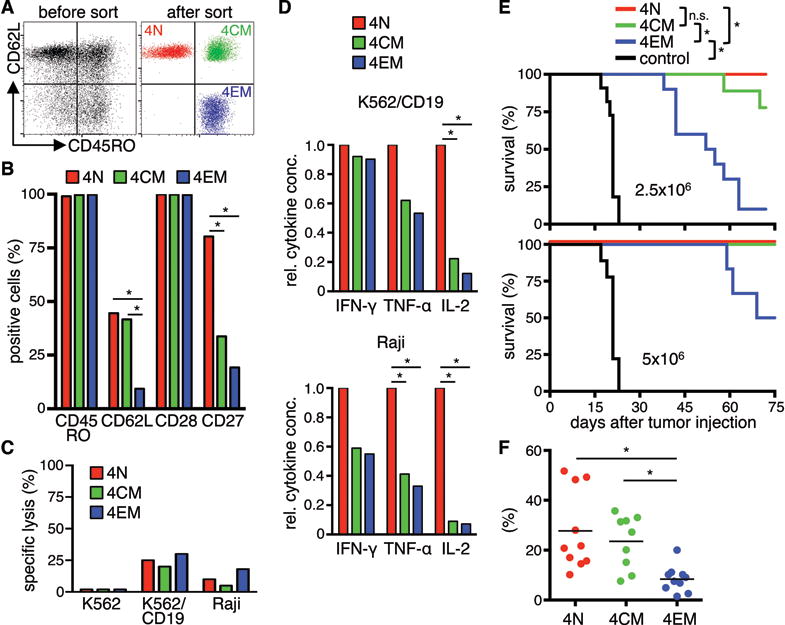
(A) CD4+ TN, TCM, and TEM cells were isolated by FACS. (B) Analysis of cell surface expression of CD45RO, CD62L, CD27, and CD28 on CD4+ CD19 CAR-T-cells derived from different subsets. Data from three independent experiments with T-cells prepared from different donors were combined. (C) Cytolytic activity of CAR-T-cells against 51Cr-labeled K562, K562/CD19, and Raji cells at an E:T ratio of 30:1 after 4 h. (D) Relative cytokine production by CD19 CAR-T-cells after co-culture with CD19+ cells for 24 h. Data from three independent experiments with T-cells prepared from different donors were combined. Cytokine production of TN was set as 1 and relative cytokine production of TCM and TEM was calculated. (E) Survival of Raji/ffluc-bearing NSG mice treated on day seven after tumor injection with CD19 CAR-T-cells derived from different CD4+ T-cell subsets (2.5×106 or 5×106 CAR-T-cells per mouse). NSG mice that received 5×106 T-cells transduced with a vector encoding EGFRt alone (EGFRt-T-cells) were used as control. Asterisk indicates significant differences between groups (n.s.: not significant). Data from three independent experiments for the lower cell dose (2.5×106) and two for the higher cell dose (5×106) with minimum three mice per group were combined. (F) Frequency of transferred T-cells (human CD45+/CD4+) on day ten after T-cell transfer in peripheral blood of mice that had received the lower T-cell dose.
Murine effector cells derived from less differentiated TCR-transgenic CD8+ T-cell subsets are more effective in treating B16 melanoma37, however similar data have not been shown for TCR-transgenic or CAR-modified CD4+ T-cells. Thus, we analyzed the in vivo antitumor function of CD4+ CAR-T-cells from each subset in NSG mice inoculated with 5×105 firefly-luciferase (ffluc) transduced Raji cells. Titrated doses of CAR-T-cells from TN, TCM, and TEM subsets, or control EGFRt-T-cells were administered one week after tumor inoculation when mice had disseminated lymphoma. CD4+ CAR-T-cells from each subset conferred a significant antitumor effect, resulting in superior survival compared with mice that received control T-cells (Fig.3E). A hierarchy in antitumor efficacy was evident by the dose titrations, with CD4+ T-cells from TN and TCM conferring superior antitumor activity compared to TEM-derived CD4+ CAR-T-cells (Fig.3E). In vivo antitumor efficacy of the TN- and TCM-derived subsets correlated with a higher peak level of CAR-T-cells in the blood at day ten after T-cell transfer (Fig.3F). These data demonstrate that CD4+ CAR-T-cells derived from TN, TCM, and TEM subsets differ in cytokine production, and that CD4+ T-cells from TN and TCM subsets exhibit superior antitumor activity in vivo.
Analysis of CAR function in naïve and memory CD8+ T-cells
We performed a similar analysis of CAR-T-cells derived from CD8+ TN, TCM, and TEM. The transduction efficiencies were similar in all three subsets and not significantly different from unselected CD8+ T-cells (Fig.S2B). Phenotypic analysis of the CD8+ CAR-T-cells from each subset showed uniform expression of CD45RO, and higher levels of CD62L on CAR-T-cells from CD8+ TN and TCM compared with TEM (Fig.4A). CAR-T-cells derived from CD8+ TN, TCM, and TEM mediated near-equivalent specific lysis of CD19+ tumor cells (Fig.4B), and TN-derived CAR-T-cells produced more cytokines than TCM- and TEM-derived CAR-T-cells in response to stimulation with CD19+ tumor cells (Fig.4C). Notably, the amount of IL-2 produced by all CAR-T-cells derived from CD8+ T-cell subsets (mean IL-2 concentration (pg/ml) after stimulation with Raji; TN: 716, TCM: 36, TEM 30) was substantially less than by CAR-T-cells derived from the corresponding CD4+ T-cells (TN: 2798, TCM: 369, TEM 315).
Figure 4. In vitro and in vivo analysis of CD19 CAR-expressing naïve and memory CD8+ T-cell subsets.
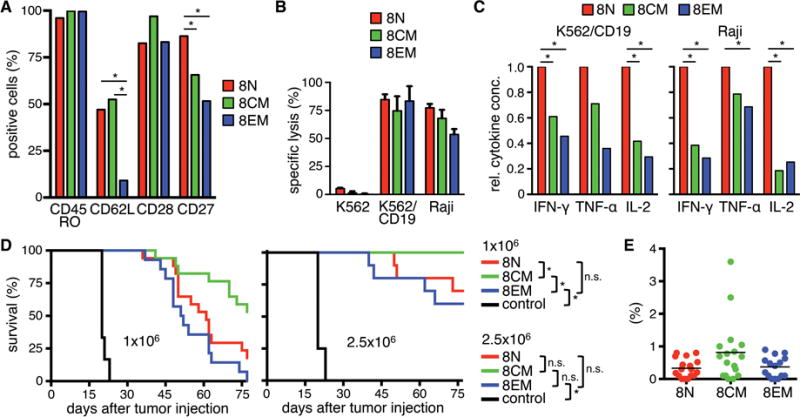
(A) Analysis of cell surface expression of CD45RO, CD62L, CD27, and CD28 on CD8+ CD19 CAR-T-cells derived from naïve and memory subsets. Data from three independent experiments with T-cells prepared from different donors were combined. (B) Cytolytic activity of CD19 CAR-T-cells against 51Cr-labeled K562, K562/CD19, and Raji cells at an E:T ratio of 30:1 after 4 h. (C) Relative cytokine production by CD19 CAR-T-cells after co-culture with CD19+ cells for 24 h. Data from three independent experiments with T-cells prepared from different donors were combined. Cytokine production of TN was set as 1 and relative cytokine production of TCM and TEM was calculated. (D) Survival of Raji/ffluc-bearing NSG mice treated on day seven after tumor injection with different doses of CD19 CAR-T-cells derived from different CD8+ T-cell subsets (1×106 and 2.5×106). NSG mice that received 2.5×106 EGFRt-T-cells were used as control. Asterisk indicates significant differences between groups (n.s.: not significant). For the lower cell dose (1×106) four and for the higher cell dose (2.5×106) three experiments with minimum three mice per group are combined. (E) Frequency of transferred T-cells (human CD45+/CD8+) on day ten after T-cell transfer in peripheral blood of mice that had received the lower T-cell dose.
We analyzed the antitumor function of CD8+ CAR-T-cells from each subset at two dose levels in the NSG/Raji/ffluc model. At the lower dose (1×106), CD8+ CAR-T-cells from TCM conferred the longest survival, whereas CD8+ CAR-TN and TEM were less effective (Fig.4D). This hierarchy was confirmed at the higher dose (2.5×106) where all mice treated with TCM-derived CAR-T-cells survived tumor-free. The superior efficacy of CD8+ CAR-T-cells derived from TCM was associated with greater proliferation in vivo (Fig.4E), consistent with our previous observations and other pre-clinical models38,39. Thus, similar to CD4+ T-cells, CD19 CAR-T-cells from CD8+ TN, TCM, and TEM exhibit differences in effector functions in vitro and antitumor efficacy in vivo. It is noteworthy that the peak frequency of CAR-T-cells derived from all CD4+ T-cell subsets, including CD4+ TEM that was incapable of eradicating tumor in a majority of mice, exceeded that of a curative dose of CD8+ TCM-derived CAR-T-cells, illustrating the greater potency of CD8+ CAR-T-cells derived from this subset (Fig.3F,4E; and data not shown).
CD4+ CAR-T-cells exhibit synergistic antitumor activity with CD8+ CAR-T-cells
In addition to direct tumor recognition, the production of abundant IL-2 by CD4+ CAR-T-cells might augment proliferation, survival, and efficacy of CD8+ CAR-T-cells. Synergistic activity of CD4 and CD8 CAR-T-cells has been observed in murine models40,41, although derivation from memory and naïve subsets was not studied. Thus, we examined potential synergy between adoptively transferred CD4+ CAR-T-cells and CD8+ CAR-T-cells derived from TCM, which was the most efficacious CD8 subset alone. We first evaluated whether CD4+ CD19 CAR-T-cells derived from TN, TCM, and TEM subsets augmented in vitro proliferation of CD8+ TCM-derived CAR-T-cells because peak numbers of CAR-T-cells in vivo correlated with antitumor efficacy in our tumor model, and has been an important parameter in clinical trials. We determined proliferation of CD8+ TCM-derived CAR-T-cells alone and combined with CD4+ CAR-T-cells by CFSE dye dilution after stimulation with CD19+ tumor cells. CD4+ CAR-T-cells derived from TN, TCM, and TEM subsets of normal donors enhanced the proliferation of CD8+ TCM-derived CAR-T-cells in response to stimulation with CD19+ JeKo-1 cells, with CD4+ TN providing the greatest enhancement (Fig.5A). Even after stimulation with CD80+/CD86+ Raji cells, the proliferation of CD8+ TCM-derived CAR-T-cells was increased by the addition of CD4+ CAR-T-cells derived from the TN subset.
Figure 5. CD4+ CD19 CAR-T-cells provide help to CD8+ CD19 CAR-T-cells in vitro and in vivo.
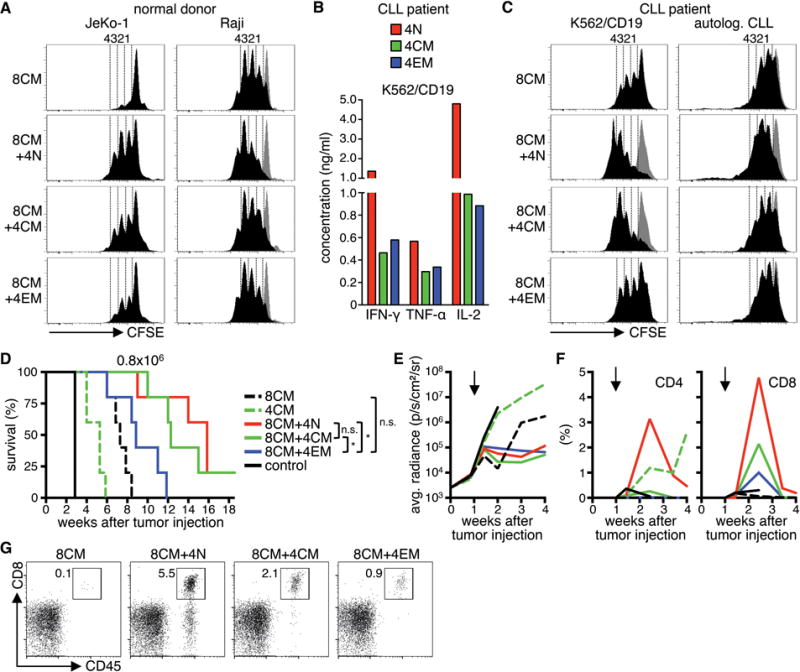
(A) Proliferation of CFSE-labeled CD8+ TCM-derived CD19 CAR-T-cells after stimulation with JeKo-1 and Raji cells for 72 h in the presence of CD4+ CD19 CAR-T-cells derived from different subsets. Histograms are gated on CD8+ T-cells. Stimulation with K562 cells is shown as a comparison in each histogram (gray graph). Data are representative for four independent experiments with T-cells generated from different healthy donors. (B) Cytokine production of CD4+ CD19 CAR-T-cells derived from different subsets from a CLL-patient after co-culture with K562/CD19 for 24 h. (C) Same as (A) with T-cells from a CLL-patient and K562/CD19 cells and autologous CLL as stimulator cells in the assay. Data in (B) and (C) are representative for two independent experiments with T-cells generated from different patients. (D) Survival of Raji/ffluc-bearing NSG mice treated on day seven after tumor injection with 8×105 CD19 CAR-T-cells derived from CD4+ TN, TCM, TEM, and CD8+ TCM (CD4/CD8 combinations: 4×105 CD4+ cells + 4×105 CD8+ cells; 5 mice per group). NSG mice that received a combination of CD4+ and CD8+ EGFRt-T-cells were used as control. (E) Bioluminescence imaging of tumor growth. Arrows mark the day of T-cell transfer. (F) Frequency of transferred CD4+ and CD8+ T-cells (human CD45+/CD4+ or CD45+/CD8+) in peripheral blood. (G) Flow cytometry data on day ten after T-cell transfer. Mice with median T-cell frequency are shown for each group.
Because CD4+ T-cells from patients with B-cell malignancies may be functionally compromised42,43, we analyzed whether helper function was preserved in CD4+ CAR-T-cells generated from CD4+ TN, TCM, and TEM subsets of CLL patients. The cytokine profile of patient CD4+ CAR-T-cells was similar to that observed in normal donors (TN>TCM/TEM) suggesting that transduction and culture of T-cells from these patients may overcome any functional defects, or result in the selective expansion of T-cells that were competent to produce cytokines (Fig.5B). As observed with normal donors, the proliferation of CD8+ TCM-derived CAR-T-cells from patients after stimulation with a tumor cell line and with the patient’s primary autologous leukemia cells was augmented when autologous CD4+ CAR-T-cells were included in the co-culture, and CD4+ CAR-T-cells from the TN subset provided the greatest enhancement of CD8+ TCM-derived CAR-T-cell proliferation (Fig.5C).
To determine whether a synergistic antitumor effect of CD8+ and CD4+ CAR-T-cells was evident in vivo, NSG mice with Raji tumors were treated with an effective but non-curative dose (8×105) of either CD8+ or CD4+ CD19 CAR-T-cells derived from the TCM subsets, or with the same total dose of CAR-T-cells but consisting of a 1:1 ratio of CD8+ TCM-derived and CD4+ CAR-T-cells derived from each of the TN, TCM, or TEM subsets. The data show that the combination of CD8+ TCM-derived CAR-T-cells with CD4+ CAR-T-cells from either the TN or TCM subsets conferred superior antitumor activity compared to CAR-T-cells derived from either CD8+ or CD4+ TCM subsets alone, or the CD8+ TCM/CD4+ TEM combination (Fig.5D,E). Analysis of the frequency of CAR-T-cells in the peripheral blood of mice showed that the improved antitumor effect in mice receiving CAR-T-cells derived from CD8+ TCM combined with CD4+ TCM or TN correlated with greater peak proliferation of CD8+ T-cells in vivo than observed with the CD8+ TCM product alone or with CD4+ TEM, consistent with the hierarchy observed in the in vitro experiments (Fig.5F,G). We also observed significant in vivo expansion of CD4+ CAR-T-cells when administered alone, however at this cell dose CD4+ CAR-T-cells were insufficient to prevent outgrowth of Raji tumors, despite sustained proliferation and persistence for more than four weeks (Fig.5F).
In healthy donors, TN and TCM are the numerically dominant CD4+ T-cell subsets (Fig.1), and we found that CAR-T-cells derived from unselected bulk CD4+ were equivalently effective in providing help as sort purified CD4+ TN and CD4+ TCM, although the enhancement of CD8+ CAR-T-cell proliferation with unselected CD4+ T-cells was less than that observed with CD4+ TN or CD4+ TCM-derived products (data not shown). Our data on the subset distribution in patients with B-cell malignancies eligible for CD19 CAR-T-cell therapy shows a marked increase in the proportion of CD4+ TEM, suggesting that improvements in potency in some patients may be achieved by selecting CD4+ TN and TCM subsets prior to preparing therapeutic products.
Patient CAR-T-cell products of defined subset composition have enhanced potency in vivo
To evaluate the potential advantages of preparing cell products of defined subset composition in a clinical setting, we prepared CD19 CAR-T-cells from two patients with non-Hodgkin’s lymphoma enrolled on an ongoing clinical trial of CAR-T-cell therapy (NCT01865617), and used these cell products to treat NSG mice engrafted with Raji/ffluc. We derived CD19 CAR-T-cells from unselected PBMC, or defined populations of sort-purified CD62L+/CD45RO+ CD8 TCM, CD62L+/CD45RO− CD4 TN and unselected CD4 T-cells (CD4 TUS) (Fig.6A). From each starting population, CD19 CAR-T-cells expanded at similar rates and to clinically useful therapeutic numbers. Phenotype analysis showed that the CAR-T-cells derived from whole PBMC was further skewed after transduction and contained mainly CD8+ T-cells and a smaller proportion of CD4+ CAR-T-cells (approx. 6:1 ratio) (Fig.6B).
Figure 6. T-cell products of defined subset composition prepared from patients have enhanced potency.
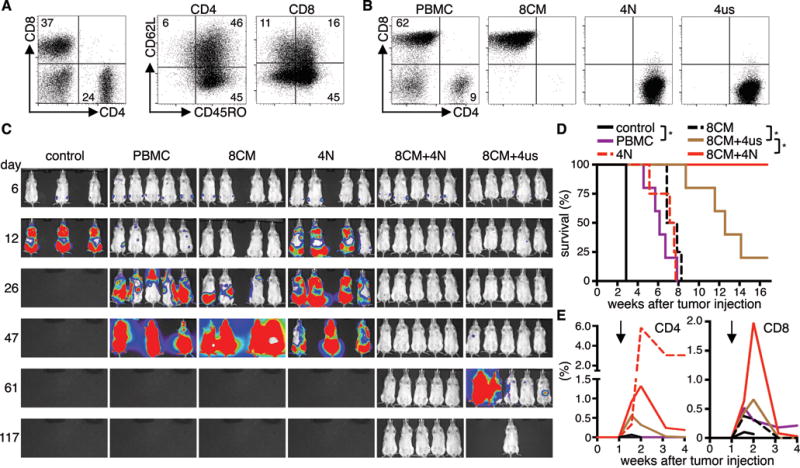
(A) T-cell subset composition in PBMC from a lymphoma patient. (B) CD4/CD8 phenotype of CD19 CAR-T-cell products derived from PBMC or from different sort-purified subsets. (C) Bioluminescence imaging data from Raji/ffluc-bearing NSG mice that had received 1×106 CD19 CAR-T-cells (CD4/CD8 ratio: 1:1, 4–5 mice per group). NSG mice that received EGFRt-T-cells were used as control (3 mice). (D) Survival and (E) persistence of transferred CD4+ and CD8+ T-cells in the blood of mice shown in (C). Asterisk indicates significant differences between groups. Arrows mark the day of T-cell transfer. Data are representative for two independent experiments with T-cells generated from different patients.
NSG mice with Raji tumors were then treated with a fixed total dose of 1×106 CAR-T-cells derived from either PBMC, CD8+ TCM, CD4+ TN, or a combined product of CD8+ TCM and CD4+ T-cells (TN or TUS) at a 1:1 ratio. Analysis of tumor progression and survival showed enhanced antitumor efficacy of cell products comprised of CD8+ TCM and either CD4+ TN or TUS compared to products from PBMC, or from CD8+ or CD4+ T-cells alone (Fig.6C,D). The combination of CD8+ TCM and CD4+ TN conferred the strongest antitumor effect, and long-term tumor-free survival was achieved in all mice in this treatment group (n=5/5). Complete tumor eradication was also observed in a subset of the mice that received the combination of CD8+ TCM and CD4+ TUS (n=1/5). All mice in the group that received PBMC-derived CAR-T-cells expired from progressive tumors. We again found a strong correlation between enhanced antitumor efficacy and superior proliferation of CD8+ CAR-T-cells (Fig.6E). The data show that formulating CD8+ and CD4+ CAR-T-cells from defined progeny can provide cell products with increased potency compared with CAR-T-cells from whole unselected PBMC or the CD8+ T-cell subset alone.
Discussion
Clinical trials in which peripheral blood T-cells were transduced with CD19 CARs containing CD28/CD3ζ or 4-1BB/CD3ζ signaling domains demonstrate a high complete remission rate in ALL and a significant response rate in CLL and non-Hodgkin’s lymphoma3–10,35,44. Not all patients respond and toxicities, including cytokine release syndrome, disseminated intravascular coagulopathy, and neurotoxicity can occur after infusing CAR-T-cells3,6. The follow-up in most trials is short, and the durability of responses remains to be determined. Comparing efficacy and toxicity outcomes in different studies is difficult because of patient heterogeneity and differences in vector design, culture methods, and cell doses3–10,35,44. Furthermore, our data show that the frequencies of CD4+ and CD8+ T-cells, and the proportions of naïve and memory cells in each subset, are skewed in patients with B-cell malignancies that have received cytotoxic chemotherapy. Thus, in the absence of selection of defined populations, the phenotype and function of genetically modified cell products prepared from patients will be diverse and reflective of the proportions of CD4+ and CD8+ T-cells and memory and naïve subsets in the blood. Indeed, clinical trials of CD19 CAR-T-cells have administered products that diverge markedly in the proportions of CD4+ and CD8+ T-cells, and it is impossible to retrospectively determine the fraction of transduced T-cells that were derived from naïve or memory T-cell precursors3,35.
The efficacy of T-cell therapy is predicated on the ability of transferred cells to proliferate in vivo to overcome numerically larger tumor burdens, migrate to tumor sites, persist, and mediate effector functions that destroy tumor cells. Independent of subset derivation, in vitro activated T-cells acquire cytotoxicity and cytokine production. However, prior studies in mice and non-human primates have shown that the persistence and function of adoptively transferred effector CD8+ T-cells differs depending on the T-cell subsets from which they were derived. The transfer of cytomegalovirus-specific CD8+ T-cell clones derived from TCM but not TEM provided persistent immunity for more than four years38,45; and TCR-transgenic T-cells derived from the TN and TCM subsets conferred superior antitumor responses to melanoma than those derived from TEM37,46. Pharmacologic activation of Wnt signaling during the culture of CD8+ TN arrests differentiation at a phenotype intermediate between TN and TCM, and these arbitrarily termed “memory stem cells” have potent antitumor activity in animals47. Human T-cells with this phenotype are present in blood as a minor population and when engineered with CARs and administered with CD4+ T-cells are effective in tumor xenograft models19. However, strategies for purification of this subset for clinical use are not available.
Unlike oncology drugs where dose/response relationships are readily measurable, CAR-T-cells prepared from unselected cell populations might vary widely in potency. To provide insight into how variation in common subsets might affect therapy with CAR-T-cells, we transduced individual CD8+ and CD4+ T-cell subsets from normal donors and patients with B-cell malignancies with a CD19 CAR. All CD4+ and CD8+ T-cell subsets mediated cytolysis, secreted cytokines, and proliferated in response to tumor cells in vitro. Unselected CD4+ CAR-T-cells and those derived from TN, TCM, and TEM subsets were less cytolytic and produced higher levels of cytokines than unselected CD8+ CAR-T-cells. CAR-T-cells derived from CD4+ TN produced more cytokines than those derived from CD4+ TCM and TEM, and a similar hierarchy of cytokine production was evident in CAR-T-cells derived from CD8+ T-cells. These differences in functional properties were evident with T-cells from normal donors and from patients with B-cell malignancies, demonstrating that although phenotypic composition was skewed in patients, subset intrinsic functional properties were retained when the T cells were signaled through the CAR.
Differences in antitumor efficacy depending on the phenotype and subset derivation of CAR-T-cells were demonstrated by experiments in NSG mice engrafted with CD19+ Raji tumors. The efficacy of CD4+ CAR-T-cells derived from different subsets has not previously been examined. Our data show that CD4+ CAR-T-cells derived from TN and TCM were more effective than those from TEM, which is the subset that is most increased in frequency in patients. CD8+ CAR-T-cells derived from TCM and TN were also more effective than those derived from CD8+ TEM. There was a correlation between antitumor activity and peak proliferation of CD4+ and CD8+ CAR-T-cells, consistent with observations in patients.
A notable finding in our studies of human CD19 CAR-T-cell products that extends prior work in mice with bulk CD4/CD8 T cell subsets40,41 was that the potency was dramatically enhanced by formulating CD8+ CAR-T-cells derived from TCM with CD4+ CAR-T-cells, and especially with CD4+ CAR-T-cells derived from TN. Formulating cell products in a 1:1 CD4/CD8 ratio resulted in complete eradication of Raji lymphoma at cell doses that were ineffective with either subset alone. Efficacy correlated with markedly greater proliferation of CD8+ CAR-T-cells, suggesting that IL-2 produced by CD4+ T-cells (TN>TCM>TEM) was important in sustaining the CD8+ T-cell response. The relevance of these findings to clinical applications was demonstrated by the enhanced potency of CD19 CAR-T-cells prepared from defined T-cell subsets compared with unselected PBMC obtained from patients with B-cell lymphoma. Based on the compelling results in the preclinical experiments reported here, we have initiated a clinical trial of CD19 CAR-T-cells of defined subset composition (https://clinicaltrials.gov/ct2/show/NCT01865617).
CAR-T-cells represent a new and promising approach to cancer therapy, however heterogeneity of cell products used in different clinical trials makes it difficult to interpret the basis for therapeutic success or failure. Adoptive therapy with defined cell products will require GMP compliant cell selection methods that do not leave bound reagents on the T-cells. Advances in cell selection methodologies have recently been made36, and it is likely that automated devices will be developed to improve producing T-cell products of defined composition. Identifying the most effective compositions of CAR-T-cells will require evaluation in carefully designed clinical trials and may be most critical in solid tumors where target molecules are expressed at lower densities than CD19, and immunosuppressive tumor microenvironments pose an obstacle to tumor eradication. Our results suggest that defining the composition of T-cells used in adoptive therapy can enhance antitumor activity, lower the T-cell doses needed for therapeutic efficacy, and reduce a major variable that currently complicates comparison of results in individual patients and across studies.
Supplementary Material
1
2
Acknowledgments
The authors would like to thank Melissa Comstock (Shared Resources, FHCRC) for expertise in performing the mouse experiments. This work was supported by grants from the National Institutes of Health CA136551, CA18029, and CA114536 (S.R.R.). D.S. and M.H. were supported by the German Research Foundation (DFG, Deutsche Forschungsgemeinschaft, SO1214/1-1, HU1668/1-1, 1-2,). M.H. was supported by the Leukemia and Lymphoma Society (LLS, 5520-11), the German Cancer Aid (Deutsche Krebshilfe e.V., Max Eder Program 110313), and the University of Würzburg (Interdisziplinäres Zentrum für Klinische Forschung, IZKF, Z-4/109 and D-244).
Footnotes
Conflict of interest statement:
M.H. and S.R.R. are inventors on a patent application (PCT/US1013/055862) related to this work that has been filed by the Fred Hutchinson Cancer Research Center (FHCRC) and licensed by Juno Therapeutics. S.R.R. is founder and shareholder of Juno Therapeutics.
Supplementary information is available at Leukemia’s website.
References
- 1.Jensen MC, Riddell SR. Design and implementation of adoptive therapy with chimeric antigen receptor-modified T cells. Immunol Rev. 2014 Jan;257(1):127–144. doi: 10.1111/imr.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kochenderfer JN, Rosenberg SA. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nature reviews Clinical oncology. 2013 May;10(5):267–276. doi: 10.1038/nrclinonc.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci Transl Med. 2014 Feb 19;6(224):224ra225. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013 Apr 18;368(16):1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011 Aug 10;3(95):95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012 Mar 22;119(12):2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011 Aug 25;365(8):725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014 Oct 16;371(16):1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. The Journal of clinical investigation. 2011 May;121(5):1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011 Nov 3;118(18):4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brentjens RJ, Santos E, Nikhamin Y, Yeh R, Matsushita M, La Perle K, et al. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin Cancer Res. 2007 Sep 15;13(18 Pt 1):5426–5435. doi: 10.1158/1078-0432.CCR-07-0674. [DOI] [PubMed] [Google Scholar]
- 12.Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. 2009 Mar 3;106(9):3360–3365. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, et al. Receptor Affinity and Extracellular Domain Modifications Affect Tumor Recognition by ROR1-Specific Chimeric Antigen Receptor T Cells. Clin Cancer Res. 2013 Jun 15;19(12):3153–3164. doi: 10.1158/1078-0432.CCR-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.James SE, Greenberg PD, Jensen MC, Lin Y, Wang J, Till BG, et al. Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J Immunol. 2008 May 15;180(10):7028–7038. doi: 10.4049/jimmunol.180.10.7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N, et al. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006 Nov 15;66(22):10995–11004. doi: 10.1158/0008-5472.CAN-06-0160. [DOI] [PubMed] [Google Scholar]
- 16.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Molecular therapy: the journal of the American Society of Gene Therapy. 2009 Aug;17(8):1453–1464. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaech SM, Hemby S, Kersh E, Ahmed R. Molecular and functional profiling of memory CD8 T cell differentiation. Cell. 2002 Dec 13;111(6):837–851. doi: 10.1016/s0092-8674(02)01139-x. [DOI] [PubMed] [Google Scholar]
- 18.Graef P, Buchholz VR, Stemberger C, Flossdorf M, Henkel L, Schiemann M, et al. Serial transfer of single-cell-derived immunocompetence reveals stemness of CD8(+) central memory T cells. Immunity. 2014 Jul 17;41(1):116–126. doi: 10.1016/j.immuni.2014.05.018. [DOI] [PubMed] [Google Scholar]
- 19.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011 Oct;17(10):1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stemberger C, Huster KM, Koffler M, Anderl F, Schiemann M, Wagner H, et al. A single naive CD8+ T cell precursor can develop into diverse effector and memory subsets. Immunity. 2007 Dec;27(6):985–997. doi: 10.1016/j.immuni.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 21.Buchholz VR, Flossdorf M, Hensel I, Kretschmer L, Weissbrich B, Graf P, et al. Disparate individual fates compose robust CD8+ T cell immunity. Science. 2013 May 3;340(6132):630–635. doi: 10.1126/science.1235454. [DOI] [PubMed] [Google Scholar]
- 22.Gerlach C, Rohr JC, Perie L, van Rooij N, van Heijst JW, Velds A, et al. Heterogeneous differentiation patterns of individual CD8+ T cells. Science. 2013 May 3;340(6132):635–639. doi: 10.1126/science.1235487. [DOI] [PubMed] [Google Scholar]
- 23.Rocha B, Tanchot C. Towards a cellular definition of CD8+ T-cell memory: the role of CD4+ T-cell help in CD8+ T-cell responses. Current opinion in immunology. 2004 Jun;16(3):259–263. doi: 10.1016/j.coi.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 24.Mitsuyasu RT, Anton PA, Deeks SG, Scadden DT, Connick E, Downs MT, et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood. 2000 Aug 1;96(3):785–793. [PubMed] [Google Scholar]
- 25.Walker RE, Bechtel CM, Natarajan V, Baseler M, Hege KM, Metcalf JA, et al. Long-term in vivo survival of receptor-modified syngeneic T cells in patients with human immunodeficiency virus infection. Blood. 2000 Jul 15;96(2):467–474. [PubMed] [Google Scholar]
- 26.Mackall CL, Fleisher TA, Brown MR, Andrich MP, Chen CC, Feuerstein IM, et al. Age, thymopoiesis, and CD4+ T-lymphocyte regeneration after intensive chemotherapy. N Engl J Med. 1995 Jan 19;332(3):143–149. doi: 10.1056/NEJM199501193320303. [DOI] [PubMed] [Google Scholar]
- 27.Hakim FT, Cepeda R, Kaimei S, Mackall CL, McAtee N, Zujewski J, et al. Constraints on CD4 recovery postchemotherapy in adults: thymic insufficiency and apoptotic decline of expanded peripheral CD4 cells. Blood. 1997 Nov 1;90(9):3789–3798. [PubMed] [Google Scholar]
- 28.Terakura S, Yamamoto TN, Gardner RA, Turtle CJ, Jensen MC, Riddell SR. Generation of CD19-chimeric antigen receptor modified CD8+ T cells derived from virus-specific central memory T cells. Blood. 2012 Jan 5;119(1):72–82. doi: 10.1182/blood-2011-07-366419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer immunology research. 2015 Feb;3(2):125–135. doi: 10.1158/2326-6066.CIR-14-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zola H, MacArdle PJ, Bradford T, Weedon H, Yasui H, Kurosawa Y. Preparation and characterization of a chimeric CD19 monoclonal antibody. Immunology and cell biology. 1991 Dec;69:411–422. doi: 10.1038/icb.1991.58. [DOI] [PubMed] [Google Scholar]
- 31.Whitlow M, Bell BA, Feng SL, Filpula D, Hardman KD, Hubert SL, et al. An improved linker for single-chain Fv with reduced aggregation and enhanced proteolytic stability. Protein engineering. 1993 Nov;6(8):989–995. doi: 10.1093/protein/6.8.989. [DOI] [PubMed] [Google Scholar]
- 32.Wang X, Chang WC, Wong CW, Colcher D, Sherman M, Ostberg JR, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011 Aug 4;118(5):1255–1263. doi: 10.1182/blood-2011-02-337360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hudecek M, Schmitt TM, Baskar S, Lupo-Stanghellini MT, Nishida T, Yamamoto TN, et al. The B-cell tumor-associated antigen ROR1 can be targeted with T cells modified to express a ROR1-specific chimeric antigen receptor. Blood. 2010 Nov 25;116(22):4532–4541. doi: 10.1182/blood-2010-05-283309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leisegang M, Engels B, Meyerhuber P, Kieback E, Sommermeyer D, Xue SA, et al. Enhanced functionality of T cell receptor-redirected T cells is defined by the transgene cassette. J Mol Med. 2008 May;86(5):573–583. doi: 10.1007/s00109-008-0317-3. [DOI] [PubMed] [Google Scholar]
- 35.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015 Feb 7;385(9967):517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stemberger C, Dreher S, Tschulik C, Piossek C, Bet J, Yamamoto TN, et al. Novel serial positive enrichment technology enables clinical multiparameter cell sorting. PloS one. 2012;7(4):e35798. doi: 10.1371/journal.pone.0035798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hinrichs CS, Borman ZA, Cassard L, Gattinoni L, Spolski R, Yu Z, et al. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc Natl Acad Sci U S A. 2009 Oct 13;106(41):17469–17474. doi: 10.1073/pnas.0907448106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. The Journal of clinical investigation. 2008 Jan;118(1):294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X, Berger C, Wong CW, Forman SJ, Riddell SR, Jensen MC. Engraftment of human central memory-derived effector CD8+ T cells in immunodeficient mice. Blood. 2011 Feb 10;117(6):1888–1898. doi: 10.1182/blood-2010-10-310599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moeller M, Haynes NM, Kershaw MH, Jackson JT, Teng MW, Street SE, et al. Adoptive transfer of gene-engineered CD4+ helper T cells induces potent primary and secondary tumor rejection. Blood. 2005 Nov 1;106(9):2995–3003. doi: 10.1182/blood-2004-12-4906. [DOI] [PubMed] [Google Scholar]
- 41.Moeller M, Kershaw MH, Cameron R, Westwood JA, Trapani JA, Smyth MJ, et al. Sustained antigen-specific antitumor recall response mediated by gene-modified CD4+ T helper-1 and CD8+ T cells. Cancer Res. 2007 Dec 1;67(23):11428–11437. doi: 10.1158/0008-5472.CAN-07-1141. [DOI] [PubMed] [Google Scholar]
- 42.Riches JC, Davies JK, McClanahan F, Fatah R, Iqbal S, Agrawal S, et al. T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production. Blood. 2013 Feb 28;121(9):1612–1621. doi: 10.1182/blood-2012-09-457531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kiaii S, Clear AJ, Ramsay AG, Davies D, Sangaralingam A, Lee A, et al. Follicular lymphoma cells induce changes in T-cell gene expression and function: potential impact on survival and risk of transformation. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013 Jul 20;31(21):2654–2661. doi: 10.1200/JCO.2012.44.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-Refractory Diffuse Large B-Cell Lymphoma and Indolent B-Cell Malignancies Can Be Effectively Treated With Autologous T Cells Expressing an Anti-CD19 Chimeric Antigen Receptor. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2015 Feb 20;33(6):540–549. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berger C, Berger M, Anderson D, Riddell SR. A non-human primate model for analysis of safety, persistence, and function of adoptively transferred T cells. Journal of medical primatology. 2011 Apr;40(2):88–103. doi: 10.1111/j.1600-0684.2010.00451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A. 2005 Jul 5;102(27):9571–9576. doi: 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009 Jul;15(7):808–813. doi: 10.1038/nm.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1
2