Pathogenic mechanism of recurrent mutations of SCN8A in epileptic encephalopathy (original) (raw)
Abstract
Objective
The early infantile epileptic encephalopathy type 13 (EIEE13, OMIM #614558) results from de novo missense mutations of SCN8A encoding the voltage‐gated sodium channel Nav1.6. More than 20% of patients have recurrent mutations in residues Arg1617 or Arg1872. Our goal was to determine the functional effects of these mutations on channel properties.
Methods
Clinical exome sequencing was carried out on patients with early‐onset seizures, developmental delay, and cognitive impairment. Two mutations identified here, p.Arg1872Leu and p.Arg1872Gln, and two previously identified mutations, p.Arg1872Trp and p.Arg1617Gln, were introduced into Nav1.6 cDNA, and effects on electrophysiological properties were characterized in transfected ND7/23 cells. Interactions with FGF14, G‐protein subunit G_βγ_, and sodium channel subunit _β_1 were assessed by coimmunoprecipitation.
Results
We identified two patients with the novel mutation p.Arg1872Leu and one patient with the recurrent mutation p.Arg1872Gln. The three mutations of Arg1872 and the mutation of Arg1617 all impaired the sodium channel transition from open state to inactivated state, resulting in channel hyperactivity. Other observed abnormalities contributing to elevated channel activity were increased persistent current, increased peak current density, hyperpolarizing shift in voltage dependence of activation, and depolarizing shift in steady‐state inactivation. Protein interactions were not affected.
Interpretation
Recurrent mutations at Arg1617 and Arg1872 lead to elevated Nav1.6 channel activity by impairing channel inactivation. Channel hyperactivity is the major pathogenic mechanism for gain‐of‐function mutations of SCN8A. EIEE13 differs mechanistically from Dravet syndrome, which is caused by loss‐of‐function mutations of SCN1A. This distinction has important consequences for selection of antiepileptic drugs and the development of gene‐ and mutation‐specific treatments.
Introduction
Early infantile epileptic encephalopathy type 13 (EIEE13, OMIM # 614558) is a newly recognized syndrome caused by de novo missense mutations of SCN8A. Clinical features include seizure onset before 18 months of age, intellectual disability, and developmental delay.1, 2, 3, 4, 5 Movement disorders are common and 50% of affected individuals are nonambulatory.1, 3, 6, 7, 8, 9, 10, 11, 12 SUDEP (sudden unexpected death in epilepsy) is reported in 10% of cases.1, 2, 4, 5
SCN8A encodes the pore‐forming voltage‐gated sodium channel subunit Nav1.6, which is widely expressed in neurons of the CNS and PNS.13 Nav1.6 is concentrated at the axon initial segment and at nodes of Ranvier.14, 15 Nav1.6 is composed of four homologous domains with six transmembrane segments each, and the cytoplasmic N‐terminal and C‐terminal domains (Fig. 1A).
Figure 1.
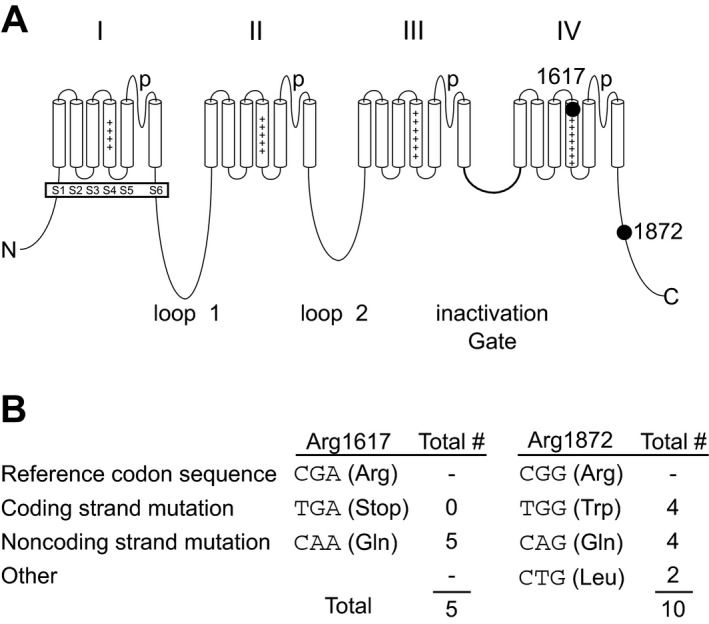
Recurrent mutations of arginine residues 1617 and 1872 in SCN8A. (A) Four‐domain structures of the voltage‐gated sodium channel α subunit. The positions of the recurrently mutated residues Arg1617 (the outermost charged residue in the S4 transmembrane segment of domain IV) and Arg1872 (near the middle of the cytoplasmic C‐terminal domain) are shown. (B) CpG mutation hot spots in the codons for Arg1617 and Arg1872.
Two amino acid residues, Arg1617 and Arg1872, are the sites of mutation in 20% of EIEE13. Arg1617 is located in the voltage‐responsive S4 transmembrane segment of domain IV (Fig. 1A). The substitution p.Arg1617Gln has occurred in five unrelated individuals.1, 2, 3, 16, 17 Arg1872 is located in the cytoplasmic C‐terminal domain (Fig. 1A). Two missense mutations of Arg1872 were described in seven patients1, 3, 11, 18 and we describe a third substitution in two patients, making this the most frequently mutated residue. The CpG dinucleotides in the codons for Arg1617 and Arg1872 are mutational hotspots (Fig. 1B). Recurrence in unrelated patients and absence from unaffected individuals (http://exac.broadinstitute.org) provides strong evidence that these mutations are pathogenic. Our data demonstrate that these mutations result in impaired channel inactivation that underlies neuronal hyperexcitability and seizures.
Material and Methods
Clinical exome sequencing
The c.5615G>T mutation (p.Arg1872Leu) in Case 1 was identified by whole exome sequencing at the Genomic Medicine Institute at the Cleveland Clinic (Cleveland, OH, USA). The c.5615G>T mutation (p.Arg1872Leu) in Case 2 was identified by whole exome sequencing at the Johns Hopkins Medical Institute (Baltimore, MD, USA). The c.5615G>A mutation (p.Arg1872Gln) in Case 3 was identified in the clinical exome sequencing laboratory at Radboud University (Nijmegen, the Netherlands).
Standard protocol approvals, registrations, and patient consents
An institutional review board (IRB) approval was obtained and the parent or legal guardian of each patient gave informed consent.
Site‐directed mutagenesis of the Nav1.6 cDNA and C‐terminal constructs
The amino acid substitutions p.Arg1617Gln, p.Arg1872Leu, and p.Arg1872Trp were introduced into the tetrodotoxin (TTX)‐resistant derivative of the full‐length Nav1.6 cDNA clone19 as described previously.4 Site‐directed mutagenesis was carried out with the QuikChange II XL kit (Agilent Technologies, Santa Clara, CA, USA). The entire 6‐kb open reading frame was sequenced to confirm the absence of other mutations.
The pHA‐Nav1.6‐CT cDNA construct containing the 213 amino acid C‐terminal domain of Nav1.6 with an N‐terminal HA epitope tag20 was kindly provided by Dr. David Ornitz, Washington University. The substitution p.Arg1872Trp was introduced as above and the open reading frame was completely sequenced.
Plasmids encoding sodium channel–interacting proteins
The sodium channel β subunit cDNA construct pNav_β_1‐EYFP was kindly provided by Dr. Jeanne Nerbonne.21 The fibroblast growth factor 14 cDNA construct pFGF14‐1b‐myc tag was a gift from Dr. David Ornitz.22 Plasmids pG_β_ 2IRES‐YFP and pG_γ_ 3IRES‐cyan encoding the G‐protein subunits G_β_ 2 and G_γ_ 3 were kindly provided by Dr. Massimo Mantegazza.23
Cell culture
DRG‐neuron–derived ND7/23 cells (Sigma Aldrich, St. Louis, MO, USA) were grown in a humidified atmosphere of 5% CO2 and 95% air at 37°C in Dulbecco's Modified Eagle's Medium (DMEM, 1X) supplemented with 10% fetal bovine serum (FBS), non‐essential amino acids (NEAA) and sodium pyruvate. Cells were plated onto Petri dishes 48 h prior to transfection and transfected for 5 h in non‐supplemented DMEM using Lipofectamine 3000 according to the manufacturer's instruction (Life Technologies, Carlsbad, CA, USA) with 5 _μ_g of Nav1.6 α subunit cDNA and 0.5 _μ_g of the fluorescent m‐Venus bioreporter. Electrophysiological recordings of fluorescent cells were made 48 h after transfection.
Electrophysiology
Recordings were carried out in the presence of 500 nmol/L TTX to block endogenous sodium currents in the neuron‐derived ND7/23 cells. Currents were recorded using the whole‐cell configuration of the patch clamp recording technique as described previously.24 The intracellular recording solution contained (in mmol/L): 140 CsF, 2 MgCl2, 1 EGTA, 10 HEPES, 4 Na2ATP, 0.3 NaGTP (pH adjusted to 7.3 with CsOH, osmolarity adjusted to 290 mOsm with sucrose). Cultured ND7/23 cells were bathed in solution containing (in mmol/L): 130 NaCl, 4 KCl, 1 CaCl2, 5 MgCl2, 5 HEPES, 5 Glucose (pH adjusted to 7.4 with NaOH, osmolarity adjusted to 300 mOsm with sucrose). Experiments were performed at room temperature (20–22°C). After establishing whole cell, a minimum series resistance compensation of 75% was applied. Capacitive and leak currents were subtracted using the P/N‐4 protocol for all experiments, except steady‐state inactivation protocols. The current–voltage relationship was determined using a 100‐msec voltage pulse from −80 to +70 mV in steps of 5 mV from a holding potential of −120 mV at 2 sec intervals. Conductance as a function of voltage was derived from the current–voltage relationship and fitted by a Boltzmann function as described previously.24 Decays of macroscopic currents were fitted to a single exponential function and time constants were determined.25 For steady‐state inactivation, neurons were held at −120 mV and test potentials from −115 mV to −10 mV for 1 sec at 5 mV increments were applied. The second pulse to −10 mV for 40 msec was used to assess channel availability. Currents during the second pulse were normalized for each cell with the largest current as 1.0 and fit to the Boltzmann function. For recovery from inactivation, neurons were held at −120 mV and depolarized to a test potential of 0 mV for 1 sec to inactivate the Na channels. Recovery was determined at times between 1 msec and 60 sec at a test potential of −90 mV. A 40 msec pulse to −10 mV was subsequently applied to assess the extent of channel recovery. For each cell, current amplitudes during this test pulse were normalized so that the largest current during the conditioning potential was 1.0. Data were fit to a double exponential function:
y=A1(1−exp[−t/T1])+A2(1−exp[−t/T2])
where A 1 and A 2 are the coefficients for the fast and slow exponentials, t is the time (in msec), and Ƭ 1 and Ƭ 2 are the fast and slow time constants, respectively.
Coimmunoprecipitation and western blotting
HEK293 cells were transfected with 3 _μ_g of cDNA encoding the C‐terminal domain of Nav1.6 and 3 _μ_g of the test construct. Cell extracts were prepared and immunoprecipitated as described previously.26 Cells were lysed in 1 mL of buffer containing 20 mmol/L Tris‐HCl, pH 7.5, 137 mmol/L NaCl, and 1% NP‐40 with protease inhibitors (Roche complete mini, EDTA‐free, Indianapolis, IN, USA). Lysates were incubated with 20 μ_L anti‐HA antibody‐coated agarose beads (sc‐7392AC, Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight at 4°C. Immune complexes on the beads were washed four times and eluted by boiling in 2X electrophoresis sample buffer at 95°C for 5 min. Western blotting was carried out as described previously.27 Blots were immunostained with rabbit polyclonal anti‐HA (H6908, Sigma Aldrich), rabbit polyclonal anti‐GFP (G1544, Sigma Aldrich), rabbit polyclonal anti‐myc (C3956, Sigma Aldrich), or rabbit polyclonal anti‐G_β 2 (sc‐380, Santa Cruz Biotechnology).
Results
Clinical features of three patients with mutations of arginine 1872
The clinical features of three new cases are summarized in Table 1. All three patients were born after full‐term pregnancies. Clinical exome sequencing identified the heterozygous mutation c.5615G>T resulting in the novel amino acid substitution p.Arg1872Leu in patients 1 and 2. Patient 3 carried the SCN8A mutation c.5615G>A resulting in the amino acid substitution p.Arg1872Gln. None of the mutations were present in either parent, demonstrating de novo origin.
Table 1.
Clinical features of three new patients with amino acid substitution of arginine residue 1872
| Patient (sex) | 1 (F) | 2 (F) | 3 (M) |
|---|---|---|---|
| Amino acid substitution | p.Arg1872Leu | p.Arg1872Leu | p.Arg1872Gln |
| Seizure onset | 2 weeks | 6 weeks | 4 months |
| Current age | 17 years | 1.5 years | 2 years |
| Seizure types | GTC | GTC with apnea | GTC, A, T with bradycardia, SE |
| Development and motor abnormalities | Walking at 12‐15 months, then regression with hypotonia progressing to hypertonia and quadriplegia | Sat 12 months, hypotonia | Speech arrest 10 months, sat 13 months, walked 16 months |
| Intellectual disability | Severe | Moderate | Severe |
| EEG | Generalized slowing, severe diffuse encephalopathy | Normal | Diffuse encephalopathy |
| MRI | Cerebella and cerebral atrophy | Normal | Bilateral flat insular cortex |
| Drug response | PHT and VNS partially effective | TPM and LEV temporarily effective, OXC, LTG, VPA, and CLN effective | ZNS and LEV temporarily effective, PHT and LEV effective |
The features of these patients are consistent with the recently described clinical spectrum of SCN8A encephalo‐pathy.1 Two patients exhibited very early onset at 2 and 6 weeks of age. Sodium channel blockers were partially effective (Table 1). Detailed clinical histories are provided as an Appendix.
Functional effects of mutations in arginine residue 1872
Arginine residue 1872 is located near the center of the cytoplasmic C‐terminal domain of Nav1.6 (Fig. 1A). This site was implicated in direct interaction with the inactivation gate of the channel, mediated by interaction between positively and negatively charged residues.28, 29 Loss of the positive charge of arginine residue 1872 is therefore predicted to impair channel inactivation.
Patient mutations substituting leucine, glutamine, or tryptophan for arginine 1872 were introduced into the full‐length Nav1.6 cDNA, which was transfected into the neuron‐derived cell line ND7/23. Representative currents from ND7/23 cells transfected with wild‐type (WT) and mutant Nav1.6 channels are shown in Figure 2A. Peak current density was elevated for p.Arg1872Trp, but was not significantly changed for p.Arg1872Leu or p.Arg1872Gln (Fig. 2B, Table 2). To determine the kinetics of open state inactivation, macroscopic current decay was fit to single exponential functions and the fast time constant (τ fast) was plotted as a function of voltage. Transition from open state to the inactivated state was significantly delayed by all three mutations (Fig. 2C), consistent with prediction. The slowing of fast inactivation is evident when the peaks of the current traces are aligned so that inactivation kinetics can be compared directly (Fig. 2D).
Figure 2.
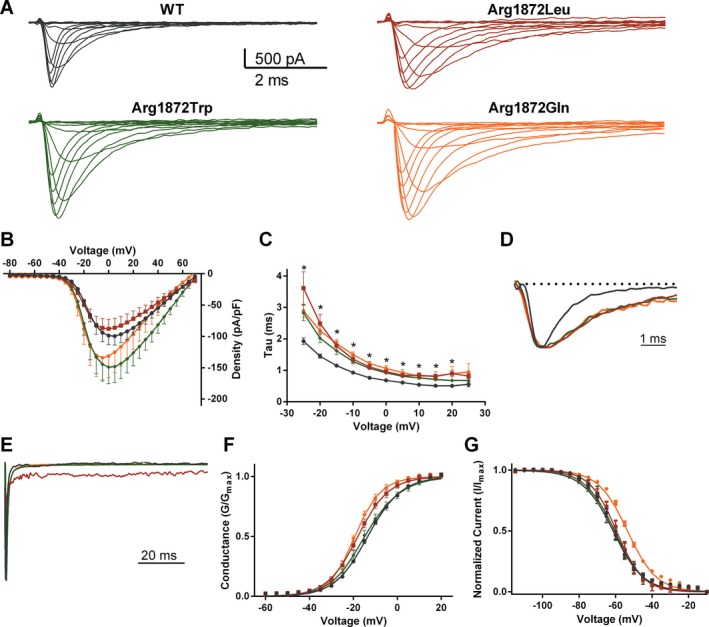
Biophysical effects of missense mutations at residue Arg1872 in Nav1.6. (A) Representative traces of families of Na currents from ND7/23 cells transfected with the indicated Nav1.6 cDNAs. (B) Averaged current–voltage (I‐V) relation for cells expressing WT and mutant Nav1.6. Peak currents were normalized to cell capacitance. (C) Average fast time constant obtained from single exponential fits to macroscopic current decays as a function of voltage (mV). (D) Representative traces of normalized currents evoked by a −20 mV stimulus from a holding potential of −120 mV illustrate delays in macroscopic current decay. (E) Representative normalized current traces recorded during a 100‐msec depolarizing pulse from −120 mV to 0 mV illustrating the presence of elevated persistent sodium current for p.Arg1872Leu. (F) Voltage dependence of channel activation. (G) Voltage dependence of steady‐state inactivation. Smooth lines correspond to the least squares fit when average data were fit to a single Boltzmann equation. Data are mean ± SEM. Statistical significance: *P < 0.05. Black, wild type; red, p.Arg1872Leu; orange, p.Arg1872Gln; green, p.Arg1872Trp.
Table 2.
Biophysical properties of SCN8A mutants
| Peak Current (pA/pF) | n | Persistent | Activation (mV) | Inactivation (mV) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ratio (Ip/Ipeak) | n | V 1/2 | k | n | V 1/2 | k | n | |||
| WT | −101 ± 15 | 30 | 1.8 ± 0.4 | 8 | −13.3 ± 0.5 | −7.3 ± 0.2 | 30 | −61.5 ± 1.1 | 8.0 ± 0.4 | 19 |
| Arg1872Leu | −89 ± 15 | 10 | 4.7 ± 1.5* | 6 | −17.9 ± 1.2*** | −6.7 ± 0.4 | 10 | −60.0 ± 1.3 | 7.5 ± 0.8 | 10 |
| Arg1872Trp | −153 ± 27† | 21 | 2.7 ± 0.4 | 11 | −14.2 ± 1.4 | −7.2 ± 0.4 | 21 | −61.4 ± 1.6 | 8.6 ± 0.6 | 18 |
| Arg1872Gln | −135 ± 35 | 8 | 1.6 ± 0.5 | 17 | −19.2 ± 0.8*** | −5.8 ± 0.5** | 8 | −53.5 ± 0.5*** | 7.9 ± 0.3 | 18 |
| Arg1617Gln | −73 ± 15 | 10 | 5.5 ± 1.7* | 5 | −17.1 ± 2.0* | −7.8 ± 0.3 | 10 | −52.6 ± 1.7*** | 12.5 ± 0.6*** | 8 |
At 100 msec after stimulus, p.Arg1872Leu exhibited elevated persistent current normalized to the peak of the macroscopic current (I persistent/I peak) (Fig. 2E, Table 2). Ramp currents evoked using a slow ramp stimulus did not differ from WT for any of the mutants (data not shown).
Analysis of steady‐state kinetics demonstrates that p.Arg1872Leu and p.Arg1872Gln cause a small but significant hyperpolarizing shift in the voltage dependence of activation (P < 0.05) (Fig. 2F, Table 2). In addition, p.Arg1872Gln caused a depolarizing shift in steady‐state inactivation (Fig. 2G, Table 2). The values for the slope (k) were unchanged.
Functional effects of the recurrent mutation p.Arg1617Gln
Arginine residue 1617 is the outermost positively charged residue in the voltage‐responsive transmembrane segment DIV‐S4 and is implicated in coupling of channel activation with subsequent inactivation.
Representative currents from ND7/23 cells transfected with the p.Arg1617Gln mutant of Nav1.6 are shown in Figure 3A. Peak current densities for p.Arg1617Gln were not different from WT (Fig. 3B). A profound change in the kinetics of transition from open state to the inactivated state was observed. At voltages above −20 mV, the WT channel exhibits faster decay time constants with increasing depolarizing voltage steps, but this response is missing for p.Arg1617Gln (Fig. 3C). Sample traces reveal the striking difference in current decay between WT and mutant channel (Fig. 3D). The apparent uncoupling of activation to fast inactivation likely accounts for the large increase in persistent current from 1% of peak transient current for the WT channels to 6% of peak transient current for the p.Arg1617Gln channels (Fig. 3E). The elevation of persistent current is evident in the ratio of persistent current to peak current for p.Arg1617Gln (Table 2).
Figure 3.
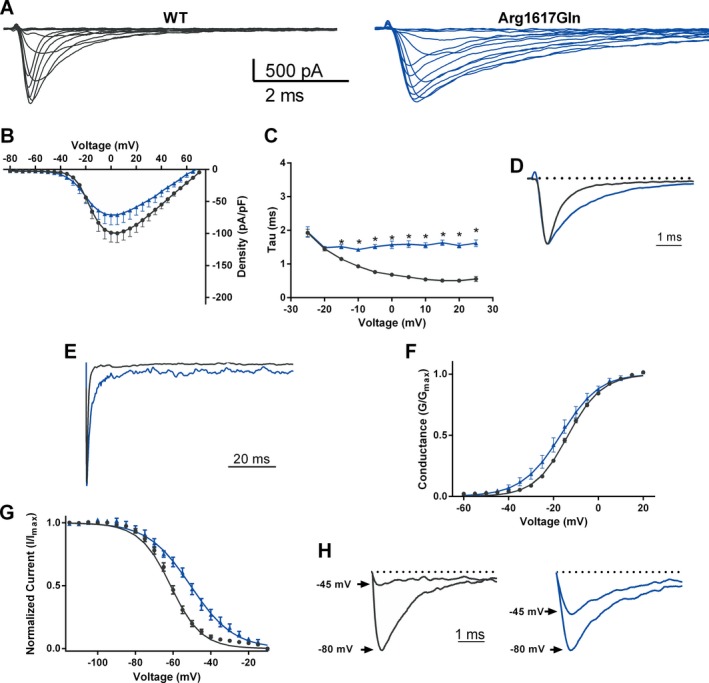
Biophysical effects of the missense mutation p.Arg1617Gln in Nav1.6. (A) Representative traces of families of Na currents from ND7/23 cells transfected with the indicated Nav1.6 cDNA. (B) Averaged current–voltage (I‐V) relation for cells expressing WT and p.Arg1617Gln. Peak currents were normalized to cell capacitance. (C) Average fast time constant obtained from single exponential fits to macroscopic current decays as a function of voltage (mV). (D) Representative traces of normalized currents evoked by a depolarizing step to −20 mV from a holding potential of −120 mV illustrate significant delay in macroscopic current decay in p.Arg1617Gln (blue) compared to WT (black). (E) Representative normalized current traces recorded during a 100‐msec depolarizing pulse from −120 mV to 0 mV illustrating the presence of elevated persistent sodium current. (F) Voltage dependence of channel activation. (G) Voltage dependence of steady‐state inactivation. (H). Representative inactivation traces recorded after a 1‐sec prepulse of −100 mV (larger amplitude) and −45 mV (smaller amplitude). Smooth lines in (F) and (G) correspond to the least squares fit when average data were fit to a single Boltzmann equation. Data are mean ± SEM. Statistical significance: *P < 0.05. Black, wild type; blue, p.Arg1617Gln.
The inactivation parameters for p.Arg1617Gln were significantly shifted in the depolarizing direction and the value of the slope was increased, demonstrating disruption in the normal process of channel inactivation (Fig. 3G, Table 2). The difference in the extent of inactivation at −45 mV is shown in Figure 3H. There was also a small hyperpolarizing shift in the voltage dependence of activation (Fig. 3F, Table 2).
Protein interactions with the C‐terminal domain of Nav1.6
In addition to its role in channel inactivation, the C‐terminal domain of Nav1.6 interacts with several proteins that regulate channel trafficking or activity.20, 23, 30 To determine whether mutations at Arg1872 disrupt these interactions, we tested the effect of the p.Arg1872Trp mutation with substitution of the large uncharged tryptophan residue.
HEK cells were cotransfected with cDNAs encoding the HA‐tagged WT or mutant C‐terminal fragment of Nav1.6 and cDNAs encoding the G‐protein subunits G_β_ 2 and G_γ_ 3. Lysates were incubated with anti‐HA‐coated beads and bound proteins were eluted and examined by western blotting. The yield of G_β_ 2 was comparable with mutant and WT C‐terminus (Fig. S1A). Similarly, the mutation did not impair binding of the sodium channel _β_1 subunit (Fig. S1B) and the fibroblast growth factor isoform FGF14b (Fig. S1C).
Discussion
Unlike protein termination mutations, the pathogenic impact of missense mutations is difficult to predict without functional tests. Due in part to the multiple intramolecular interactions within the sodium channel α subunit,31, 32 substitution of a single amino acid often has multiple effects on sodium channel function.
Mutations at Arg1872 and Arg1617 impair channel inactivation by different intramolecular mechanisms
The three allelic mutations that eliminate the positively charged residue Arg1872 from the C‐terminus of Nav1.6 all result in a significant delay in transition from the open state to the inactivated state. This observation is consistent with previous studies demonstrating a direct role of the C‐terminal domain in fast inactivation.29, 33 Interaction of the C‐terminus of sodium channel Nav1.5 with its inactivation gate was shown to stabilize the inactivated state.31, 34 Exchange of C‐terminal domains between sodium channels Nav1.2, Nav1.5, and Nav1.6 results in partial transfer of inactivation properties.28, 33 The interaction between the C‐terminus and the inactivation gate appears to involve electrostatic interactions, since inactivation of Nav1.2 was slowed by reversing the charge of Glu1880 (corresponding to Glu1870 in Nav1.6) by replacement with lysine.29 These data support the view that loss of the positively charged residue arginine 1872 destabilizes interaction with the inactivation gate and delays entry into the inactivated state. This is predicted to increase the fraction of channels that can reopen during sustained depolarization, resulting in increased channel activity and epileptogenic persistent current. The different effects on current density and channel kinetics of these allelic substitutions can be related to the chemical structure of the substituted residue. Substitution of the corresponding arginine residue of Nav1.2 by leucine or glutamine also results in epileptic encephalopathy.18, 35
Replacement of arginine residue 1617 by glutamine appears to interfere with the initiation of fast inactivation from the open state. Arginine 1617 is the outermost charged residue in the S4 voltage sensor segment of DIV, which is thought to initiate channel inactivation through a conformational change.36 In the crystal structure of the single domain bacterial channel NaChBac, the outermost arginine of S4 interacts with the negatively charged glutamate residue in segment S1 in the open state, and the negatively charged aspartate in segment S2 in the closed state.37 Loss of the positively charged arginine by p.Arg1617Gln would disrupt these electrostatic interactions, consistent with the observed defect in coupling of activation and inactivation (Fig. 2C). We also observed a small depolarizing shift in the voltage dependence of inactivation due to p.Arg1617Gln. The alterations in V 1/2 and the slope of the steady‐state inactivation curve are consistent with impaired allosteric interactions between the DIV‐S1 sensor segment that initiates activation and the DIV‐S4 sensor segment that initiates inactivation.
Comparison with previously described pathogenic mutations of SCN8A
Through different effects on intramolecular interactions, the recurrent mutations of Arg1617 and Arg1872 lead to disruption of channel inactivation in 15 independent patients. Along with the original mutation, p.Asn1768Asp, this work indicates that impaired inactivation of Nav1.6 is the predominant pathogenic mechanism in EIEE13. A contrasting mechanism, premature channel activation, was observed for two mutations located in transmembrane segments of domain II, pThr767Ile and p.Asn984Lys.6, 9 The other two functionally characterized SCN8A mutations caused reduction in peak current density (p.Gly1451Ser)6 and decreased protein stability (p.Arg223Gly)7; the role of these effects in neuronal hyperexcitability is unclear.
Functional alterations of SCN8A and clinical severity
There is considerable heterogeneity in the clinical features of patients with mutation of Arg1872, even among patients with identical mutations (Table S1). With respect to age of onset, intellectual disability, and motor function, the least severe course of disease was seen in patients with p.Arg1872Gln. More severe impairment, extending to inability to initiate purposeful movement and one case of SUDEP, was seen in the patients with p.Arg1872Leu and p.Arg1872Trp. p.Arg1872Gln led to bradycardia in patient 3, consistent with our recent observation of cardiac arrhythmia in a mouse model of EIEE13 (Frasier et al., unpubl. ms.). The expression of Nav1.6 in cardiomyocytes38 may contribute to risk of SUDEP in EIEE13 patients.
The five previously described patients with the p.Arg1617Gln mutation also displayed considerable clinical heterogeneity, with seizure onset between 3 and 12 months of age, and variation in drug responses.5 One patient became nonambulatory and one had SUDEP.
The features of patients with mutations of Arg1617 and Arg1872 exhibit considerable overlap, consistent with their shared effect on channel inactivation. Seizure onset within the first month is more common in patients with mutations of Arg1872, but there is no difference in the proportion of patients whose seizures are refractory to drug treatment, or in the frequency of nonambulatory status. The variation within each group appears as great as the difference between groups. Variation between patients with identical mutations may be accounted for by differences in genetic background, including genetic variation in other channel genes,39 as well as stochastic variation during the development of the nervous system. The course of drug treatment itself may influence clinical outcome.5
Development of targeted therapy for EIEE13
Sodium channel blockers have some efficacy in this patient population, presumably by countering the effects of gain‐of‐function mutations causing channel hyperactivity (Tables 1, S1). This beneficial response contrasts to the response of patients with Dravet syndrome (EIEE6), in which the effects of loss‐of‐function mutations of SCN1A are exacerbated by administration of sodium channel blockers. Unfortunately, response to antiepileptic drugs in EIEE13 is often transient. The development of Nav1.6‐specific channel blockers has been hampered by the structural similarity among members of the voltage‐gated sodium channel gene family. A mouse epilepsy model with impaired inactivation of Scn2a was effectively treated with the cardiac drugs ranolazine and GS967, which are somewhat selective inhibitors of persistent sodium current.40 Activation of potassium channels to compensate for sodium channel hyperactivity is another potential approach.
Recurrent missense mutations at SCN8A residues Arg1617 and Arg1872 result in elevated activity of Nav1.6 as a result of impaired channel inactivation. This work confirms the important role of gain‐of‐function mutations in EIEE13 and demonstrates that impaired channel inactivation is a prominent pathogenic mechanism.
Author Contributions
The study was designed by J. L. W., M. H. M., and M. K. P. Clinical descriptions were provided by C. A. H., A. S., T. M., S. P., and R. D. M. Experimental data were collected by J. L. W., B. S. B., and J. A. H. and interpreted by J. L. W., B. S. B., J. A. H., M. K. P., and M. H. M. The manuscript was drafted by J. L. W., M. H. M., and M. K. P., and revised by all authors.
Conflict of Interest
None declared.
Supporting information
Figure S1. Substitution of tryptophan for Arg1872 of Nav1.6 does not impair coimmunoprecipitation of interacting proteins. (A) G_β_ 2, (B) GFP‐tagged sodium channel subunit _β_1, (C) myc‐tagged FGF14b.
Table S1. Phenotypes of 10 individuals with amino acid substitutions at arginine residue 1872.
Acknowledgments
We thank the families for participating in this study. We are grateful to Sulayman Dib‐Hajj and Stephen Waxman for providing the Nav1.6 cDNA. We also thank David Ornitz, Jeanne Nerbonne, and Massimo Mantegazza for cDNA constructs, and Jaclyn Walker and Rosie Bunton‐Stasyshyn for technical assistance and helpful discussions. This work was supported by the National Institutes of Health (NIH) grants R01‐NS34509 (to M. H. M.) and R01‐NS075157 (to M. K. P.). J. L. W. is recipient of a postdoctoral fellowship from the Dravet Syndrome Foundation.
Appendix 1.
Clinical Descriptions
Patient 1 experienced onset of generalized tonic–clonic seizures at 2 weeks of age that were refractory to treatment. A vagus nerve stimulator placed at 4 years of age reduced seizure frequency. Phenytoin reduced seizures, while carbamazepine, oxcarbazepine, lamotrigine, and topirimate were not effective. The patient was able to stand and walk with support at 12–15 months, but regressed and became nonambulatory, with generalized hypotonia at 5 years, contractures and spasticity at 13 years, and generalized hypertonia and spastic quadriplegia at 16 years. MRI revealed moderate cerebellar and mild cerebral atrophy. At 17 years, the patient is nonverbal with severe intellectual disability.
Patient 2 experienced three prolonged seizures between 6 and 8 weeks of age. No abnormalities were detected by MRI or prolonged EEG. She responded well to levetiracetam and topiramate for 2 months before the return of generalized tonic–clonic seizures with stiffening of the extremities, twitching, and eye fluttering. Seizures were often accompanied by apnea and oxygen desaturation that resolved after the seizure. The patient was transitioned from levetiracetam and topirimate to oxcarbazepine and lamotrigine with good response until seizure frequency increased at 11 months of age. With the addition of valproate and clonazepam, seizures were well controlled from 12 months to the current age of 18 months. Developmental delay was mild until 11 months, when increased seizure frequency resulted in acute developmental regression including loss of head and trunk control, loss of eye contact, and placement of a feeding tube. Control of seizures at 12 months of age resulted in improvement including regained eye contact and head control and the ability to sit with support.
Patient 3 experienced seizures beginning at 4 months of age, usually during sleep or on waking, accompanied by tonic posturing of the head and severe bradycardia, followed by flexing of the arms and extension of the legs. Levetiracetam and zonisamide controlled seizures for 1 month before daily seizures reoccurred. Elevating zonisamide reduced seizures but caused severe (nonepileptic) encephalopathy, reversible upon lowering zonisamide. Addition of phenytoin reduced seizures to ≤1 per week. Gradual reduction of phenytoin resulted in increased seizure frequency. After an episode of status epilepticus at 19 months, seizure frequency stabilized at 5–10 per day, with development of asymmetric and absence seizures. Complete seizure control was obtained at 20 months with phenytoin and levetiracetam, and continued through the current age of 24 months. EEG was normal initially, but 6 weeks after seizure onset a hypofunctional area was detected in the left temporal lobe, from which abundant spike‐and‐wave configurations and polyspikes emerged. EEGs at 6 and 9 months showed severe diffuse encephalopathy and multifocal epileptic discharges without hypsarrhythmia. MRI at 12 months showed a slightly underdeveloped cortex. Development was normal prior to seizure onset. Speech arrested at 10 months of age. While seizures were partially controlled between 11 and 17 months of age, he began sitting and walking with support. After status epilepticus at 19 months there was regression, but by 24 months he recovered unassisted walking and improved social interaction without improvement in development of speech.
References
- 1.Larsen J, Carvill GL, Gardella E, et al. The phenotypic spectrum of SCN8A encephalopathy. Neurology 2015;84:480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kong W, Zhang Y, Gao Y, et al. SCN8A mutations in Chinese children with early onset epilepsy and intellectual disability. Epilepsia 2015;56:431–438. [DOI] [PubMed] [Google Scholar]
- 3.Ohba C, Kato M, Takahashi S, et al. Early onset epileptic encephalopathy caused by de novo SCN8A mutations. Epilepsia 2014;55:994–1000. [DOI] [PubMed] [Google Scholar]
- 4.Veeramah KR, O'Brien JE, Meisler MH, et al. De novo pathogenic SCN8A mutation identified by whole‐genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet 2012;90:502–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wagnon JL, Meisler MH. Recurrent and non‐recurrent mutations of SCN8A in epileptic encephalopathy. Front Neurol 2015;6:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blanchard MG, Willemsen MH, Walker JB, et al. De novo gain‐of‐function and loss‐of‐function mutations of SCN8A in patients with intellectual disabilities and epilepsy. J Med Genet 2015;52:330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Kovel CG, Meisler MH, Brilstra EH, et al. Characterization of a de novo SCN8A mutation in a patient with epileptic encephalopathy. Epilepsy Res 2014;108:1511–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Epi4K Consortium, Epilepsy Phenome/Genome Project , Allen AS, et al. De novo mutations in epileptic encephalopathies. Nature 2013;501:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Estacion M, O'Brien JE, Conravey A, et al. A novel de novo mutation of SCN8A (Nav1.6) with enhanced channel activation in a child with epileptic encephalopathy. Neurobiol Dis 2014;69:117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh R, Jayapal S, Goyal S, et al. Early‐onset movement disorder and epileptic encephalopathy due to de novo dominant SCN8A mutation. Seizure 2015;26:69–71. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi S, Yamamoto S, Okayama A, et al. Electroclinical features of epileptic encephalopathy caused by SCN8A mutation. Pediatr Int 2015;57:758–762. [DOI] [PubMed] [Google Scholar]
- 12.Vaher U, Noukas M, Nikopensius T, et al. De novo SCN8A mutation identified by whole‐exome sequencing in a boy with neonatal epileptic encephalopathy, multiple congenital anomalies, and movement disorders. J Child Neurol 2014;29:NP202–NP206. [DOI] [PubMed] [Google Scholar]
- 13.O'Brien JE, Meisler MH. Sodium channel SCN8A (Nav1.6): properties and de novo mutations in epileptic encepha‐lopathy and intellectual disability. Front Genet 2013;4:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caldwell JH, Schaller KL, Lasher RS, et al. Sodium channel Na(v)1.6 is localized at nodes of Ranvier, dendrites, and synapses. Proc Natl Acad Sci USA 2000;97:5616–5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lorincz A, Nusser Z. Cell‐type‐dependent molecular composition of the axon initial segment. J Neurosci 2008;28:14329–14340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dyment DA, Tetreault M, Beaulieu CL, et al. Whole‐exome sequencing broadens the phenotypic spectrum of rare pediatric epilepsy: a retrospective study. Clin Genet 2015;88:34–40. [DOI] [PubMed] [Google Scholar]
- 17.Rauch A, Wieczorek D, Graf E, et al. Range of genetic mutations associated with severe non‐syndromic sporadic intellectual disability: an exome sequencing study. Lancet 2012;380:1674–1682. [DOI] [PubMed] [Google Scholar]
- 18.Carvill GL, Heavin SB, Yendle SC, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet 2013;45:825–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herzog RI, Cummins TR, Ghassemi F, et al. Distinct repriming and closed‐state inactivation kinetics of Nav1.6 and Nav1.7 sodium channels in mouse spinal sensory neurons. J Physiol 2003;551:741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laezza F, Lampert A, Kozel MA, et al. FGF14 N‐terminal splice variants differentially modulate Nav1.2 and Nav1.6‐encoded sodium channels. Mol Cell Neurosci 2009;42:90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marionneau C, Carrasquillo Y, Norris AJ, et al. The sodium channel accessory subunit Navbeta1 regulates neuronal excitability through modulation of repolarizing voltage‐gated K(+) channels. J Neurosci 2012;32:5716–5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laezza F, Gerber BR, Lou JY, et al. The FGF14(F145S) mutation disrupts the interaction of FGF14 with voltage‐gated Na+ channels and impairs neuronal excitability. J Neurosci 2007;27:12033–12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mantegazza M, Yu FH, Powell AJ, et al. Molecular determinants for modulation of persistent sodium current by G‐protein betagamma subunits. J Neurosci 2005;25:3341–3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hargus NJ, Nigam A, Bertram EH III, Patel MK. Evidence for a role of Nav1.6 in facilitating increases in neuronal hyperexcitability during epileptogenesis. J Neurophysiol 2013;110:1144–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ko SH, Lenkowski PW, Lee HC, et al. Modulation of Na(v)1.5 by beta1– and beta3‐subunit co‐expression in mammalian cells. Pflugers Arch 2005;449:403–412. [DOI] [PubMed] [Google Scholar]
- 26.O'Brien JE, Sharkey LM, Vallianatos CN, et al. Interaction of voltage‐gated sodium channel Nav1.6 (SCN8A) with microtubule‐associated protein Map1b. J Biol Chem 2012;287:18459–18466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wagnon JL, Korn MJ, Parent R, et al. Convulsive seizures and SUDEP in a mouse model of SCN8A epileptic encephalopathy. Hum Mol Genet 2015;24:506–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee A, Goldin AL. Role of the amino and carboxy termini in isoform‐specific sodium channel variation. J Physiol 2008;586:3917–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen HM, Goldin AL. Sodium channel carboxyl‐terminal residue regulates fast inactivation. J Biol Chem 2010;285:9077–9089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spampanato J, Kearney JA, de Haan G, et al. A novel epilepsy mutation in the sodium channel SCN1A identifies a cytoplasmic domain for beta subunit interaction. J Neurosci 2004;24:10022–10034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Motoike HK, Liu H, Glaaser IW, et al. The Na+ channel inactivation gate is a molecular complex: a novel role of the COOH‐terminal domain. J Gen Physiol 2004;123:155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Payandeh J, Scheuer T, Zheng N, Catterall WA. The crystal structure of a voltage‐gated sodium channel. Nature 2011;475:353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mantegazza M, Yu FH, Catterall WA, Scheuer T. Role of the C‐terminal domain in inactivation of brain and cardiac sodium channels. Proc Natl Acad Sci USA 2001;98:15348–15353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cormier JW, Rivolta I, Tateyama M, et al. Secondary structure of the human cardiac Na+ channel C terminus: evidence for a role of helical structures in modulation of channel inactivation. J Biol Chem 2002;277:9233–9241. [DOI] [PubMed] [Google Scholar]
- 35.Baasch AL, Huning I, Gilissen C, et al. Exome sequencing identifies a de novo SCN2A mutation in a patient with intractable seizures, severe intellectual disability, optic atrophy, muscular hypotonia, and brain abnormalities. Epilepsia 2014;55:e25–e29. [DOI] [PubMed] [Google Scholar]
- 36.Chanda B, Asamoah OK, Bezanilla F. Coupling interactions between voltage sensors of the sodium channel as revealed by site‐specific measurements. J Gen Physiol 2004;123:217–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yarov‐Yarovoy V, DeCaen PG, Westenbroek RE, et al. Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc Natl Acad Sci USA 2012;109:E93–E102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Noujaim SF, Kaur K, Milstein M, et al. A null mutation of the neuronal sodium channel NaV1.6 disrupts action potential propagation and excitation‐contraction coupling in the mouse heart. FASEB J 2012;26:63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klassen T, Davis C, Goldman A, et al. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell 2011;145:1036–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anderson LL, Thompson CH, Hawkins NA, et al. Antiepileptic activity of preferential inhibitors of persistent sodium current. Epilepsia 2014;55:1274–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Substitution of tryptophan for Arg1872 of Nav1.6 does not impair coimmunoprecipitation of interacting proteins. (A) G_β_ 2, (B) GFP‐tagged sodium channel subunit _β_1, (C) myc‐tagged FGF14b.
Table S1. Phenotypes of 10 individuals with amino acid substitutions at arginine residue 1872.