TEtranscripts: a package for including transposable elements in differential expression analysis of RNA-seq datasets (original) (raw)
Abstract
Motivation: Most RNA-seq data analysis software packages are not designed to handle the complexities involved in properly apportioning short sequencing reads to highly repetitive regions of the genome. These regions are often occupied by transposable elements (TEs), which make up between 20 and 80% of eukaryotic genomes. They can contribute a substantial portion of transcriptomic and genomic sequence reads, but are typically ignored in most analyses.
Results: Here, we present a method and software package for including both gene- and TE-associated ambiguously mapped reads in differential expression analysis. Our method shows improved recovery of TE transcripts over other published expression analysis methods, in both synthetic data and qPCR/NanoString-validated published datasets.
Availability and implementation: The source code, associated GTF files for TE annotation, and testing data are freely available at http://hammelllab.labsites.cshl.edu/software.
Contact: mhammell@cshl.edu.
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Transposable elements are mobile DNA elements that constitute a large fraction of most eukaryotic genomes. These parasitic genetic elements propagate by multiplying within the genomes of host cells and can be passed from generation to generation through the germline lineage. Although the majority of TE copies are non-functional, a subset has retained the ability to transcribe and mobilize (Beck et al., 2010; Bennett et al., 2008; Hancks and Kazazian, 2012; Honma et al., 1993; Huang et al., 2012; Kano et al., 2009; Mills et al., 2007). Although retrotransposons require an RNA intermediate to transpose, both DNA and RNA transposons are transcribed from the genome, and they can accumulate in conditions such as cancer (Criscione et al., 2014; Lamprecht et al., 2012; Lee et al., 2012; Sciamanna et al., 2013; Sciamanna et al., 2014; Shukla et al., 2013; Tubio et al., 2014), neurodegenerative diseases (Bundo et al., 2014; Li et al., 2013; Reilly et al., 2013), as well as during embryogenesis (Fadloun et al., 2013; Macia et al., 2011; Peaston et al., 2004), neural development (Coufal et al., 2009; Coufal et al., 2011; Faulkner et al., 2009; Muotri et al., 2005; Perrat et al., 2013; Thomas et al., 2012) and aging (De Cecco et al., 2013; Li et al., 2013; Sedivy et al., 2013). However, TE-associated reads are often discarded in sequencing data analyses because of the uncertainty in attributing ambiguously mapped reads to these regions, despite some previous attempts to integrate them in downstream analyses (Chung et al., 2011; Day et al., 2010; Rosenfeld et al., 2009; Treangen and Salzberg, 2011; Tucker et al., 2011; Wang et al., 2010). Here, we present a program called TEtranscripts that allows users to analyze both gene- and TE-associated reads concurrently in one simplified workflow.
2 Input data
The input data for TEtranscripts consists of alignment files in either the SAM or BAM format (Li et al., 2009), and two annotation files in the General Transfer Format (GTF) (http://mblab.wustl.edu/GTF22.html) for genes and TEs, respectively. For the purposes of this article, we will use the terms, unique-reads and multi-reads, to designate the reads that have a unique alignment in the genome or map to multiple loci with equal quality, respectively. The utilization of multi-reads for TE quantification is critical, as a read originating from a TE could align to multiple instances (insertions) of that element in the genome. Many aligners support multi-reads alignments, and provide limits for the maximum number of multiple alignments per reads to output, e.g. bowtie -m (Langmead et al., 2009). To optimally set this parameter, we recommend a saturation analysis on the multi-read alignments as described in the Supplementary Materials (Supplementary Fig. S1). TEtranscripts also supports strand-specific read counting, and applies it to both genes and TEs. GTF files of transposable element annotations were generated from the RepeatMasker (Smit et al., 1996, http://www.repeatmasker.org) tables obtained from the UCSC genome database (Karolchik et al., 2003), or from annotations provided by the maize MIPS (Nussbaumer et al., 2013) and MTEC databases (http://maizetedb.org/∼maize/). The annotation tables were parsed to filter out low complexity and simple repeats, rRNA, scRNA, snRNA, srpRNA and tRNA. Each TE insertion in the table was given a unique identifier. The genomic location, element name, as well as family and class information were also extracted from the table and included in the GTF file. TEtranscripts can also utilize custom TE annotations, such as those generated from de novo TE insertion analysis, as long as they conform to the format described earlier and are consistent with the genome sequencing files used for the alignment.
3 Methods
TEtranscripts estimates both gene and TE transcript abundances in RNA-seq data and conducts differential expression analysis on the resultant count tables. The general workflow of TEtranscripts is given in Figure 1. Read assignment and statistical modeling is discussed in detail in this section.
Fig. 1.

TEtranscripts flow chart. Reads mapping to TEs are assigned in two different modes: uniq (reads mapping uniquely in the genome), and multi (reads mapping to multiple insertions of TEs). In the multi mode, an iterative algorithm is used to optimally distribute ambiguously mapped reads
3.1 Index genomic features
To quickly find all genes/TEs that overlap with any given read alignment, TEtranscripts builds two independent index structures on gene and TE annotations, respectively. The gene/TE index structure consists of a hash table with reference sequence names and a list of interval trees as key-value pairs. For each chromosome, there is an interval tree generated based on gene/TE insertions annotated on that chromosome. It allows the SAM/BAM read alignment to be rapidly matched with the genome intervals in GTF annotations, especially when there are a large number of TE insertions.
3.2 Read assignment
The next step involves distributing the mapped reads among the annotated genes and TEs that overlap those genomic alignments. Unique-reads, which represent most gene-associated reads, but only a subset of TE-associated reads, are comparatively simple to distribute. For a multi-read, the task is more difficult. TEtranscripts takes advantage of the sequence similarities at the different levels of the hierarchy of TEs in order to optimally distribute reads amongst closely related TE sequences. Based on the definitions and nomenclature provided by RepBase (Jurka et al., 2005), TE ‘insertions’ (loci within the genome) are grouped into ‘elements’, which are sub-families of TEs that are highly related at the sequence level and relatively distinct from other elements. For example, Repbase and RepeatMasker report 16 293 insertions for the L1Md_A element in the mouse reference genome (mm9), all of which are more similar to each other than they are to other elements of the L1 family (such as L1Md_T). By estimating combined abundances for all insertions of an element, we obtain more reliable and reproducible results than analyses that attempt to pin down the exact genomic instance of the TE being transcribed. Thus, TEtranscripts performs estimation of expression abundances on the element level by default, which is the recommended setting. TEtranscripts parses the alignment file only once, processing genes and TEs at the same time. Given a uniquely mappable read, the algorithm first searches for overlapping gene exons; if it is a multi-read, overlap with TEs will be first computed. For TE-associated reads, the user can choose whether to count only unique-reads or all reads, i.e. uniq mode and multi mode.
Under multi mode, it is important to assign weight to the contribution of the ambiguously mapped reads at each mapped locus, so that no double counting of reads occurs. Given all available alignments of a read, every alignment is assigned a weight of 1/n, where n is the number of alignments. Therefore, the total contribution of a multi-read to the library size is the same as a unique-read. This is important in maintaining the library size for each sample (calculated based on the total number of mapped reads), as it is heavily utilized for normalization when comparing between multiple libraries.
3.3 TE transcript estimation
After the read assignment step, an expectation-maximization (EM) algorithm is used to determine the maximum-likelihood estimates of multi-reads assignments to all TE transcripts. The unique-reads are not used as a prior for the initial abundance estimates in the EM procedure to reduce potential bias to certain TEs. Specifically, active TEs, which tend to be younger elements, have accumulated far fewer polymorphisms than older TEs, and thus have far fewer reads mapping uniquely to these elements. Using uniquely mapped TE reads in the optimization step will bias read assignment away from the youngest TE sub-families and toward the older related TE subfamilies with higher uniquely mappable content.
3.3.1 Expectation maximization
The EM algorithm alternates between computing the fractional distribution of each multi-read to each mapped TE instance (E-step) and estimating the relative abundances of all TE transcripts (M-step), until the estimated relative abundances converge. The initial estimation of relative abundance on multi-reads, ρ, of each TE transcript, t, is computed by Equation (1).
T is the set of all TE transcripts; Ft is the set of multi-reads assigned to t; l˜t denotes the effective length of transcript t, l˜t=lt−m+1, where m is the fragment length and lt is the length of transcript t. The fragment length is calculated from the paired-end alignment input file, or provided as a parameter by the user for single-end samples.
The E-step computes the fraction of each multi-read allocated to each TE transcript. Suppose that a multi-read f maps to a set of TE transcripts Ti. According to the initial assignment, the fraction of f attributed to any TE transcript t in Ti, α0(f,t), is the relative abundance of t over the sum of relative abundance of all TE transcripts in Ti, α0(f,t)=ρt0∑t′∈Tiρt′0. This allocation will then be used in the M-step of the algorithm to compute the relative abundance of each TE transcript. As described in Equations (2) and (3), these two steps will run alternatively for a specified number of iterations, k, until the program converges or as set by the user.
| αk(f,t)=ρt(k−1)∑t′∈Tiρt′(k−1) | (2) |
|---|
| ρtk=∑∀f→tαk(f,t)l˜t∑s∈T∑∀f′→sαk(f′,s)l˜s | (3) |
|---|
After the EM procedure, the estimated relative abundance of each TE transcript from the multi-reads is integrated with the unique-read counts to compute the total relative abundance. The element level abundances are then computed by summing up all instances of each TE subfamily.
3.4 Differential analysis
Following the generation of a count table for gene and TE transcripts, the differential expression analysis closely follows the DESeq package (Anders and Huber, 2010) for modeling the counts data with a negative binomial distribution and computing adjusted _P_-values. In addition to the standard transcript abundance normalization approach used by the DESeq package, TEtranscripts offers two additional options, reads per mapped million (RPM) and Quantile normalization. All other procedures exactly follow the DESeq method. TEtranscripts runs the DESeq method with a default set of general parameters. When there are no (or very few) replicates, we use the blind method for variance estimation and fit-only for SharingMode. Otherwise, we use pooled or per-condition methods and maximum SharingMode, as suggested by the DESeq package. In all scenarios, we use the parametric fitting model (fitType). The R code used for differential expression analysis is generated as part of the output to allow users to further customize the DESeq parameters and re-calculate differential expression statistics.
3.5 Implementation
TEtranscripts is written in Python. The SQUAREM (Varadhan and Roland, 2008) procedure is used during EM iterations to improve the convergence speed. TEtranscripts is available as an open-source program under a standard GPLv3 open source license and has been developed and tested on Linux and Macintosh OSX. The software package and associated TE GTF files can be found at http://hammelllab.labsites.cshl.edu/software. The TE GTF files currently include chimpanzee (panTro4), fly (dm3), maize (Zea mays RefGen v2), mouse (mm9 and mm10), rat (rn5 and rn6) and human (hg18, hg19/GRCh37, and hg38/GRCh38).
4 Results
To examine the accuracy and performance of TEtranscripts, we compared it with HTSeq-count version 0.5.4p3 (Anders et al., 2014), Cufflinks version v2.1.1 (Trapnell et al., 2010, 2012) and RepEnrich (Criscione et al., 2014) on both synthetic and real data. HTSeq-count was chosen as a standard method that counts only uniquely mapped reads and is nearly identical to the uniq mode in TEtranscripts. Cufflinks was chosen as a popular method for gene abundance estimation that works from pre-mapped BAM files and includes options to handle multi-reads, but is not specifically designed for TEs. To the best of our knowledge, only one other published method has been designed for TE expression analysis from RNA-seq data, RepEnrich, but this method does not work with pre-mapped BAM files. There is a recently published pipeline set piPipes (Han et al., 2015) to study piRNAs and TE-derived RNAs. Because it uses HTSeq-count and Cufflinks for quantification and Cuffdiff for differential analysis, we did not include comparisons to piPipes separately.
For all comparisons, TE abundance measurements were given at the resolution of the element level (e.g. L1Md_A). For the synthetic datasets, accuracy was quantified as the proportion of abundances accurately recovered by each method for each TE element. For published datasets, accuracy was determined by agreement between the quantitative validation measurements (e.g. Q-PCR, NanoString) and the expression estimations computed by each software package.
4.1 TE recovery in synthetic data
Simulated datasets were used to investigate the TE abundance recovery rate by each approach, paying particular attention to the recovery of TEs known to be active in the mouse genome, obtained from a study by Molaro et al . (2014). FluxSimulator v1.2.1 (Griebel et al., 2012) was used to generate multiple RNA-seq datasets from the mouse genome (mm9), consisting of 76 bp single-end reads from transcripts that include both annotated genes and TEs (see Supplementary Table S1 for parameters used). Each dataset consisted of 24 million reads in total, with 17% of all transcripts derived from TEs of varying abundances. STAR (Dobin et al., 2012) was used to map the simulated reads with maximum multiple alignments of no more than 100, using the variables –winAnchorMultimapNmax 100 and –outFilterMultimapNmax 100. Based on these parameters, we found that 87% of the reads were mapped onto the mouse genome, while TE reads had an average mappability of 70%. Furthermore, roughly half of the TE reads generated were uniquely mappable, comparable to what is observed in published transcriptome datasets. The simulated TE reads had a smaller mappability rate than gene-associated reads largely due to reads aligning to more than 100 genomic locations.
The aligned read files were then used as input for the four abundance estimation approaches used in this study: HTSeq-count, Cufflinks, RepEnrich, and TEtranscipts. HTSeq-count was run in intersection-nonempty mode, and using a GTF file that contains both TE and gene annotations for abundance estimation. Because HTSeq-count considers only uniquely mapped reads, any multi-mapped TE reads will be discarded by this approach. Cufflinks was run with the settings of rescue method for multi-reads (−u), which takes into account both unique-reads and multi-reads, and uniformly divide each multi-read to all the positions initially. When multi-read correction is enabled, Cufflinks will reassign each multi-mapped read probabilistically based on the initial abundance estimations, with the uniquely mapping reads used to inform the likely distribution of the multi-reads (Trapnell et al., 2010, 2012). To run RepEnrich, we built the peseudogenome of TEs using the RepeatMasker file on mm9 as described in the RepEnrich tutorial. Unique-reads and multi-reads were derived based on the STAR output. TEtranscripts was run in multi mode with EM optimization invoked. The accuracy of each method in estimating abundances of TE expression was computed and displayed as the frequencies of the rate of recovered abundances (Fig. 2; Supplementary Fig. S2).
Fig. 2.
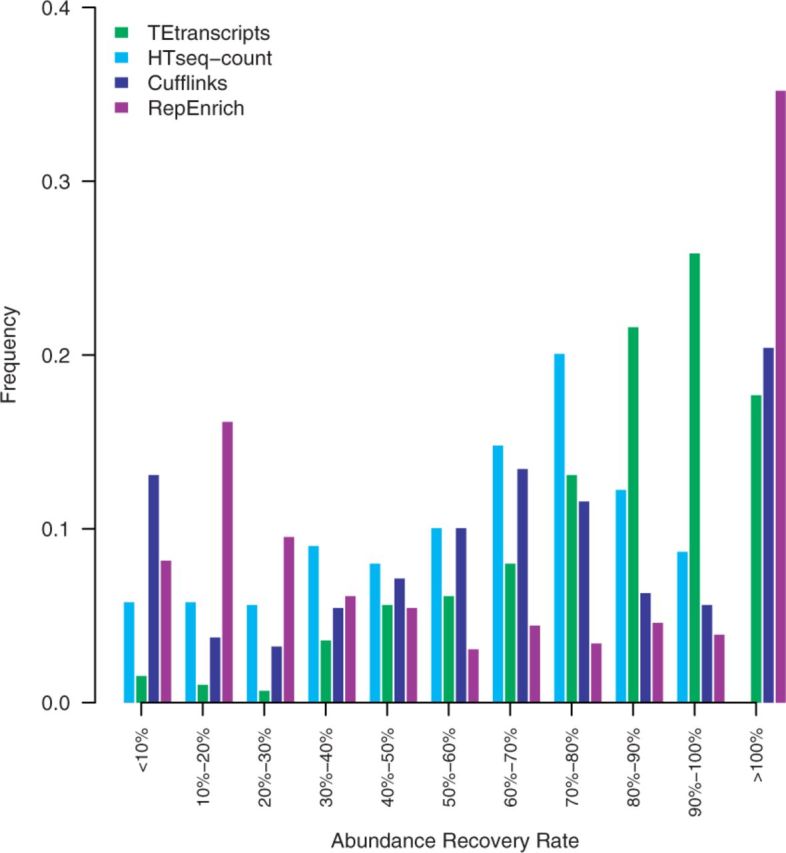
Comparison of TE abundance recovery. Displayed are the distributions of the rate of measured abundances versus true abundances by each method. HTSeq-count abundance recovery rates shown in light blue, Cufflinks recovery rates shown in blue, TEtranscripts with multi mode recovery rates shown in green, and RepEnrich recovery rates shown in purple
In general, TEtransctripts outperforms HTSeq-count, Cufflinks and RepEnrich in terms of abundance recovery rate, both for non-functional and active TEs. The overall average recovery rate for TEtranscripts was 88.84%, with 53.74% for HTSeq-count, 43.72% for Cufflinks and 59.8% for RepEnrich. The fraction of TEs for which the estimated abundance is within 15% of the true abundance was: 41.7% for TEtranscripts, 14.4% for HTSeq-count, 9.3% for RepEnrich and 16.8% for Cufflinks. In this dataset, 14.5% of the detected TEs are active TEs, and all three approaches were able to capture some of them. TEtranscripts was able to recover >80% of the reads for 77% of the active TEs; HTSeq-count was not able to recover >80% of the reads for any of the active elements; Cufflinks recovered at least 80% of the reads for 8% of the active TEs; RepEnrich recovered >80% of reads for 75% of the active TEs, but over-counted 57.69% of the active TEs. The fraction of active TEs for which the estimated abundance is within 15% of the true abundance was: 61.5% for TEtranscripts, 0% for HTSeq-count, 23.1% for RepEnrich, and 3.8% for Cufflinks. TEtranscripts and RepEnrich over-counted some TEs, which is displayed as the 15% of elements whose abundances were estimated to be >100% of their actual values at the far right of Figure 2. Please refer to Supplementary Figure S4 for recovery rates with and without the EM optimization option in TEtranscripts.
Figure 3 shows the abundances of active TEs estimated by each software package as compared with the ground truth (denoted with red dots). In most cases, TEtranscripts (green dots) is within 90% of the actual value, while the other two methods, HTSeq-count and Cufflinks, frequently under-estimate the abundance of TE transcripts for young, active TEs. RepEnrich (purple dots) shows more variations, nearly 30% under-estimation and 40% over-estimation. HTSeq-count (light blue dots) was expected to underestimate the counts, since the discarded multi-reads often constitute nearly 50% of the TE-associated reads. Surprisingly, Cufflinks also under-estimates the abundances for many active TEs, despite incorporating multi-reads in its analysis. Both under- and over-estimating the TE transcript abundances will affect both the ability to accurately calculate fold changes between samples and the power with which to calculate _P_-values for any associated changes. Please refer to Supplementary Figure S3 for the distribution of abundance recovery rates on active TEs of four approaches. In the next section, we assess the ability of these methods to return accurate fold changes and significant _P_-values for TEs known to have altered expression in published datasets that used a quantitative validation of their RNA-seq data.
Fig. 3.

Active TE expression estimation. Reads associated with active TE elements are more likely to be missed by algorithms that rely heavily on unique reads
4.2 TE recovery in published data
To determine the usefulness of our algorithm on experimentally generated results, we tested TEtranscripts on previously published RNA-seq datasets of Drosophila and mouse transcriptomes. We then compared TEtranscripts to other approaches, such as HTSeq-count, Cufflinks and RepEnrich, each combined with DESeq. We applied DESeq for differential expression analysis on the outputs of the three approaches to directly compare the effects of quantification on _P_-value estimation. Both HTSeq-count and RepEnrich output raw counts that can be input to DESeq without transformation. The output of Cufflinks has to be converted to raw counts before running DESeq. To convert Cufflinks output, we first calculated raw counts assigned to each isoform by multiplying the length over 1000 to the Fragments Per Kilobase of exon per Million reads mapped (FPKM) value of each isoform, and then summarized them on genes or transposable elements.
Although the Cufflinks package includes its own differential expression analysis software, Cuffdiff, it required at least 550 GB of memory to run on these datasets, and performed no better in terms of fold change estimation or _P_-value concordance than the results displayed below (Supplementary Fig. S5).
4.2.1 Drosophila melanogaster transcriptome
The Drosophila dataset comes from a study by Ohtani et al. (2013) that observed the de-repression of transposable elements upon alteration of DmGTSF1, which works with the Piwi-associated silencing complex (piRISC) to silence TEs in the Drosophila ovary. This dataset was chosen because they assessed TE expression levels with RNA-seq, followed by validation through Q-PCR. This will allow us to compare the estimated fold changes from the four approaches, HTSeq-count, Cufflinks, RepEnrich, and TEtranscripts, with their Q-PCR results. We obtained the raw FASTQ data from Gene Expression Omnibus (accession no. GSE47006) and mapped with STAR, as described earlier, onto the D. melanogaster genome (dm3). Although TEtranscripts, HTSeq-count and Cufflinks were able to perform quantification directly from the BAM alignment output, RepEnrich requires independent alignment and TE quantification. We built a pseudogenome for dm3 TEs using the RepeatMasker open-4.0.5 release file (Smit et al., 1996) download from the following link: http://www.repeatmasker.org/species/dm.html.
Figure 4 shows the TE expression changes between Piwi knock-down and wild type. The log2 fold change (log2FC) calculated by TEtranscripts in multi mode closely resembles the Q-PCR results in most of the TEs interrogated. HTSeq-count performs well on most elements, but not as well as TEtranscripts on others (e.g. mdg1). Cufflinks reports similar values to HTSeq-count for most TEs, but performs better than TEtranscripts and HTSeq-count on the roo element. Surprisingly, RepEnrich deviates substantially from the qPCR validation results, and DESeq identified no differentially expressed TEs.
Fig. 4.

Comparing Drosophila TE expression estimation. TEtranscripts was compared with HTSeq-count, Cufflinks, and RepEnrich. Log2 fold changes of Piwi knock-down versus wild type are shown here. ‘NA’ denotes circumstances where expression could not be estimated. The asterisk symbol represents the level of significances, ‘***’ adjusted _P_-value <1e−5, ‘**’ adjusted _P_-value < 0.01, ‘*’ adjusted _P_-value < 0.05
4.2.2 Mouse transcriptome
In order to evaluate the utility of TEtranscripts on a mammalian genome with higher TE content, we selected a recently published study in mouse from Gnanakkan et al . (2013). In this study, they provided NanoString quantification of several TEs, comparing a previously published RNA-seq dataset (GEO accession number GSE30352) (Brawand et al., 2011), to their microarray-based tool, TE-array. We performed similar analyses as described in the previous section, except that we map the reads to the mouse genome (mm10) and mouse TE pseudogenome (for RepEnrich).
Figure 5 shows the comparison between TEtranscripts with multi mode and other quantification approaches. Similar to the results on Drosophila data, TEtranscripts outperforms HTSeq-count on many TEs (e.g. L1Mm, _ERBV_4, _MMVL_30 and _ERVB_2). Cufflinks (in multi-reads rescue mode) performs comparably with TEtranscripts on some TEs, but often fails to return abundance counts on others (as indicated by ‘NA’ in the plot). RepEnrich again deviates from the expected values (nanoString), and fails to identify any differentially expressed TEs from its quantification results.
Fig. 5.

Comparing mouse TE expression estimation. We selected TEs that show significant differential expression between testis and somatic tissues. The somatic tissue sample is the integration of RNA-seq data from multiple organs: liver, heart, brain and kidney. The same figure legend was used as Figure 4
4.2.3 TEtranscripts quantification in other published studies
Finally, the methodology utilized in TEtranscripts has also been applied in studying transposable elements mis-regulation in TDP-43-mediated neurodenerative disorders (Li et al., 2013) and the roles of Piwi in the piRNA pathway and transposon repression (Rozhkov et al., 2013). In both studies, we were able to demonstrate significant differential expression of TEs that was consistent with the biological phenotypes and with the set of TEs altered in independent experiments for those studies.
4.3 Running time and memory usage
We tested the running time and memory usage of TEtranscripts on simulated RNA-seq data. A variety of library sizes ranging from 20 to 100 M reads were generated based on the mouse genome (mm9), with each sample having 10% of the reads coming from TEs. Although TEtranscripts takes additional time and memory to distribute reads between the millions of TE instances in the genome as compared with other gene expression analysis packages, it is still relatively efficient, with a typical memory requirement of 8 GB and run times on the order of 1–2.5 h for datasets with 20–100 million reads per sample (Fig. 6). All the experiments were run on a server with 128 GB memory and Xeon E5-2665 processors running at 2.40 GHz (16 cores). The running time was measured using the built-in bash date command.
Fig. 6.

Running time and memory usage. RepEnrich has a pre-requisite preparation step of building the pseudogenome of transposable elements, which was not included in this plot
5 Discussion
Transcripts derived from TEs form a small but important subset of all transcriptomic datasets. Often thought of as junk transcripts with little importance for biological phenotypes, TEs can play a large and unexpected role in important processes such as stem cell identity and reprogramming (Kelley and Rinn, 2012; Lu et al., 2014; Ohnuki et al., 2014; Wang et al., 2014), and in human diseases (Bundo et al., 2014; Lamprecht et al., 2012; Li et al., 2013; Reilly et al., 2013; Sciamanna et al., 2013, 2014; Shukla et al., 2013; Tucker et al., 2011). Although TE-derived transcripts should be included as part of standard expression analyses, there have previously been few tools that allow the easy inclusion of TE-associated reads. TEtranscripts allows users to simultaneously analyze gene- and TE-derived transcripts in a simple expression analysis framework that works with aligned (BAM) files and annotation files (GTF).
Using simulated reads as well as published datasets that include independent validations, we have shown that TEtranscripts outperforms all other published methods in abundance estimation, and concordance between statistical significance estimation and validated alterations in expression. In simulated datasets, we show that TEtranscripts performs particularly well at estimating the abundance of young TEs, which are more likely to be mobile and active in cells. In published datasets for both fly and mouse genomes, we show that alterations in TE expression estimated from RNA-seq data by TEtranscripts show better overall concordance with external validation data. TEtranscripts particularly outperforms other methods for complex mammalian genomes, such as the mouse, which has many more insertions per TE than flies, and a larger diversity in TE families.
As with all approaches that quantify RNA expression from alignment data, TEtranscripts is highly dependent on the quality of the genomic alignment and annotation data (for genes and transposable elements). This is especially problematic when working with strains or cultivars whose DNA sequences and transposable element content have diverged significantly from the ‘reference’ genome and annotations. TEtranscripts mitigates this limitation by providing flexibility in the input files provided by the user. Our software is agnostic to the genomic aligner and mapping parameters used to generate the input alignment files, as long as it complies with the SAM/BAM format. This enables users to optimize genome alignment parameters according to the characteristics of their experimental system before analysis with TEtranscripts. TEtranscripts can also utilize user-defined annotations for both gene and transposable elements during quantification. Although we have provided transposable element annotation files for a few common genomes, our software will process any TE annotations in the GTF format described earlier. These could include TE annotations that have been manually curated for a specific strain, or those identified by bioinformatics tools searching for de-novo transposable elements. This will allow users to provide the best annotation data suitable for their experiment, and maximizes the quality of analysis produced by TEtranscripts.
Supplementary Material
Supplementary Data
Acknowledgement
We would like to thank Josh Dubnau and members of his lab for helpful discussions. We would also like to thank the Bioinformatics Shared Resource at Cold Spring Harbor Laboratory, which is supported by the National Institutes of Health.
Funding
This work was supported by the Rita Allen Foundation, of which MH is a scholar, and by a grant from the National Science Foundation [MCB-1159098> to MH]. This work was also supported by a CSHL Cancer Center Support Grant from the National Institutes of Health (NIH-NCI-CA-045508).
Conflict of Interest: none declared.
References
- Anders S., Huber W. (2010) Differential expression analysis for sequence count data. Genome Biol., 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S., et al. (2014) Htseq—a python framework to work with high-throughput sequencing data. Bioinformatics, 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck C.R., et al. (2010) LINE-1 retrotransposition activity in human genomes. Cell, 141, 1159–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett E.A., et al. (2008) Active alu retotransposons in the human genome. Genome Res., 18, 1875–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brawand D., et al. (2011) The evolution of gene expression levels in mammalian organs. Nature, 478, 343–348. [DOI] [PubMed] [Google Scholar]
- Bundo M., et al. (2014) Increased L1 retrotransposition in the neuronal genome in schizophrenia. Neuron, 81, 306–313. [DOI] [PubMed] [Google Scholar]
- Chung D., et al. (2011) Discovering transcription factor binding sites in highly repetitive regions of genomes with multi-read analysis of ChIP-Seq data. PLoS Comput. Biol., 7, e1002111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coufal N.G., et al. (2009) L1 retrotransposition in human neural progenitor cells. Nature, 460, 1127–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coufal N.G., et al. (2011) Ataxia telangiectasia mutated (ATM) modulates long interspersed element-1 (l1) retotransposition in human neural stem cells. Proc. Natl. Acad. Sci., 108, 20382–20387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscione S., et al. (2014) Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genomics, 15, 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day D.S., et al. (2010) Estimating enrichment of repetitive elements from high-throughput sequence data. Genome Biol., 11, R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cecco M., et al. (2013) Transposable elements become active and mobile in the genomes of aging mammalian somatic tissues. Aging, 5, 867–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., et al. (2012) STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadloun A., et al. (2013) Chromatin signatures and retrotransposon profiling in mouse embryos reveal regulation of LINE-1 by RNA. Nat. Struct. Mol. Biol., 20, 332–338. [DOI] [PubMed] [Google Scholar]
- Faulkner G.J., et al. (2009) The regulated retrotransposon transcriptome of mammalian cells. Nat. Genet., 41, 563–571. [DOI] [PubMed] [Google Scholar]
- Gnanakkan V.P., et al. (2013) TE-array-a high throughput tool to study transposon transcription. BMC Genomics, 14, 869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griebel T., et al. (2012) Modelling and simulating generic RNA-seq experiments with the flux simulator. Nucleic Acids Res., 40, 10073–10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B.W., et al. (2015) piPipes: a set of pipelines for piRNA and transposon analysis via small RNA-seq, RNA-seq, degradome- and CAGE-seq, ChIP-seq and genomic DNA sequencing. Bioinformatics, 31, 593–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancks D.C., Kazazian H.H., Jr. (2012) Active human retrotransposons: variation and disease. Curr. Opin. Genet. Dev., 22, 191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honma M.A., et al. (1993) High-frequency germinal transposition of DsALS in Arabidopsis. Proc. Natl. Acad. Sci., 90, 6242–6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.R., et al. (2012) Active transposition in genomes. Annu. Rev. Genet., 46, 651–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J., et al. (2005) Repbase update, a database of eukaryotic repetitive elements. Cytogent. Genome Res., 110, 462–467. [DOI] [PubMed] [Google Scholar]
- Kano H., et al. (2009) L1 retrotransposition occurs mainly in embryogenesis and creates somatic mosaicism. Genes Dev., 23, 1303–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D., et al. (2003) The UCSC genome browser database. Nucleic Acids Res., 31, 51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley D., Rinn J. (2012) Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol., 13, R107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamprecht B., et al. (2012) Derepression of an endogenous long terminal repeat activates the CSF1R proto-oncogene in human lymphoma. Nat. Med., 16, 571–579. [DOI] [PubMed] [Google Scholar]
- Langmead B., et al. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol., 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E., et al. (2012) Landscape of somatic retrotransposition in human cancers. Science, 337, 967–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., et al. (2012) Transposable elements in TDP-43-mediated neurodegenerative disorders. PLoS One, 7, e44099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., et al. (2013) Activation of transposable elements during aging and neuronal decline in drosophila. Nat. Neurosci., 16, 529–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X., et al. (2014) The retrovirus HERVH is a long noncoding RNA required for human embryonic stem cell identity. Nat. Struct. Mol. Biol., 21, 423–425. [DOI] [PubMed] [Google Scholar]
- Macia A., et al. (2011) Epigenetic control of retrotransposon expression in human embryonic stem cells. Mol. Cell Biol., 31, 300–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills R.E., et al. (2007) Which transposable elements are active in the human genome?. Trends Genet., 23, 183–191. [DOI] [PubMed] [Google Scholar]
- Molaro A., et al. (2014) Two waves of de novo metylation during mouse germ cell development. Genes Dev., 28, 1544–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muotri A.R., et al. (2005) Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature, 35, 903–910. [DOI] [PubMed] [Google Scholar]
- Nussbaumer T., et al. (2013) MIPS PlantsDB: a database framework for comparative plant genome research. Nucleic Acids Res., 41, 1144–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnuki M., et al. (2014) Dynamic regulation of human endogenous retroviruses mediates factor-induced reprogramming and differentiation potential. Proc. Natl. Acad. Sci., 111, 12426–12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani H., et al. (2013) DmGTSF1 is necessary for Piwi-piRISC-mediated transcriptional transposon silencing in the drosophila ovary. Genes Dev., 27, 1656–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peaston A.E., et al. (2004) Retrotransposons regulate host genes in mouse oocytes and preimplantation embryos. Dev. Cell, 7, 597–606. [DOI] [PubMed] [Google Scholar]
- Perrat P.N., et al. (2013) Transposition-driven genomic heterogeneity in the drosophila brain. Science, 340, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly M.T., et al. (2013) The role of transposable elements in health and diseases of the central nervous system. J. Neurosci., 33, 17577–17586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld J.A., et al. (2009) Investigating repetitively matching short sequencing reads: the enigmatic nature of H3K9me3. Epigenetics, 4, 476–486. [DOI] [PubMed] [Google Scholar]
- Rozhkov N., et al. (2013) Multiple roles for Piwi in silencing drosophila transposons. Genes Dev., 27, 400–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciamanna I., et al. (2013) A tumor-promoting mechanism mediated by retrotransposon-encoded reverse transcriptase is active in human transformed cell lines. Oncotarget, 4, 2271–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciamanna I., et al. (2014) Regulatory roles of LINE-1-encoded reverse transcriptase in cancer onset and progression. Oncotarget, 5, 8039–8051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedivy J.M., et al. (2013) Death by transposition—the enemy within? Bioessays, 35, 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla R., et al. (2013) Endogenous retrotransposition activates oncogenic pathways in hepatocellular carcinoma. Cell, 153, 101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit A., et al. (1996–2010) Repeatmasker Open-3.0, <http://www. repeatmasker.org>. [Google Scholar]
- Thomas C.A., et al. (2012) LINE-1 retotransposition in the nervous system. Annu. Rev. Cell Dev. Biol., 28, 555–573. [DOI] [PubMed] [Google Scholar]
- Trapnell C., et al. (2010) Transcript assembly and quantification by RNASeq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotech., 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., et al. (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat. Protocols, 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treangen T.J., Salzberg S.L. (2011) Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet., 13, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubio J.M., et al. (2014) Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science, 345, 1251343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker B.A., et al. (2011) Exome sequencing and analysis of induced pluripotent stem cells identify the cilia-related gene male germ cell-associated kinase (MAK) as a cause of retinitis pigmentosa. Proc. Natl. Acad. Sci., 108, E569–E576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadhan R., Roland C. (2008) Simple and globally convergent methods for accelerating the convergence of any EM algorithm. Scand. J. Stat., 35, 335–353. [Google Scholar]
- Wang J., et al. (2010) A Gibbs sampling strategy applied to the mapping of ambiguous short-sequence tags. Bioinformatics, 26, 2501–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., et al. (2014) Primate-specific endogenous retrovirus-driven transcription defines naive-like stem cells. Nature, 516, 405–409. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data