Cleaning out the Litterbox of Proteomic Scientists’ Favorite Pet: Optimized Data Analysis Avoiding Trypsin Artifacts (original) (raw)
Abstract
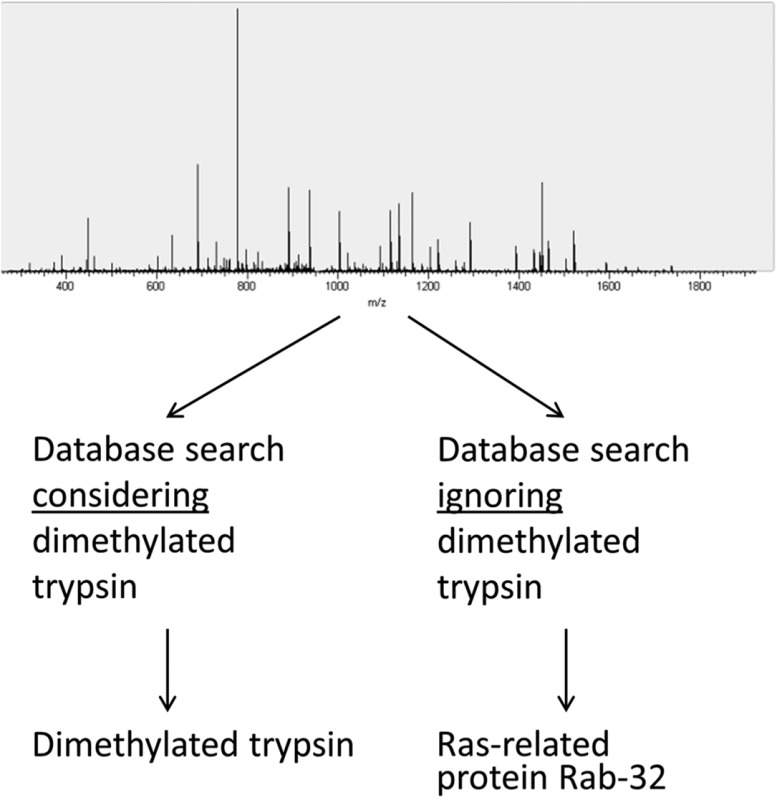
Chemically modified trypsin is a standard reagent in proteomics experiments but is usually not considered in database searches. Modification of trypsin is supposed to protect the protease against autolysis and the resulting loss of activity. Here, we show that modified trypsin is still subject to self-digestion, and, as a result, modified trypsin-derived peptides are present in standard digests. We depict that these peptides commonly lead to false-positive assignments even if native trypsin is considered in the database. Moreover, we present an easily implementable method to include modified trypsin in the database search with a minimal increase in search time and search space while efficiently avoiding these false-positive hits.
Keywords: proteomics, autolysis protected trypsin, database search, search space restriction, misassigned spectra, false positives
Introduction
Many proteomics experiments are performed on the peptide level (namely, bottom-up proteomics). For this purpose, the proteome is enzymatically digested, and the peptide and peptide fragment masses are measured by tandem mass spectrometry after online separation by reverse-phase liquid chromatography (LC–MS/MS). For the analysis of LC–MS/MS data, peptides are statistically scored by search engines such as Mascot,1 SEQUEST,2 OMSSA,3 MaxQuant,4 X!Tandem,5 MS-GF+,6 MS Amanda,7 MyriMatch,8 or COMET.9 The detected peptides are subsequently used to infer source proteins.10 All of these tools compare input peptide MS/MS spectra to theoretical spectra generated by the in silico digest of protein databases. The peptides can, dependent on the settings of the search engine, contain missed cleavage sites and variable or fixed amino acid modifications. All of the in silico generated spectra constitute the so-called search space (i.e., spectra of all peptides and variants thereof that are considered a potential match to the input spectrum).11 The search engine settings and database used are crucial for the quality of the results obtained because discrepancies between biological sample content and peptides present in the search space can lead to several undesirable effects. For the statistical evaluation of the results, in most cases, false discovery rates (FDR) are calculated. FDR is defined as the ratio of false positives to the total number of assignments (false positives + true positives) and is often estimated with the help of decoy databases (i.e., randomized or reversed databases).12
Usually, most of the proteins not contained in the target database are sample contaminants from different species, either introduced by accident or as a reagent during sample preparation. The former can be reduced by careful sample handling, and among the best known examples is human keratin. The latter type of common contaminants in proteomics experiments includes proteases, which are added to the sample to digest proteins to the more readily analyzed peptides.
The most widely used protease is trypsin due to its specificity, efficiency, availability, and relatively low cost. Trypsin cleaves the amino acid sequences’ C-terminal to lysine and arginine residues (with the exception of the occurrence of N-terminal proline) and gives rise to peptides of suitable length and charge for LC–MS/MS analysis.13 However, trypsin is also subject to autolysis because it contains 11 lysine and 4 arginine residues.14 Because trypsin is used in high quantities to promote efficient digestion (trypsin to sample protein ratio: 1:20–1:100), trypsin-derived peptides are highly abundant and result in high-quality spectra during MS/MS. In the case where the trypsin sequence is not present in the database, these spectra might be incorrectly assigned to peptides of other proteins. It is therefore widely accepted in the community to concatenate a contaminant list that includes trypsin with sample specific databases for the reasons given above. The groups of Ron Beavis (http://www.thegpm.org/cRAP/) and Matthias Mann (http://www.maxquant.org/downloads.htm) make their list of contaminants publicly available. Although the database should cover all possible components of a sample, care should be taken not to choose a too-large database (e.g., the entire UniProt TrEMBL database) because large databases not only increase the search time but also, more importantly, increase the number of random peptide hits.15
For the prevention of the autolysis of trypsin and the associated loss of activity,16 the protease is often modified by acetylation (New England Biolabs) or reductive methylation (Promega, Sigma-Aldrich) of lysines, the latter being the most common approach. Both modifications yield a trypsin that is more resistant to autolysis while retaining its catalytic function.17
Here, we show that the most widely employed trypsin preparations are chemically modified and that including native trypsin in the contaminant database is not sufficient to prevent false-positive identifications of trypsin-derived spectra. Instead, it is necessary to add modified trypsin to the list of contaminants. We present a new database search strategy with a minimal increase in search space and the concomitant loss of statistical significance. Finally, this approach can be easily implemented into existing workflows for many widely used search engines.
Experimental Section
Databases
Swiss-Prot_Yeast database was downloaded on June 10, 2015. Swiss-Prot_Human database was downloaded on August 11, 2015. The list of common Repository of Adventitious Proteins (cRAP) was downloaded from ftp://ftp.thegpm.org/fasta/cRAP (Version: January 30, 2015). The protein identifiers were then uploaded to UniProt Retrieve/ID mapping tool to generate a FASTA file in UniProtKB format (February 2, 2015).
Databases for Separate Decoy Searches
Databases from search strategies A–E were reversed using the CompOmics dbToolkit 4.2.3.18
Search Strategy A
Swiss-Prot_Human or Swiss-Prot_Yeast was used as the database; carbamidomethylation of cysteines was set as a fixed modification. Oxidation of methionine and protein N-terminal acetylation were set as variable modifications.
Search Strategy B
Swiss-Prot_Human or Swiss-Prot_Yeast with concatenated cRAP was used as the database. Carbamidomethylation of cysteines was set as a fixed modification. Oxidation of methionine and protein N-terminal acetylation were set as variable modifications.
Search Strategy C
Swiss-Prot_Human or Swiss-Prot_Yeast with concatenated cRAP was used as the database. Carbamidomethylation of cysteines was set as a fixed modification. Oxidation of methionine and protein N-terminal acetylation were set as variable modifications. For data set 2, acetylation of lysine was allowed as an additional variable modification; for all other data sets, dimethylation of lysine was allowed as an additional variable modification.
Search Strategy D
Swiss-Prot_Human or Swiss-Prot_Yeast with concatenated cRAP was used as the database. Carbamidomethylation of cysteines was set as a fixed modification. Oxidation of methionine and protein N-terminal acetylation were set as variable modifications. Monomethylation and dimethylation of lysine were allowed as an additional variable modification.
Search Strategy E
Swiss-Prot_Human or Swiss-Prot_Yeast with concatenated cRAP was used as the database. Furthermore, modified trypsin was added to the database (where all lysines (K) were replaced by dimethylated lysines (J) to detect dimethylated lysines in trypsin). Carbamidomethylation of cysteines was set as a fixed modification. Oxidation of methionine, protein N-terminal acetylation, loss of one methylation of dimethylated lysines (J) (to detect monomethylated lysines in trypsin), and loss of dimethylation of dimethylated lysines (J) (to detect unmethylated lysines in trypsin) were set as variable modifications.
Data Sets
Data set 1: Saccharomyces cerevisiae; PXD002726
Data set 2: Phosphorylation sites of Numb (human cell line) with mouse recombinant protein Numb; PXD00099719
Data set 3: Human Breast Cancer Study; PXD00024620
Data set 4: Identified spectra from 209 public human data sets from the PRIDE repository (see Supporting Table S4)
Data set 5: Human Brain 6–11 reference map; PRD00015121
Data Analysis
Database search was performed using Mascot Ver. 2.4.1 or MS Amanda Standalone_Windows_1.0.0.3948 customized to contain dimethylated lysine J = 156.125710 (kindly provided by Viktoria Dorfer). Data were searched against human and S. cerevisiae Swiss-Prot database as specified in the database section. For FDR estimation, a target–decoy database search was used. Peptides were matched using trypsin as a digestion enzyme. Peptides’ mass tolerance was set to 10 ppm (or 100 ppm for data set 5) and fragment mass tolerance to 0.8 Da. A maximum of two missed cleavages was allowed. Carbamidomethylation of cysteine was set as a fixed modification, and oxidation of methionine was set as a variable modification. To detect peptides containing peptide c-terminal artificial amino acid J, we changed the cleavage specificity for trypsin to J, K, and R, not after P. Furthermore, to detect mixed (i.e., peptides including methylated and unmodified lysine) and monomethylated peptides, we added the loss of monomethylation (14.0153759 Da) and loss of dimethylation (28.0307517 Da) of the artificial amino acid (J) as variable modifications.
Data set 4 was searched using X!Tandem5 (version: “Piledriver” (2015.04.01.1)). Because X!Tandem restricts the amino acids that may be redefined, “O” was used to represent dimethylated lysine. The mass of O was set to 156.12571 using the “protein, modified residue mass file” option. Carbamidomethylation of cysteine was set as a fixed modification, and oxidation of methionine, N-terminal acetylation and a loss of 28.0307517 Da on ‘O’ representing loss of dimethylation and a loss of 14.0153759 Da on ‘O’ representing monomethylated lysines were set as variable modifications. Parent-mass error was set to 2 Da, and fragment mass error was set to 0.4 Da. A total of two missed cleavages were allowed, the specificity of trypsin was adapted as described before, and refinement mode was disabled. All results were filtered to reach an FDR of 1%.
Statistical Analysis
Statistical analysis was done in R using the Kruskal–Wallis test and Dunn posthoc test if not stated otherwise.
Calculation of FDR for Histograms
FDR for separate target–decoy searches (data set 3) was calculated for Mascot ion scores as FDR being equal to the number of decoy hits divided by the number of target hits, with score increments of 0.01 until FDR was smaller than 1%.
Experimental Details: Data Set 1
Proteins from S. cerevisiae lysates were separated with SDS-PAGE. Proteins were stained using Coomassie Brilliant Blue. Gel bands were cut out and destained using 50% acetonitrile in 100 mM ammonium bicarbonate. After dehydration with 100% acetonitrile, gel pieces were dried and then reduced with 10 mM dithiothreitol for 30 min at 56 °C and subsequently alkylated with 55 mM iodoacetamide for 20 min at 37 °C in the dark. Gel pieces were dehydrated and dried again and then incubated with 0.19 μg of sequencing-grade modified trypsin (Promega, V5111) overnight. Peptides were extracted in four sequential steps: 15 μL of 25 mM ammonium bicarbonate, 150 μL of acetonitrile, 40 μL of 5% formic acid, and 150 μL of acetonitrile. Supernatants were combined and dried under vacuum. The dried peptide extracts were resuspended in 5% acetonitrile and 0.1% trifluoroacetic acid, and peptides were separated on an Acclaim PepMap RSLC C18, 2 μm, 100 Å, 75 μm i.d. × 50 cm column employing a Dionex Ultimate 3000 RSLC coupled to a LTQ-Orbitrap Velos (Thermo Fisher Scientific). The LC method consisted of a 70 min gradient. Data-dependent Top 10 CID MS and MS/MS data were acquired in the Orbitrap analyzer at a resolution of 60 000 and in the ion trap employing normal scan rate settings, respectively. The software version of XCalibur (Thermo Fisher Scientific) was 3.0. The mass spectrometry proteomics data was deposited to the ProteomeXchange Consortium22 (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the data set identifier PXD002726 and 10.6019/PXD002726.
Results
Incompleteness of Protease Modification and Inhibition of Autoproteolytic Activity
Despite the above-mentioned chemical modifications, peptides originating from trypsin are among the most commonly high-scoring identified peptides in bottom up searches. This can be seen when looking at the PRIDE Cluster resource,23 where many of the largest clusters containing several thousand spectra represent peptides from trypsin (e.g., “LGEHNIDVLEGNEQFINAAK” identified 21722 times). These unmodified peptides are the fraction of peptides that were not modified during reductive dimethylation, which could be due to the native state of trypsin during the modification. Assuming that the majority of accessible lysines are methylated but still many cleavage events occur, it is reasonable to expect peptides containing methylated lysines upstream of the unmodified lysine at the cleavage site.
We therefore reanalyzed several of our own data sets as well as publicly deposited data sets to assess the presence of methylated peptides and the impact of different search strategies. Search strategies and data sets are described in detail in the Experimental section. The first data set we examined was a set of in gel digests from S. cerevisiae prepared in our laboratory (data set 1, PXD002726), which was analyzed with different search strategies (Table 1). Initially, we searched only against the Swiss-Prot_Yeast database without accounting for potential contaminants (search strategy A). Second, we included the list of common contaminants (search strategy B). And third, to find dimethylated trypsin, we allowed dimethylation of lysine as a variable modification (search strategy C). Search C identified porcine trypsin with a higher score than search B because more peptides could be identified. We also identified numerous peptides with dimethylated lysines at their C-termini, challenging the assumption of complete inhibition of tryptic activity at the C-terminal of dimethylated lysines (Figure 1). Moreover, we performed a search with the additional variable modification monomethylation of lysine (search strategy D) to check for incomplete conversion during chemical stabilization. Indeed, peptides harboring monomethylated lysines were identified, being a fraction of roughly 30% of total methylated lysines (Supporting Table S1). When analyzing a trypsin self-digest, we could identify the same peptides, confirming our assumption that the methylated peptides indeed originated from trypsin (Supporting Table S2). Using a similar approach, we also found acetylated peptides from trypsin in a human epithelial cell data set19 (data set 2, PXD000997) (Supporting Table S3).
Table 1. Protein Top Identifications’ Dependence on Database and Search Settingsa.
| | search strategy | | | | | | | | | | ------------------ | ------------ | ------- | ------------- | -------- | ------------- | -------- | ------------- | -------- | | hit no. | A | B | C | D | | | | | | | protein | score | protein | score | protein | score | protein | score | | | 1 | HSP72_YEAST | 6551 | K2C1_HUMAN | 7212 | TRYP_PIG | 6794 | TRYP_PIG | 7377 | | 2 | HSP77_YEAST | 2283 | HSP72_YEAST | 6460 | K2C1_HUMAN | 6736 | K2C1_HUMAN | 6789 | | 3 | HS104_YEAST | 1731 | TRYP_PIG | 5878 | HSP72_YEAST | 5909 | HSP72_YEAST | 5830 | | 4 | TRP_YEAST | 1575 | K1C10_HUMAN | 3779 | K1C10_HUMAN | 3473 | K1C10_HUMAN | 3443 | | 5 | HSP76_YEAST | 1439 | HSP77_YEAST | 2261 | HSP77_YEAST | 2032 | HSP77_YEAST | 2000 | | 6 | VATA_YEAST | 1049 | HS104_YEAST | 1709 | HS104_YEAST | 1498 | HS104_YEAST | 1466 |
Figure 1.
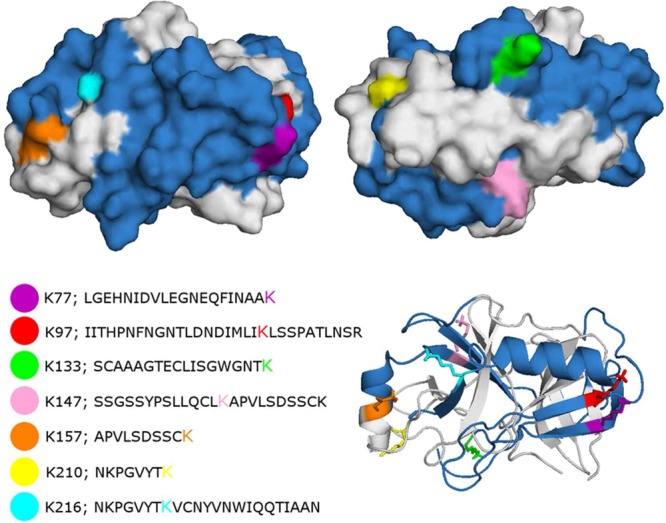
Methylated lysine residues identified on autolysis peptides of reductively methylated porcine trypsin. Blue: sequence coverage of peptides identified in data set 1 by search C. Peptides are listed in Supporting Table S1. Figures were rendered with PyMOL 0.99rc6 based on PDB entry 1EP. Gray: not identified peptides. Other colors: methylated lysine.
New Search Strategy Employing Artificial Amino Acids
In searches C and D (by allowing variable modifications of lysine), we identified both methylated lysines and unmodified lysines in trypsin-derived peptides. Although the strategy led to an increased score of porcine trypsin (Table 1), this approach has a severe drawback. Every addition of a global variable modification increases the search space dramatically. This combinatorial expansion of search space has various unwanted effects. The most severe is the impact on result statistics because more random matches will occur when all possible permutations are tested. Enlargement of the database leads to a rise of false-positive hits, increasing both the number of false positives but also false negatives when applying the same FDR threshold.15 However, ignoring the modified peptides can have the detrimental effect of leading to the identification of false positives.
To circumvent this, we devised a method that considers the chemically modified tryptic autolysis peptides without unnecessarily inflating search space. We envisaged introducing dimethylated lysine as an artificial amino acid into the database to be able to limit its occurrence to the actually modified protein in the database. Mascot allows the easy introduction of artificial amino acids via the unimod.xml file and the two capital letters J and O for user-defined amino acids. We therefore introduced the artificial amino acid J, having a total monoisotopic mass of 156.125710 Da (J = K + dimethyl (+ C2H4, + 28.0313)). For a copy of the porcine trypsin entry, we then replaced all lysines (K) with dimethylated lysines (J) (fictional accession number 012345, TRY_ART) and added it to the database including the contaminants. In addition, the cleavage specificity of trypsin was set to the C-terminal of K, R, and J (except when followed by P). Using the database including artificial trypsin and the search settings explained above, we successfully identified peptides of trypsin containing dimethylated and unmodified lysines in data set 1. To identify mixed peptides, including both dimethylated and unmodified lysines within one peptide, we chose a rather counterintuitive approach: we included the loss of dimethylation of dimethylated lysines as a variable modification. This approach limits the increase of search space to the smallest degree necessary (i.e., only peptides that contain J are multiplied while still covering all possible combinations of dimethylated and unmethylated lysines). The same approach can also be used to identify monomethylated lysines by allowing the loss of one methyl group for J (because J minus methyl is equal to K plus methyl). In contrast to the addition of two global variable modifications, we could not observe any impact on search specificity with this strategy (search strategy E), as is detailed in the next section.
Detrimental Effects of Increased Search Space
We next assessed the influence of the different search parameters on peptide spectrum matches (PSMs), peptide scores, and protein scores using five raw files from a human breast-cancer data set20 (data set 3, PXD000246). The FDR was kept constant at 1% for all searches. Search strategy A identified a lower number of peptides because it was unable to assign spectra to contaminants. Search strategy B, representing the community standard to employ a contaminant database without considering modifications of trypsin, yielded a higher number of total PSMs as well as more PSMs after applying a 1% FDR cutoff. To visualize the impact of the increased search space in search strategies C and D, we depict histograms of Mascot ion scores of a representative raw file (Site1_Cell_line_ERneg_HER2neg_Pool1_1D_Rep1.raw) from data set 3 in Figure 2. It is clearly visible that both the decoy and the target populations contain more hits when employing search strategies C and D. Less obviously, the mean of the log(2) decoy scores is shifted to slightly higher values (1.47 (A, B, and E) versus 1.64 (C)). These effects are further amplified when adding another variable modification (search strategy D; Figure 2; mean 1.95). Because of the bimodal distribution of the target populations even after log(2) transformations, the Kruskal–Wallis test was employed for statistical testing and showed highly significant changes (p < 10–9) in both score distributions for search strategies C and D. Calculating the 1% FDR score threshold for the target–decoy pairs, search strategies A, B, and E required an ion score of 20.50 to pass the cutoff, while this threshold was increased to 20.94 in search strategy C and 22.21 in search strategy D. Although search strategies C and D lead to the highest number of total PSMs, the number of PSMs passing the 1% FDR threshold decreases significantly (see Figure 2, Supporting Figure S1). Naturally, this also reduces the average protein score (average protein scores were 262.0, 262.3, 256,6, 252,5 and 262.7 for searches A, B, C, D, and E, respectively, with only the scores from search C and D being significantly lower than the average scores from all other searches) (Figure S1). In summary, the use of global variable modifications leads to more total PSMs but at the expense of specificity, as can be seen after applying 1% FDR.
Figure 2.

Log(2) score distribution for separate decoy and target database searches of data set 3 using different search strategies. Left panel: decoy database searches; right panel: target database searches. A: Search strategy A, Swiss-Prot_Human; B: search strategy B, Swiss-Prot_Human plus list of common contaminants; C: search strategy C, Swiss-Prot_Human plus common contaminants and additional variable modification dimethyl (K); D: search strategy D, Swiss-Prot_Human plus common contaminants and additional variable modifications methyl (K) and dimethyl (K). E: search strategy E, Swiss-Prot_Human plus common contaminants and methylated lysines of trypsin considered as artificial amino acids. Marked in black are peptides that pass the FDR 1% cutoff (Mascot ion score of 20.50 for searches A, B, and E, 20.94 for search C, and 22.21 for search D). *** indicates _p_-value of <10–9, Kruskal–Wallis test; # psm indicates number of peptide spectrum matches.
The use of the artificial amino acid based search strategy E did not yield a statistically significant improvement over search strategy B in either total PSMs or PSMs after applying 1% FDR. However, individual spectra were incorrectly assigned to unrelated proteins by search strategy B rather than modified trypsin, as revealed by search strategy E. This is depicted in Figure 3, showing which fractions of false positives of search strategy A can be explained by the common contaminant approach (i.e., search strategy B (23%) and the artificial amino acid search strategy E (additional 7%, total 30%)). To rule out search-engine-specific effects, we processed the same data set with a customized version of MS Amanda,7 a scoring algorithm especially suited for data sets with high-resolution MS/MS spectra. Employing search strategy B, MS Amanda yielded the same misassignments as Mascot. However, with both search engines, search strategy E resulted in much higher probabilities for the dimethylated trypsin derived peptide to be the correct identification, as shown for one example spectrum in Figure 4.
Figure 3.

Fractions of false positives explained by different search strategies (data set 3). Search strategy B reveals that 23% of all false positives from search strategy A are caused by common-contaminant-derived peptides (including unmodified trypsin). Search strategy E identifies an additional 7% in this data set (overall range 0–66% and mean 6.6% in all human PRIDE data sets (Supporting Table S4)), which are exclusively caused by methylated trypsin peptides.
Figure 4.

Individual spectrum misassigned by two separate search engines despite including contaminants in the database (data set 3). * indicates that the accession number for dimethylated trypsin can be set at discretion, avoiding occupied accession numbers.
False-Positive Assignments Despite Considering Common Contaminants
Depicted in Figure 4 is an exemplary case, which may easily lead to identifications passing the 1% FDR threshold and subsequently increases the chance of wrong protein inference using search strategy B. We want to emphasize that search B was done using a database of appropriate size (Swiss-Prot_Human) and included common contaminants. This widely used approach results in statistically valid but still incorrect assignments, which were revealed to actually represent dimethylated trypsin derived peptides with a very high probability, employing search strategy E. To assess the prevalence of these incorrectly assigned spectra, we reprocessed the identified spectra from 209 public human data sets (data set 4) stored in the PRIDE database24 (see Supporting Table S4). The extracted identified spectra were identified using X!Tandem and the new artificial amino acid based search strategy (search strategy E). We were able to identify methylated trypsin peptides in 38 out of these 209 experiments. For the 171 experiments in which we could not identify methylated trypsin, it is unclear whether unmodified trypsin was used. It has to be noted that most publications only specify the trypsin vendor, and most vendors offer different types of trypsin preparations.
The number of spectra identified as dimethylated trypsin peptides ranged from 1 to 600 per project (see Supporting Table S4). We want to point out that these spectra constitute a substantial amount of all false-positive peptides in community gold standard search strategy B. At an FDR of 1%, the spectra reassigned by search strategy E to methylated peptides originating from trypsin correspond to up to 66% of all false positive hits (mean of projects containing methylated trypsin 6.6%). The distribution of unmodified to monomethylated to dimethylated trypsin derived peptides was on average 25:35:40%. It is also important to highlight that we only reanalyzed the submitted, identified spectra because only these may have led to incorrect identifications. Because of the way data is submitted to PRIDE, it is often not possible to know whether all identifications were submitted or only the ones passing the FDR threshold applied by the authors. Nevertheless, methylated trypsin peptides passing 1% FDR of the cluster search do occur in a considerable number of public data sets and thus lead to incorrect identifications. In total, 3960 modified trypsin peptides were identified from wrongly assigned spectra (listed in Table S4).
Pseudoisobaric Amino Acids Arginine and Dimethyl Lysine
One interesting example was found in data set 5 from human brain tissue21 (PRD000151). When we first reanalyzed this sample set with search strategy B, we were unable to reproduce the original assignment of a spectrum to human trypsin isoform 2. Comparing the search settings, we found that the original publication used a higher parent-mass tolerance, namely 100 ppm for Orbitrap data. With the same parent-mass tolerance, we were also able to identify the peptide from human trypsin isoform 2, with a δ parent mass of approximately 25 mmu. Search E yielded a peptide from dimethylated porcine trypsin and from human trypsin isoform 2 with almost identical scores for both Mascot and MS Amanda searches (see Figure 5). Comparing the sequences of the two peptides, we found that the human isoform contains an arginine instead of the lysine at the same position in the porcine trypsin. Interestingly, arginine and dimethylated lysine have a mass difference of 24.6 mmu, almost perfectly fitting the mass difference of expected to measured mass for the human isoform 2 peptide. Because of the expected instrument accuracy and the biological origin of the sample (brain), we are inclined to propose the dimethylated peptide (with mass difference 1 ppm) as the correct match. A precursor mass δ of 25 mmu as for the initial match might exclude a peptide from the list of potential candidates in search strategies in which stringent precursor mass filtering is applied before peptide matching. However, when using wide parent-mass windows for the initial search, as suggested recently by Bonzon-Kulichenko et al. for database-dependent scoring functions (e.g., Mascot expect value),25 other matches can actually outscore and outrank the correct match (as shown in Figure 5). It therefore has to be ensured to include all potential matches irrespective of score and rank until applying post-filtering according to precursor mass when using this approach.
Figure 5.
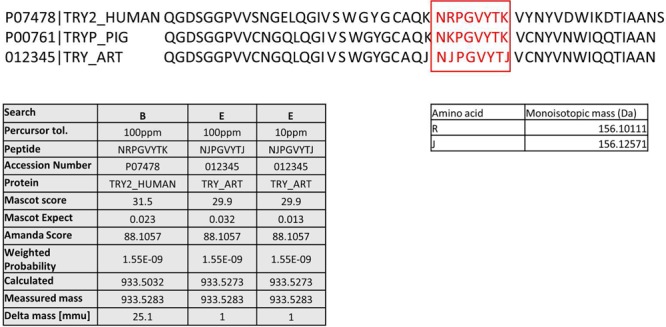
Peptide originating from dimethylated trypsin assigned to human trypsin isoform 2 (data set 5). Even though the score for dimethylated trypsin is lower in the Mascot search, the mass difference and the biological origin of the sample implicate that the dimethylated trypsin is the correct assignment. Analysis with MS Amanda showed identical scores and probabilities for both dimethylated porcine trypsin and human trypsin isoform 2.
Discussion
Methylation of trypsin is employed to protect against autolysis and concomitant enzymatic activity loss.17 As expected, our results show that the enzyme is not completely modified because the modification of the natively folded protease can only take place at lysine residues accessible to the reagent. More interestingly, we also show that methylation does not completely protect against autolysis. In many of the analyzed samples, we identified peptides of trypsin that were actually methylated directly at the cleavage site.
The use of appropriate databases in proteomics is crucial for identifying correct peptides and avoiding false-positive peptide hits that might result in inferring wrong proteins.26 Even if common contaminants are included in the database, methylated trypsin derived peptides are not considered in the search space, and the presence of these modified peptides leads to false-positive peptide identifications. Unfortunately, the most intuitive solution, to consider methylated lysines in the search (i.e., adding variable modifications), has been reported to result in reduced specificity of a search.27 While this approach is able to efficiently reduce the number of unassigned spectra, it also increases the peptide score threshold at a constant FDR because of the considerably larger search space, as demonstrated in this study. Peptides filtered out due to the now more stringent score cutoff also cannot add to protein scores, which results in a lower average protein score (Supporting Figure S1).
The same effect can be observed whenever considering variable modifications, which might occur naturally or during sample preparation (e.g., phosphorylations, carbamidomethylation artifacts, and nontryptic cleavages). Therefore, one has to carefully balance the beneficial effect of additionally assigned spectra to the detrimental effects of increased search space on a sample specific basis. This is depicted in Figure 2, where either two or three global variable modifications plus one variable protein N-terminal modification are used. Additional variable modifications lead to a further combinatorial expansion of search space, reducing search specificity and resulting in a net loss of statistically valid peptide hits. The same is true to an even bigger extent when allowing for semi- or nonspecific enzymatic cleavages (Supporting Figure S1).
Thus, we suggest including the modification of the employed digestion enzymes into the contaminant database as an artificial amino acid. This approach does not have significant drawbacks regarding the search space, as demonstrated in this study, and can be used alongside other sample-specific search parameters without a loss of search specificity. Several search engines such as Mascot, X!Tandem, and COMET (see Box 1) allow for the easy introduction of non-natural amino acid, but for some, all amino acid letters and masses are hard-coded. Examples for the latter are the (discontinued) OMSSA engine, Andromeda (MaxQuant), Sequest, and MS Amanda. Sequest, for example, does in our hands not accept additional amino acids defined in the unimod.xml, and MaxQuant in its current version has no option to add non-natural amino acids. In our opinion, it would be useful to either include a special letter for the omnipresent case of dimethylated lysine from trypsin in each search engine or to make the amino acid code customizable by the user. Adaption of the employed contaminant databases to include an additional trypsin copy in which lysines are replaced by the letter of choice is usually trivial. However, care has to be taken that the letter is not used as an ambiguity symbol or for special amino acids in the employed database (e.g., J is used for I/L in NCBInr). In general, it might be worth considering increasing the namespace for amino acids to be more flexible when searching for nonstandard amino acids. At the moment, one is usually limited to the 26 letters of the Latin alphabet, 20 of them being already used for the major amino acids, with the employed search engines. In Mascot, four of the remaining six letters are hard-coded, which leaves only two letters for the users to add amino acids such as selenocysteine, pyrrolysine, or, in our case, dimethyl-lysine.
Box 1. Adding Nonstandard Amino Acids to Selected Search Engines.
Mascot: Nonstandard amino acids can be defined via the unimod.xml file; letters J and O are available for modification; http://www.matrixscience.com/blog/non-standard-amino-acid-residues.html
X!Tandem: The protein, modified residue mass file can be used to change masses associated with individual letters except the ambiguity letters B, J, X, Z, leaving letters O and U available to define nonstandard amino acids; http://thegpm.org/TANDEM/api/pmrmf.html
COMET: User amino acids can be added by using the static modifications parameter with available letters B, J, U, X, Z, (e.g., add_B_user_amino_acid =156.125710); http://comet-s.sourceforge.net/parameters/parameters_201501/
An alternative approach could rely on the ability of more recent versions of search engines to provide the functionality to use multiple databases in one search. If the search engine would allow the setting of different variable modifications for the individual source databases, one could then include a database containing only trypsin and set mono- and dimethylation as variable modifications for just this one database, resulting in the same search space as with our artificial amino acid method. Another feasible approach would be to combine the in silico generated search space with measured spectral libraries from contaminants. Finally, the likely ideal solution would be to store known post-translational modifications directly in the sequence database file, as suggested for the HUPO PSI Extended Fasta Format (PEFF). This would not only remove false positives caused by a well-defined subset of peptides but also directly allow the assigning of all peptides with known post-translational modifications even without the use of variable modifications. Unfortunately, this file format is not yet supported by any of the search engines used in this study.
Conclusion
Here, we show that the current state-of-the-art standard proteomic data analysis has severe drawbacks and suggest a new strategy that improves the accuracy of proteomics experiments. Autolysis-protected trypsin (a standard tool for proteomics) leads to false-positive peptide identifications. When reanalyzing all publically available human proteomic data sets (PRIDE database), we found hundreds of statistically significant wrong assignments in more than 18% of the data sets because of methylated peptides originating from trypsin, corresponding to up to 66% of all false positive hits. Furthermore, we present a simple yet efficient search strategy accounting for this problem that can be readily implemented in many widely used search engines. We have demonstrated that our adapted search strategy employing an artificial amino acid for modified trypsin yields more accurate results than the community gold standard employing contaminant databases solely. This is accomplished by limiting the inflation of search space to a minimum while efficiently avoiding false-positive peptide identifications.
Acknowledgments
We thank the MS Amanda development team and especially Viktoria Dorfer for supplying a customized version of MS Amanda that assigns the mass of dimethylated lysine to the database letter J.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.5b01105.
- Figures showing the impact of different search strategies on total PSM, statistically significant PSM after applying 1% FDR and resulting protein scores and fractions of false positives explained by different search strategies employing semitryptic digest. Tables showing peptides originating from methylated porcine trypsin in a S. cerevisiae digest, peptides originating from a Promega modified trypsin self-digest, and peptides originating from acetylated trypsin in data set 2. (PDF)
- A table showing a summary of PRIDE cluster search results. (XLS)
Author Contributions
∥ M.S. and K.F. contributed equally. M.S., K.F., and R.B.-G. devised the study and wrote the manuscript. M.S., K.F., and L.L. performed the experiments and data analysis. PRIDE cluster analysis was done by J.G. All authors have given approval to the final version of the manuscript.
This work was supported by Austrian Science Fund (FWF) projects P26074 and KLI425 and the doctoral school “DK Metabolic and Cardiovascular Disease” (W1226). J.G. is funded by a grant of the Vienna Science and Technology Fund (WWTF) (project LS11-045).
The authors declare no competing financial interest.
Supplementary Material
References
- Perkins D. N.; Pappin D. J.; Creasy D. M.; Cottrell J. S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20 (18), 3551–67. . [DOI] [PubMed] [Google Scholar]
- Eng J. K.; McCormack A. L.; Yates J. R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5 (11), 976–89. 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Geer L. Y.; Markey S. P.; Kowalak J. A.; Wagner L.; Xu M.; Maynard D. M.; Yang X.; Shi W.; Bryant S. H. Open mass spectrometry search algorithm. J. Proteome Res. 2004, 3 (5), 958–64. 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- Cox J.; Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26 (12), 1367–72. 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Craig R.; Beavis R. C. TANDEM: matching proteins with tandem mass spectra. Bioinformatics 2004, 20 (9), 1466–7. 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- Kim S.; Pevzner P. A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 2014, 5, 5277. 10.1038/ncomms6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorfer V.; Pichler P.; Stranzl T.; Stadlmann J.; Taus T.; Winkler S.; Mechtler K. MS Amanda, a universal identification algorithm optimized for high accuracy tandem mass spectra. J. Proteome Res. 2014, 13 (8), 3679–84. 10.1021/pr500202e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabb D. L.; Fernando C. G.; Chambers M. C. MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res. 2007, 6 (2), 654–61. 10.1021/pr0604054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng J. K.; Jahan T. A.; Hoopmann M. R. Comet: an open-source MS/MS sequence database search tool. Proteomics 2013, 13 (1), 22–4. 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii A. I.; Aebersold R. Interpretation of shotgun proteomic data: the protein inference problem. Mol. Cell. Proteomics 2005, 4 (10), 1419–40. 10.1074/mcp.R500012-MCP200. [DOI] [PubMed] [Google Scholar]
- Alpi E.; Griss J.; da Silva A. W.; Bely B.; Antunes R.; Zellner H.; Rios D.; O’Donovan C.; Vizcaino J. A.; Martin M. J. Analysis of the tryptic search space in UniProt databases. Proteomics 2015, 15 (1), 48–57. 10.1002/pmic.201400227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H.; Nesvizhskii A. I. False discovery rates and related statistical concepts in mass spectrometry-based proteomics. J. Proteome Res. 2008, 7 (1), 47–50. 10.1021/pr700747q. [DOI] [PubMed] [Google Scholar]
- Olsen J. V.; Mann M. Improved peptide identification in proteomics by two consecutive stages of mass spectrometric fragmentation. Proc. Natl. Acad. Sci. U. S. A. 2004, 101 (37), 13417–22. 10.1073/pnas.0405549101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestling M. M.; Murphy C. M.; Fenselau C. Recognition of trypsin autolysis products by high-performance liquid chromatography and mass spectrometry. Anal. Chem. 1990, 62 (21), 2391–4. 10.1021/ac00220a025. [DOI] [PubMed] [Google Scholar]
- Jeong K.; Kim S.; Bandeira N. False discovery rates in spectral identification. BMC Bioinf. 2012, 13 (16), S2. 10.1186/1471-2105-13-S16-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice R. H.; Means G. E.; Brown W. D. Stabilization of bovine trypsin by reductive methylation. Biochim. Biophys. Acta, Protein Struct. 1977, 492 (2), 316–21. 10.1016/0005-2795(77)90082-4. [DOI] [PubMed] [Google Scholar]
- Fraenkel-Conrat H.; Bean R. S.; Lineweaver H. Essential groups for the interaction of ovomucoid, egg white trypsin inhibitor, and trypsin, and for tryptic activity. J. Biol. Chem. 1949, 177 (1), 385–403. [PubMed] [Google Scholar]
- Martens L.; Vandekerckhove J.; Gevaert K. DBToolkit: processing protein databases for peptide-centric proteomics. Bioinformatics 2005, 21 (17), 3584–5. 10.1093/bioinformatics/bti588. [DOI] [PubMed] [Google Scholar]
- Krieger J. R.; Taylor P.; Moran M. F.; McGlade C. J. Comprehensive identification of phosphorylation sites on the Numb endocytic adaptor protein. Proteomics 2015, 15 (2–3), 434–46. 10.1002/pmic.201400232. [DOI] [PubMed] [Google Scholar]
- Kennedy J. J.; Abbatiello S. E.; Kim K.; Yan P.; Whiteaker J. R.; Lin C.; Kim J. S.; Zhang Y.; Wang X.; Ivey R. G.; Zhao L.; Min H.; Lee Y.; Yu M. H.; Yang E. G.; Lee C.; Wang P.; Rodriguez H.; Kim Y.; Carr S. A.; Paulovich A. G. Demonstrating the feasibility of large-scale development of standardized assays to quantify human proteins. Nat. Methods 2014, 11 (2), 149–155. 10.1038/nmeth.2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus C. A.; Polden J.; Cotter D. R.; Dunn M. J. Two-dimensional reference map for the basic proteome of the human dorsolateral prefrontal cortex (dlPFC) of the prefrontal lobe region of the brain. Proteomics 2010, 10 (13), 2551–5. 10.1002/pmic.200900705. [DOI] [PubMed] [Google Scholar]
- Vizcaino J. A.; Deutsch E. W.; Wang R.; Csordas A.; Reisinger F.; Rios D.; Dianes J. A.; Sun Z.; Farrah T.; Bandeira N.; Binz P. A.; Xenarios I.; Eisenacher M.; Mayer G.; Gatto L.; Campos A.; Chalkley R. J.; Kraus H. J.; Albar J. P.; Martinez-Bartolome S.; Apweiler R.; Omenn G. S.; Martens L.; Jones A. R.; Hermjakob H. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32 (3), 223–6. 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griss J.; Foster J. M.; Hermjakob H.; Vizcaino J. A. PRIDE Cluster: building a consensus of proteomics data. Nat. Methods 2013, 10 (2), 95–6. 10.1038/nmeth.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaino J. A.; Cote R. G.; Csordas A.; Dianes J. A.; Fabregat A.; Foster J. M.; Griss J.; Alpi E.; Birim M.; Contell J.; O’Kelly G.; Schoenegger A.; Ovelleiro D.; Perez-Riverol Y.; Reisinger F.; Rios D.; Wang R.; Hermjakob H. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013, 41 (D1), D1063–9. 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonzon-Kulichenko E.; Garcia-Marques F.; Trevisan-Herraz M.; Vazquez J. Revisiting peptide identification by high-accuracy mass spectrometry: problems associated with the use of narrow mass precursor windows. J. Proteome Res. 2015, 14 (2), 700–10. 10.1021/pr5007284. [DOI] [PubMed] [Google Scholar]
- Knudsen G. M.; Chalkley R. J. The Effect of Using an Inappropriate Protein Database for Proteomic Data Analysis. PLoS One 2011, 6 (6), e20873. 10.1371/journal.pone.0020873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell J. S. Protein identification using MS/MS data. J. Proteomics 2011, 74 (10), 1842–51. 10.1016/j.jprot.2011.05.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.