Robust detection of alternative splicing in a population of single cells (original) (raw)
Abstract
Single cell RNA-seq experiments provide valuable insight into cellular heterogeneity but suffer from low coverage, 3′ bias and technical noise. These unique properties of single cell RNA-seq data make study of alternative splicing difficult, and thus most single cell studies have restricted analysis of transcriptome variation to the gene level. To address these limitations, we developed SingleSplice, which uses a statistical model to detect genes whose isoform usage shows biological variation significantly exceeding technical noise in a population of single cells. Importantly, SingleSplice is tailored to the unique demands of single cell analysis, detecting isoform usage differences without attempting to infer expression levels for full-length transcripts. Using data from spike-in transcripts, we found that our approach detects variation in isoform usage among single cells with high sensitivity and specificity. We also applied SingleSplice to data from mouse embryonic stem cells and discovered a set of genes that show significant biological variation in isoform usage across the set of cells. A subset of these isoform differences are linked to cell cycle stage, suggesting a novel connection between alternative splicing and the cell cycle.
INTRODUCTION
Every cell within a multicellular organism accomplishes its specialized function through carefully coordinated spatiotemporal gene expression changes. Many eukaryotic genes exhibit alternative splicing, producing multiple types of transcripts with distinct exon combinations, which often result in distinct proteins with different functions (1). Bulk RNA-seq experiments performed on populations of cells are commonly used to obtain an aggregate picture of the splicing changes between biological conditions (2). The recent development of single cell RNA-seq protocols enabled genomewide investigation of gene expression differences at the level of individual cells, opening many new biological questions for study (3,4). However, due to the technical limitations of nascent methods for single cell RNA-seq analysis, most single-cell studies have investigated cellular expression differences at the level of genes but not isoforms (5,6).
Single cell RNA-seq experiments possess several unique properties (summarized in Supplementary Table S1), including high technical variation (7) and low coverage (8), requiring the use of methods different from bulk RNA-seq experiments (6). A single cell possesses only a very small amount of RNA and the sequencing reaction is limited by the amount of starting material; consequently, variability in ‘cell size’ (amount of biological RNA present) affects the sequencing results and must be taken into account during data analysis (7,9). Note that technical variables such as global capture efficiency (10) can also cause differences in ‘cell size’. The tiny amount of RNA in a single cell also means that much amplification is required, which introduces a high level of technical noise (7,10,11). The single molecule capture efficiency is also low (12), making single cell experiments much less sensitive than bulk RNA-seq experiments; transcripts expressed at low levels may not be detected (5).
Single cell RNA extraction protocols prime reverse transcription using the poly(A) tail. During this process, the reverse transcriptase enzyme sometimes produces short cDNAs by falling off before reaching the 5′ end of the transcript (5). The probability of RT falloff increases with distance from the 3′ end, resulting in read coverage biased toward the 3′ end. In addition, most single cells are sequenced at low coverage to maximize the number of cells surveyed (8); as many as 96 cells are usually sequenced in a single HiSeq run (13), and emerging technologies are able to sequence thousands of cells at very low coverage (14,15). Because RNA-seq produces reads that are much shorter than transcripts, inferring abundance estimates for full-length transcripts is not always possible even with bulk RNA-seq. The technical challenges of single cell RNA-seq data make abundance estimates for full-length transcripts highly unreliable (6).
Another key difference is the experimental design; most bulk RNA-seq experiments use an _n_-class design, in which two or more biological groups are compared. The problem of identifying genes and isoforms that are differentially expressed is well studied for _n_-class designs. However, many single cell RNA-seq experiments use a single group design (7). A common problem is to identify genes that vary within a supposedly homogeneous population of cells. Because variation in the expression level of a gene can come from either technical noise or biological variation, a single group design requires modeling the technical noise of single cell sequencing protocol to determine genes whose variation exceeds that expected from noise (7,10,11).
Recent papers have introduced models that describe the technical variation in expression levels of genes measured with single cell RNA-seq (7,10,11). These noise models are trained using spike-in transcripts added at known, constant amounts across a set of cells and can be used to identify genes with significant biological variation in excess of technical variation across populations of single cells. However, existing noise models are unable to detect isoform changes for two reasons: (i) an isoform switching event is a change in ratio, not necessarily absolute expression level and (ii) single cell RNA-seq data do not generally contain sufficient information to measure expression levels of full-length transcripts.
To understand the distinction between a ratio change and a change in absolute expression, consider a gene G that is transcribed into two different isoforms, A and B. If 30 transcripts of G are present in condition 1 and 60 in condition 2, G shows differential gene expression. But if the 30 copies of G in condition 1 consist of 10 A transcripts and 20 B transcripts, and the 60 copies of G in condition 2 consist of 20 A transcripts and 40 B transcripts, G does not undergo a change in isoform usage. In both conditions, isoform A makes up one-third of the transcripts from G and isoform B makes up two-thirds. To identify differences in isoform usage, we must look for a change in the proportions of the transcripts of G that come from A and B, independent of the overall gene expression level. Note that the situation may be more complicated if G has more than two isoforms; in this case, changes in isoform usage may change the contributions of multiple isoforms to the overall expression of G. However, any isoform usage change must result in a different ratio for at least one pair of isoforms. To detect differences in isoform usage, a distribution comparison metric like Jensen–Shannon Divergence can be used (16). Alternatively, the relative proportions of each pair of isoforms can be examined.
To overcome these difficulties, we developed a computational method, SingleSplice, which uses a statistical model to detect genes whose isoform usage varies more than expected from the effects of technical noise alone. Importantly, SingleSplice detects such isoform usage differences without attempting to infer expression levels for full-length transcripts. To the best of our knowledge, SingleSplice is the first method that can detect genes whose isoform usage shows significant variation across a set of single cells.
MATERIALS AND METHODS
Overview of SingleSplice
The SingleSplice method consists of three main phases. In the first phase, we compute expression levels for the longest pieces of transcripts that can be unambiguously identified using short reads (Figure 1A). We accomplish this using the DiffSplice method (16). Briefly, we construct a directed, acyclic splice graph directly from read alignments so that possible transcripts correspond to paths through the graph. Using this splice graph, we identify single-entry, single-exit modules in the graph (Figure 1A). These single-entry, single-exit portions of the graph are called alternative splicing modules (ASMs), and each path through an ASM corresponds to a piece of one or more transcripts spanning two or more exons; there may be one or more ASMs per gene. ASMs possess the important property that any alternative splicing a gene undergoes will cause a change in the ratio of at least one pair of ASM paths.
Figure 1.

Diagram of SingleSplice method. (A) SingleSplice constructs an expression-weighted splice graph directly from aligned reads (top), then identifies alternative splicing modules (ASMs) and calculates the coverage on each ASM path (indicated in black, red, yellow and green). (B) For each ASM path, a distribution is fit to capture the expected variation in coverage due to technical noise. (C) SingleSplice computes the expected variation in isoform usage by sampling repeatedly from the fitted noise distributions. The resulting sampled values are used to compute an empirical _P_-value for the null hypothesis that the observed variation in isoform usage results from technical noise alone.
The second phase of SingleSplice fits distributions describing the expected expression variation of each ASM path due to technical noise (Figure 1B). In the third phase, to determine whether a gene shows significant splicing changes across a set of cells, we sample values from the fitted noise model of each ASM path to predict the variance of isoform ratios due to technical noise alone, then use these predicted values to assess the significance of the observed variation in isoform ratio (Figure 1C). Intuitively, performing this sampling procedure (a statistical technique known as parametric bootstrapping) is like sequencing the same set of cells repeatedly to see how the isoform usage changes from technical variation alone.
Identifying alternative splicing modules and estimating coverage
To identify genes that exhibit alternative splicing, we construct an expression-weighted splice graph (ESG) directly from the genomic read alignments. An ESG is a directed, acyclic graph in which vertices are genomic coordinates, edges represent splices or contiguous transcription, and the weight on each edge corresponds to its coverage (16–18). Each gene has its own graph, and transcripts are represented as paths through the graph from a start site to an end site. A graph algorithm is subsequently used to identify ASMs (16). An ASM is defined as a subgraph of an ESG such that there is only one path into and out of the subgraph, and there is more than one path through the subgraph (16).
Intuitively, an ASM represents the longest portion of two or more distinct isoforms that can be each identified by at least one unique set of reads; an ASM path corresponds to the portion of the isoform that differs from other isoforms. To avoid isoforms expressed at very low levels, we used only splice junctions with 10 or more reads in at least 20 samples when identifying ASM structures. A probabilistic model is then fit using expectation maximization to estimate the coverage of each ASM path using the numbers of reads on both the exons and junctions of the paths (16). The strategy of identifying ASMs directly from the data as opposed to a simpler strategy such as that used by MISO (19) provides two important benefits: (i) discovery of isoforms incorporating unannotated splicing events and (ii) abundance estimation of the longest uniquely identifiable portions of transcripts rather than just the exons immediately adjacent to an alternative splicing event.
Fitting distributions to predict technical variation
In order to predict the variation of isoform ratios caused by technical noise, we first needed a model for technical variation in measured expression level. The basic idea of our approach is to learn a mean-variance relationship from a set of spike-in transcripts, as has been shown to be effective in previous studies (7,10,11). Once this mean-variance model is trained, the expected technical variation of any transcript (spike-in or endogenous) can be calculated from the mean of its measured expression levels.
Previous papers (10,11) have used negative binomial models to predict the expression-dependent variation in read counts on genes. Note that a fundamental assumption of such approaches is that the level of technical noise depends on expression level, or more precisely the number of molecules present at the beginning of the sequencing process. To accurately reflect this assumption, we developed a model for the variation in coverage, not raw read counts, because we are comparing ASM paths that may be of different lengths, so we need to normalize read counts by length. The need to normalize by length follows directly from the fact that read count is proportional to the number and length of transcripts sequenced. For a given isoform (or ASM path) t,
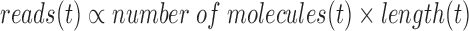
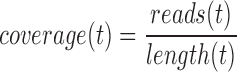
Therefore, coverage (reads per base) is proportional to the number of transcripts present, and we model expression-dependent noise variation using coverage.
Since coverage is continuous rather than count data, we used a gamma distribution – the continuous analog of the negative binomial distribution. When we attempted to fit gamma distributions to the spike-in data, we found that the gamma model worked well for highly expressed transcripts, but did not accurately predict the behavior of transcripts at low abundance. Testing the gamma fits using the Kolmogorov–Smirnov test showed that the fits were accepted for all highly expressed spike-ins but rejected for nearly all spike-ins expressed below 100 RPKMs. While looking at these low expression transcripts, we noticed frequent expression levels of 0 (a ‘dropout’ event) (10), which has an undefined probability under the gamma distribution. Dropout events can occur because of the low capture efficiency of single cell RNA-seq protocols; transcripts expressed at low levels often fail to be captured and amplified (10). We thus chose to model technical variation using the following mixture distribution (where 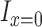 is 1 if
is 1 if 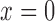 and 0 otherwise):
and 0 otherwise):
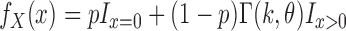
The problem of fitting a noise model then reduces to finding values for p, k and 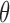 . We accomplished this by using linear regression to predict the dropout probability p and variance
. We accomplished this by using linear regression to predict the dropout probability p and variance 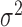 from the mean expression level
from the mean expression level 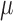 . The variance is predicted using a generalized linear model of the gamma family (Figure 2A) and the dropout probability is predicted using logistic regression (Figure 2B). Once
. The variance is predicted using a generalized linear model of the gamma family (Figure 2A) and the dropout probability is predicted using logistic regression (Figure 2B). Once 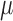 , p and
, p and 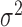 are known, k and
are known, k and 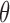 can be directly computed using the following equations (which can be easily derived from the expressions for the variance of a gamma distribution). Note that for
can be directly computed using the following equations (which can be easily derived from the expressions for the variance of a gamma distribution). Note that for 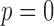 (i.e. in the absence of dropouts), these expressions reduce to the equations for gamma mean and variance in terms of k and
(i.e. in the absence of dropouts), these expressions reduce to the equations for gamma mean and variance in terms of k and 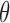 .
.
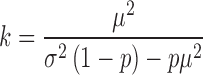
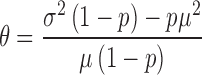
Figure 2.

Fitting a technical noise model using spike-in transcripts. (A) Gamma regression model to predict variance in coverage as a function of mean expression level. The observed data are shown as black points and the gamma fit is drawn in red. (B) Logistic regression model predicting dropout rate as a function of mean expression level. The observed data are shown as black points, and the regression line is shown in red. (C) Expected (line) and observed (histogram) ratio distributions for a pair of spike-in transcripts showing no ratio change. Note that expectation and observation match very well in this case, indicating that the model effectively predicts the effects of technical noise. (D) Expected (line) and observed (histogram) ratio distributions for a pair of spike-in transcripts showing simulated isoform switching. Note that the observed ratio values differ significantly from what is expected based on technical noise alone.
We performed the gamma regression using the glmgam.fit function from the statmod R package. Only spike-in transcripts with expression levels above a 10 RPKM certain threshold were used to fit the gamma model. Logistic regression was performed using the glm function in R.
Normalizing by cell size
Unlike bulk RNA-seq experiments, cellular variation in the amount of starting RNA (‘cell size’) is significant in single cell RNA-seq experiments. Cellular differences like cell cycle stage can affect cell size (Figure 3A). Failure to account for this variation can result in artifacts such as the one shown in Figure 3C where two spike-in transcripts whose expression levels should vary randomly are instead correlated with cell size and with each other. Since spike-ins are added at known, constant amounts, we can use the ratio of biological reads to spike-in reads as a proxy for cell size. The total number of aligned reads per cell also varies independently of cell size variations due to differences in total sequencing depth, read quality, amount of non-polyadenylated RNA that was sequenced, etc. To account for these effects, we normalize coverage both by number of aligned reads and by cell size. To normalize by cell size, we compute a ‘scale factor’ 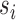 for each cell i so that the expression levels of each cell are scaled to the median cell size:
for each cell i so that the expression levels of each cell are scaled to the median cell size:
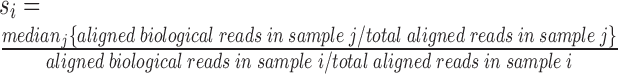
Figure 3.

Accounting for effects of cell size. (A) Variation in the relative proportions of reads mapping to spike-in transcripts and cellular transcripts indicates that the amount of cellular RNA varies reproducibly during the cell cycle. (B) Since spike-in transcripts are added at constant amounts, their measured expression levels should vary randomly across the set of cells. Instead, PCA using only reads per kilobase length per million reads (RPKMs) from spike-in transcripts before cell size normalization predicts cell cycle stage. (C) Spike-in expression levels should fluctuate randomly due to technical noise, but instead spike-in expression levels before normalization are strongly correlated with each other and with cell size. Note how closely the blue, orange and grey lines trend together. (D) Normalizing for cell size using the fraction of reads that come from spike-in versus cellular RNA removes this effect.
We normalize coverage by the total number of aligned reads, yielding a quantity similar to reads per kilobase length per million reads (RPKM), then multiply by the cell size scale factor:
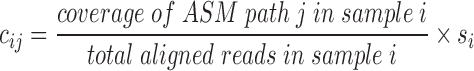
The normalized coverage no longer shows the effects of cell size (compare Figure 3B and D).
Detecting biological variation in isoform usage
We use a parametric bootstrapping approach to identify genes whose isoform usage varies more than expected based on technical variation. In the following discussion, we will refer to transcript abundance for convenience, but the values we work with are derived from ASM paths. After determining the parameters of a gamma distribution that predict technical variation in expression level of a pair of transcripts (as described above), we sample repeatedly from these distributions and calculate the proportions of each ASM path in the resulting samples. More formally, for transcript A expressed at an average level of 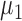 and transcript B expressed at an average level of
and transcript B expressed at an average level of 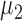 in a set of n cells, we sample n expression levels for each transcript and repeat this process 1000 times:
in a set of n cells, we sample n expression levels for each transcript and repeat this process 1000 times:
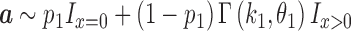
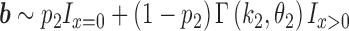
Then, for each of the 1000 sets of n values, we compute the sample variance of the isoform proportions:
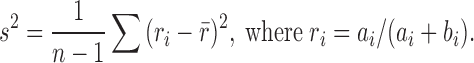
This gives the expected variation in isoform proportions due to technical noise. Intuitively, our parametric bootstrap samples simulate sequencing the same set of cells 1000 times to see how the results change due to technical noise alone. Using the set of 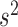 values computed in this way, we determine an empirical _P_-value—for the null hypothesis that technical noise alone accounts for the observed changes in isoform proportions—by simply counting the number of times that variation at least as great as the experimental variation is present in our simulated
values computed in this way, we determine an empirical _P_-value—for the null hypothesis that technical noise alone accounts for the observed changes in isoform proportions—by simply counting the number of times that variation at least as great as the experimental variation is present in our simulated 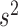 values. Note that our parametric bootstrapping approach also gives an empirical distribution for isoform proportion r; these values can be compared to the distribution observed in the population of sequenced cells (as shown in Figures 2C–D and 4A) using, for example, the Kolmogorov–Smirnov test. We therefore re-ran our true positive and true negative examples (Supplementary Figure S1) using the KS test and found that the performance was very similar whether an empirical _P_-value for ratio variance or the KS test was used, although the KS test performed slightly worse (data not shown). Note that our method is designed to predict ratio variance for a pair of ASM paths. For an ASM with more than two paths, we compare all pairs of paths; in the case of an ASM with prohibitively many paths, we look only at the k most highly expressed paths, where k is a user-specified constant.
values. Note that our parametric bootstrapping approach also gives an empirical distribution for isoform proportion r; these values can be compared to the distribution observed in the population of sequenced cells (as shown in Figures 2C–D and 4A) using, for example, the Kolmogorov–Smirnov test. We therefore re-ran our true positive and true negative examples (Supplementary Figure S1) using the KS test and found that the performance was very similar whether an empirical _P_-value for ratio variance or the KS test was used, although the KS test performed slightly worse (data not shown). Note that our method is designed to predict ratio variance for a pair of ASM paths. For an ASM with more than two paths, we compare all pairs of paths; in the case of an ASM with prohibitively many paths, we look only at the k most highly expressed paths, where k is a user-specified constant.
Figure 4.

Testing the sensitivity and specificity of SingleSplice using spike-in transcripts. (A) True negative examples are created by pairing spike-in transcripts. Any variation in the ratio of these transcripts is due to technical noise. (B) Scatter plot showing expected (SingleSplice prediction) ratio variance versus observed ratio variance for true negative test cases. Each box represents a single pair of spike-ins, and area of the box is proportional to the mean expression level. Test cases where SingleSplice correctly identified the pair of spike-ins as showing no isoform variation are colored green. (C) True positive examples are created by swapping half of the measured expression levels of a pair of spike-in transcripts. Ratio variation in these examples comes from technical noise and simulated isoform switching. (D) Scatter plot showing expected versus observed ratio variance for true positive test cases. Test cases where SingleSplice correctly identified the pair of spike-ins as showing significant isoform variation are colored green.
To find genes that showed isoform usage differences linked to cell cycle stage, we computed the isoform proportions 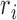 for pairs of ASM paths and used a Kruskal–Wallis test. This test allowed identification of pairs of ASM paths for which the isoform proportions observed in each phase of the cell cycle (G1, S and G2/M) are not drawn from the same distribution. The _P_-values from the Kruskal–Wallis test were adjusted using the method of Benjamini–Hochberg to correct for multiple hypothesis testing, and any pairs of ASM paths with adjusted _P_-values below 0.05 were considered to show changes linked to the cell cycle.
for pairs of ASM paths and used a Kruskal–Wallis test. This test allowed identification of pairs of ASM paths for which the isoform proportions observed in each phase of the cell cycle (G1, S and G2/M) are not drawn from the same distribution. The _P_-values from the Kruskal–Wallis test were adjusted using the method of Benjamini–Hochberg to correct for multiple hypothesis testing, and any pairs of ASM paths with adjusted _P_-values below 0.05 were considered to show changes linked to the cell cycle.
RESULTS
SingleSplice accurately predicts behavior of spike-in transcripts
We used spike-in transcripts (20) added at known, constant concentrations across a set of cells in a previously published data set (9) to calibrate our model and test the sensitivity and specificity of SingleSplice. Because we are comparing ASM paths that may be of different lengths, and the number of reads obtained from a particular ASM path depends on both initial number of molecules and length, we developed a model for the variation in coverage, not raw read counts (see Materials and Methods for a detailed discussion of this point). We used the gamma distribution—the continuous analog of the negative binomial distribution—to model coverage, since coverage is continuous rather than count data.
When we attempted to fit gamma distributions to the spike-in data, we found that the model did not accurately predict the behavior of transcripts at low abundance. These low expression transcripts frequently show expression levels of 0 (a ‘dropout’ event) (11), which has an undefined probability under the gamma distribution. We thus chose to model technical variation using a mixture of gamma and Bernoulli distributions (see Materials and Methods section for details). The problem of fitting a noise model then reduces to finding the parameters of this mixture distribution. We accomplished this by using logistic regression to predict dropout probability and gamma regression to predict variance from mean expression level (Figure 2A–B). Parametric bootstrapping using this noise model allows computation of the expected variation in ratio due to technical noise (Figure 2C–D).
In addition, we found that it was necessary to normalize expression levels by ‘cell size’, the total amount of mRNA present in each cell. Since spike-in transcripts are added at known, constant amounts, the ratio of biological to spike-in reads can be used as a proxy for cell size. In the Buettner data set that we analyzed (9), cells at different stages of the cell cycle show consistent differences in cell size (Figure 3A). As a result, PCA using only spike-in expression levels (which should show only stochastic variation across the set of cells) separates cells by cell cycle stage (Figure 3B), and the expression levels of pairs of spike-ins are strongly correlated with each other and with cell size (Figure 3C), even when total sequencing depth is taken into account. Normalizing expression levels by the proportion of reads that came from the cell rather than from spike-in transcripts removes this effect (Figure 3D).
To evaluate the performance of SingleSplice, we used two different kinds of tests constructed by pairing spike-in transcripts within each cell so that each spike-in represents an isoform of an alternatively spliced gene (Figure 4). We constructed true negative tests by simply pairing the measured expression levels of spike-in transcripts (Figure 4A). Because each spike-in transcript is added at a constant amount in every cell, the ratio between a given pair of spike-ins is also constant, technical noise being the only source of variation. The set of spike-ins consists of 96 separate transcripts, which gave 4186 pairs of spike-ins, each pair corresponding to an alternatively spliced gene, after omitting self-pairings and transcripts whose measured expression was identically zero. SingleSplice correctly identified the majority (85% specificity) of these true negative spike-in pairs as showing no significant isoform ratio change at 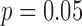 . Figure 4B shows the results of this test as a scatter plot, where the x-axis represents the ratio variance predicted by SingleSplice and the y-axis is the observed ratio variance of the spike-in pair. Each rectangle corresponds to a single pair of spike-ins, true negatives are colored green, false positives are colored black and the expression level is indicated by the size of the rectangle. Note that the SingleSplice model predicts the behavior of the isoform ratios quite well, as indicated by how the points generally lie along the dotted line.
. Figure 4B shows the results of this test as a scatter plot, where the x-axis represents the ratio variance predicted by SingleSplice and the y-axis is the observed ratio variance of the spike-in pair. Each rectangle corresponds to a single pair of spike-ins, true negatives are colored green, false positives are colored black and the expression level is indicated by the size of the rectangle. Note that the SingleSplice model predicts the behavior of the isoform ratios quite well, as indicated by how the points generally lie along the dotted line.
We next devised a set of true positive tests in which we swapped half of the measured expression levels for pairs of spike-in transcripts (Figure 4C), mimicking isoform switching across a set of cells. In these examples, variation in the ratio of pairs of spike-in transcripts comes from technical noise and simulated isoform switching. As in the true negative case, we constructed 4186 pairs of spike-in transcripts. We found that SingleSplice again performed very well (86% sensitivity). Note that, unlike the true negative test cases, the observed ratio variance generally exceeds the variance expected from technical noise alone (indicated by the dotted line). This shows that SingleSplice accurately detects biological variation in excess of technical variation. Many of the false negatives come from pairs where the spike-ins were expressed at very low levels, as shown by the small boxes in Figure 4D that are also black. This effect may be due to a detection threshold below which isoform switching is simply undetectable due to the high level of technical noise (see also the discussion of Supplementary Figure S1 below).
In addition, we note that the External RNA Controls Consortium (ERCC) spike-ins span a very wide range of concentrations, which for some spike-in pairs results in large abundance changes when we swap expression levels to simulate isoform switching. This wide range of spike-in concentrations allows us to assess the performance of SingleSplice across the full spectrum of ratio changes. However, by looking at subsets of the spike-ins we also confirmed that SingleSplice sensitivity shows graceful degradation as the expression levels of the swapped spike-ins approach each other. For spike-ins whose mean expression levels differ by at most a factor of five (mean > 10 RPKMs), sensitivity is 85%. Similarly, for spike-in pairs with fold changes of at most four, three and two, the sensitivity values are 83%, 79% and 69%, respectively. Note also that these sensitivity values vary based on the actual expression level of each spike-in; i.e. isoform switching is much easier to detect between spike-ins with mean expression of 1000 and 2000 than mean expression of 10 and 20.
To demonstrate the importance of the modeling strategies SingleSplice uses to capture expression-dependent noise behavior, we also compared the performance of SingleSplice to a baseline method. A reasonable first approach to identifying alternatively spliced genes would be to choose a threshold value c. This baseline method would then classify any genes with ratio variance greater than c as showing significant alternative splicing, and all other genes as showing no significant change. For an appropriately chosen threshold value, the baseline method is fairly effective, achieving 92% sensitivity and 81% specificity across the full set of spike-in pairs described above for 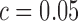 (Supplementary Figure S1A). The surprising effectiveness of this strategy is due to the separation between ratio variance for the true positive and true negative spike-in pairs (Supplementary Figure S1B). However, a key shortcoming of the baseline method is its inability to account for differences in expected ratio variance due to expression level. Based on the mean-variance relationship that describes the behavior of technical noise (see Figure 2A), we expect that pairs of transcripts expressed at low levels will show much more ratio variance than highly expressed transcript pairs. Inspecting pairs of spike-ins where both transcripts are expressed at a low level (mean < 10 RPKMs) compared to highly expressed spike-ins (mean > 1000 RPKMs) shows that the ratio variance is strongly related to expression level (Supplementary Figure S1B and S1C). This fact will systematically bias the baseline method toward calling low expression genes as alternatively spliced and identifying high expression genes as not alternatively spliced, the exact opposite of what is desirable when analyzing noisy, low coverage single cell data. For example, using the cutoff
(Supplementary Figure S1A). The surprising effectiveness of this strategy is due to the separation between ratio variance for the true positive and true negative spike-in pairs (Supplementary Figure S1B). However, a key shortcoming of the baseline method is its inability to account for differences in expected ratio variance due to expression level. Based on the mean-variance relationship that describes the behavior of technical noise (see Figure 2A), we expect that pairs of transcripts expressed at low levels will show much more ratio variance than highly expressed transcript pairs. Inspecting pairs of spike-ins where both transcripts are expressed at a low level (mean < 10 RPKMs) compared to highly expressed spike-ins (mean > 1000 RPKMs) shows that the ratio variance is strongly related to expression level (Supplementary Figure S1B and S1C). This fact will systematically bias the baseline method toward calling low expression genes as alternatively spliced and identifying high expression genes as not alternatively spliced, the exact opposite of what is desirable when analyzing noisy, low coverage single cell data. For example, using the cutoff 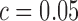 on pairs of spike-ins where both transcripts have mean expression below 10 RPKMs gives a specificity of just 25%. In contrast, SingleSplice correctly identifies 86% of these low expression true negative pairs. Conversely, the cutoff
on pairs of spike-ins where both transcripts have mean expression below 10 RPKMs gives a specificity of just 25%. In contrast, SingleSplice correctly identifies 86% of these low expression true negative pairs. Conversely, the cutoff 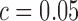 gives 71% sensitivity on highly expressed spike-in pairs compared to SingleSplice's sensitivity of 94% on the same pairs. By modeling the expected ratio variance as a function of expression level, we are able to remove the bias toward calling low expression genes as alternatively spliced. Instead, we determine the significance of splicing variation by the amount of variation expected based on the expression levels of the transcripts involved.
gives 71% sensitivity on highly expressed spike-in pairs compared to SingleSplice's sensitivity of 94% on the same pairs. By modeling the expected ratio variance as a function of expression level, we are able to remove the bias toward calling low expression genes as alternatively spliced. Instead, we determine the significance of splicing variation by the amount of variation expected based on the expression levels of the transcripts involved.
We also devised a set of tests to demonstrate SingleSplice's ability to detect alternative splicing in ASMs with more than two paths. To do this, we sampled random triples of spike-ins, then swapped half of the measured expression levels between two of the transcripts in the triple to mimic isoform switching. True negative examples were created as in the pairwise case by simply using the measured expression levels of the three chosen transcripts. Because there are more than 125 000 possible spike-in triples, we randomly sampled 10 000 rather than looking at all possible combinations as we did for the pairwise case. We then tested all 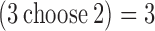 pairs of spike-ins for each triple and called the triple alternatively spliced if the _P_-value for any pair was significant. SingleSplice showed 87% sensitivity and 67% specificity on these tests. The reduction in specificity and the slight increase in sensitivity compared to the pairwise tests is likely due to the fact that a gene is called alternatively spliced if any pair shows a significant change. One strategy to mitigate the drop in specificity is to perform ‘majority voting’ and call the gene as alternatively spliced only if a majority of the pairwise comparisons are significant. Using this voting strategy on the set of 10 000 spike-in triples gives 91% specificity and 85% sensitivity. Our analysis of real data showed that most ASMs do not have more than two highly expressed paths, and SingleSplice allows the user to restrict analysis to the k most highly expressed paths. In addition, SingleSplice outputs the result of the statistical test for each pair of ASM paths, allowing the user to choose whether to use majority voting when assessing if a gene truly shows alternative splicing.
pairs of spike-ins for each triple and called the triple alternatively spliced if the _P_-value for any pair was significant. SingleSplice showed 87% sensitivity and 67% specificity on these tests. The reduction in specificity and the slight increase in sensitivity compared to the pairwise tests is likely due to the fact that a gene is called alternatively spliced if any pair shows a significant change. One strategy to mitigate the drop in specificity is to perform ‘majority voting’ and call the gene as alternatively spliced only if a majority of the pairwise comparisons are significant. Using this voting strategy on the set of 10 000 spike-in triples gives 91% specificity and 85% sensitivity. Our analysis of real data showed that most ASMs do not have more than two highly expressed paths, and SingleSplice allows the user to restrict analysis to the k most highly expressed paths. In addition, SingleSplice outputs the result of the statistical test for each pair of ASM paths, allowing the user to choose whether to use majority voting when assessing if a gene truly shows alternative splicing.
Mouse embryonic stem cells show isoform usage differences linked to cell cycle stage
Having verified the performance of SingleSplice using spike-in transcripts, we looked for genes with significant isoform usage variation across a set of mouse embryonic stem cells whose cell cycle stage had been determined experimentally before sequencing (9). In the Buettner data set, SingleSplice identified 797 genes that showed significant biological variation in isoform usage (Figure 5A; Supplementary File 1). Because the cells in this data set are all from the same cell line, this biological variation is most likely due to changes in the dynamic state of the cells rather than genetic differences. Thus, we would expect isoform usage variation to come from primarily (i) stochastic changes in transcription among cells or (ii) cell cycle differences.
Figure 5.

Discovery of splicing changes during the cell cycle. (A) Expected (line) and observed (histogram) ratio distributions for the Rbm25 gene. Note that the isoform usage differs significantly from what is expected based on technical noise alone. (B–D) The Hnrnpc, Snhg3 and Rbm25 genes show isoform usage changes during the cell cycle. The exon-intron structure (5′ to 3′ in direction of transcription) of each pair of ASM paths is shown above the corresponding plot. The ratios shown in these panels are computed with respect to the top ASM path – i.e. a ratio of 0 corresponds to only the bottom ASM path, and a ratio of 1 indicates only the top ASM path. (E) PCA using isoform ratios alone separates cells according to cell cycle stage.
To further investigate the source of the observed variation, we looked for genes whose isoform usage changes are linked to cell cycle phase. To do this, we compared the isoform proportions calculated by SingleSplice across cells in the G1, S and G2/M cell cycle phases. Using a Kruskal–Wallis test and false discovery rate (FDR) correction, we identified 124 genes that show significant isoform usage differences among cell cycle stages, including three particularly interesting examples: Hnrnpc, Snhg3 and Rbm25 (Figure 5B–D; Supplementary File 2). Hnrnpc encodes an RNA binding protein that plays a role in mRNA splicing (21), nuclear export (22) and translational regulation (23). In addition, in human cells, the protein product is known to play a crucial role in cell cycle regulation through interaction with the long noncoding RNA MALAT1 (22); is differentially phosphorylated during the cell cycle (24); and modulates translation of the c-myc protein in a cell cycle dependent manner (23). Our SingleSplice analysis revealed that Hnrnpc uses an alternative 5′ splice site that results in either a long or a short upstream exon, and the short upstream exon is used primarily in S-phase (Figure 5B, transcript structure above graph). Snhg3 is a long non-coding RNA that is conserved between mice and humans but has not been extensively studied, and little is known about its function. Snhg3 shows a cell-cycle-dependent alternative splicing change in which two short exons are replaced with a longer exon (Figure 5C). The relative abundance of the splice form containing two short exons (upper transcript structure in Figure 5C) steadily increases through G1 and S phase, peaking in G2/M phase. Rbm25 is a spliceosome-associated RNA binding protein that has been shown to regulate apoptosis by modulating alternative splicing of the BCL2L1 gene (25). Our analysis showed that exon skipping in Rbm25 produces two distinct splice variants (Figure 5D) with an expression pattern that differs strikingly between G2/M phase and G1 and S phase. Intriguingly, the distribution of these two splice variants across the set of single cells is bimodal, with modes at 0 and 1, indicating that most cells almost exclusively express either one form or the other (Figure 5D). The ASM path with two internal exons (lower transcript structure in Figure 5D) appears to be used with much greater frequency among cells that are in G2/M phase compared to the other cell cycle phases.
Principal component analysis (PCA) using only isoform proportions from these 124 genes separates cells by cell cycle stage, underscoring the strong relationship between cell cycle stage and isoform usage (Figure 5E). We also looked for gene ontology terms enriched in this set of genes to verify that the genes are involved in the cell cycle (Supplementary File 3). A number of GO terms related to the cell cycle process, including regulation of DNA replication, nuclear division and maintenance of chromosome number, are enriched, lending further credence to the hypothesis that the mRNA splicing changes we observed are likely to play a role in the cell cycle. Interestingly, the set of 124 genes is also enriched for genes involved in RNA splicing and RNA processing, suggesting that global splicing regulation may change during the cell cycle.
Although we also investigated a different data set (13), we found fewer genes with multiple isoforms detected at appreciable levels, possibly due to lower sequencing depth. In contrast, the Buettner data set was sequenced to greater depth and showed many more splice variants. We found a roughly linear relationship between the read depth per cell and the number of ASM paths detected above 10 RPKMs (Supplementary Figure S2). The majority of ASM paths that we detected occur in only a few cells, which suggests many alternative splicing events are relatively rare due to a combination of biological and technical variation. For this reason, the number of cells sequenced will likely also influence the detection rate of ASM paths. In addition, sequencing more cells increases the statistical power for detecting alternative splicing across the set of single cells by giving more chances to observe a given splicing event. Furthermore, the number of ASM paths detected in each cell at low coverage is smaller than the number of genes detected in a typical single cell RNA-seq experiment, suggesting that many of the genes are not sampled deeply enough to reveal multiple isoforms. Thus, it appears that the low coverage typically used in single cell RNA-seq studies does not completely sample the complexity of the transcriptome, and experiments investigating alternative splicing may need to use increased sequencing depth.
DISCUSSION
We have developed SingleSplice, a tool for studying alternative splicing using single cell RNA-seq data. SingleSplice models the effects of technical noise on isoform ratios, allowing investigators to detect biological variation in isoform usage across a population of single cells. We discovered a set of 797 genes that show significant isoform usage differences in mouse embryonic stem cells. One can also use SingleSplice to identify alternative splicing between pre-specified groups of single cells, as we did with cells separated by experimentally determined cell cycle stage. Alternatively, the output of SingleSplice can be used to cluster cells by their isoform ratios to discover intrinsic cell types based on their isoform ratios.
With the development of SingleSplice, a number of interesting biological questions can be investigated using single cell RNA-seq data. For example, it is not known whether every cell within a tissue generally expresses all of the isoforms that are detected in a bulk RNA-seq sample. Preliminary studies suggest that populations of cells may display different ‘modes’ of isoform usage that are blended together in bulk RNA-seq data (26). Single cell studies can provide insight into the isoform usage differences that occur during dynamic biological processes, such as differentiation (27), immune cell activation (28) or tumorigenesis (29). SingleSplice can also be used to investigate heterogeneity within healthy or diseased tissues, with the goal of characterizing previously unknown intrinsic subpopulations of cells defined by splicing differences. Ultimately, integrating other types of functional genomic assays such as single cell DNA sequencing (30), single cell Hi-C (31), single cell ATAC-seq (32) or single cell ChIP-seq (33) with single cell RNA-seq will give insights into the connections between alternative splicing and other biological processes. Our analysis here indicates that deep coverage and use of spike-in transcripts are important prerequisites for careful and detailed future studies of alternative splicing at the single cell level. Combined with the robust detection method of SingleSplice, single cell RNA-seq studies promise to generate many new insights into basic RNA biology and the ways in which cells work together to enable complex multicellular life.
Supplementary Material
SUPPLEMENTARY DATA
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
SingleSplice source code is available at https://github.com/jw156605/SingleSplice
FUNDING
National Institutes of Health (NIH) [HG06272] to JFP, NSF Graduate Research fellowship [DGE-1144081], NIH BD2K Fellowship [T32 CA201159] to JDW. Funding for open access charge: National Institutes of Health (NIH) [HG06272].
Conflict of interest statement. None declared.
REFERENCES
- 1.Nilsen T.W., Graveley B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Z., Gerstein M., Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sandberg R. Entering the era of single-cell transcriptomics in biology and medicine. Nat. Methods. 2013;11:22–24. doi: 10.1038/nmeth.2764. [DOI] [PubMed] [Google Scholar]
- 4.Shapiro E., Biezuner T., Linnarsson S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet. 2013;14:618–630. doi: 10.1038/nrg3542. [DOI] [PubMed] [Google Scholar]
- 5.Saliba A.-E., Westermann A.J., Gorski S.A., Vogel J. Single-cell RNA-seq: advances and future challenges. Nucleic Acids Res. 2014;42:8845–8860. doi: 10.1093/nar/gku555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stegle O., Teichmann S.A., Marioni J.C. Computational and analytical challenges in single-cell transcriptomics. Nat. Rev. Genet. 2015;16:133–145. doi: 10.1038/nrg3833. [DOI] [PubMed] [Google Scholar]
- 7.Brennecke P., Anders S., Kim J.K., Kołodziejczyk A.A., Zhang X., Proserpio V., Baying B., Benes V., Teichmann S.A., Marioni J.C., et al. Accounting for technical noise in single-cell RNA-seq experiments. Nat. Methods. 2013;10:1093–1095. doi: 10.1038/nmeth.2645. [DOI] [PubMed] [Google Scholar]
- 8.Streets A.M., Huang Y. How deep is enough in single-cell RNA-seq. Nat. Biotechnol. 2014;32:1005–1006. doi: 10.1038/nbt.3039. [DOI] [PubMed] [Google Scholar]
- 9.Buettner F., Natarajan K.N., Casale F.P., Proserpio V., Scialdone A., Theis F.J., Teichmann S.A., Marioni J.C., Stegle O. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat. Biotechnol. 2015;33:155–160. doi: 10.1038/nbt.3102. [DOI] [PubMed] [Google Scholar]
- 10.Grün D., Kester L., van Oudenaarden A. Validation of noise models for single-cell transcriptomics. Nat. Methods. 2014;11:637–640. doi: 10.1038/nmeth.2930. [DOI] [PubMed] [Google Scholar]
- 11.Kharchenko P.V., Silberstein L., Scadden D.T. Bayesian approach to single-cell differential expression analysis. Nat. Methods. 2014;11:740–742. doi: 10.1038/nmeth.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu A.R., Neff N.F., Kalisky T., Dalerba P., Treutlein B., Rothenberg M.E., Mburu F.M., Mantalas G.L., Sim S., Clarke M.F., et al. Quantitative assessment of single-cell RNA-sequencing methods. Nat. Methods. 2014;11:41–46. doi: 10.1038/nmeth.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Treutlein B., Brownfield D.G., Wu A.R., Neff N.F., Mantalas G.L., Espinoza F.H., Desai T.J., Krasnow M.A., Quake S.R. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. 2014;509:371–375. doi: 10.1038/nature13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Macosko E.Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A.R., Kamitaki N., Martersteck E.M., et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fan H.C., Fu G.K., Fodor S.P.A. Combinatorial labeling of single cells for gene expression cytometry. Science. 2015;347 doi: 10.1126/science.1258367. [DOI] [PubMed] [Google Scholar]
- 16.Hu Y., Huang Y., Du Y., Orellana C.F., Singh D., Johnson A.R., Monroy A., Kuan P.-F., Hammond S.M., Makowski L., et al. DiffSplice: the genome-wide detection of differential splicing events with RNA-seq. Nucleic Acids Res. 2013;41:e39. doi: 10.1093/nar/gks1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh D., Orellana C.F., Hu Y., Jones C.D., Liu Y., Chiang D.Y., Liu J., Prins J.F. FDM: a graph-based statistical method to detect differential transcription using RNA-seq data. Bioinformatics. 2011;27:2633–2640. doi: 10.1093/bioinformatics/btr458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pertea M., Pertea G.M., Antonescu C.M., Chang T.-C., Mendell J.T., Salzberg S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015;33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katz Y., Wang E.T., Airoldi E.M., Burge C.B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods. 2010;7:1009–1015. doi: 10.1038/nmeth.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang L., Schlesinger F., Davis C.A., Zhang Y., Li R., Salit M., Gingeras T.R., Oliver B. Synthetic spike-in standards for RNA-seq experiments. Genome Res. 2011;21:1543–1551. doi: 10.1101/gr.121095.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.König J., Zarnack K., Rot G., Curk T., Kayikci M., Zupan B., Turner D.J., Luscombe N.M., Ule J. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 2010;17:909–915. doi: 10.1038/nsmb.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang F., Yi F., Han X., Du Q., Liang Z. MALAT-1 interacts with hnRNP C in cell cycle regulation. FEBS Lett. 2013;587:3175–3181. doi: 10.1016/j.febslet.2013.07.048. [DOI] [PubMed] [Google Scholar]
- 23.Kim J.H., Paek K.Y., Choi K., Kim T.-D., Hahm B., Kim K.-T., Jang S.K. Heterogeneous nuclear ribonucleoprotein C modulates translation of c-myc mRNA in a cell cycle phase-dependent manner. Mol. Cell. Biol. 2003;23:708–720. doi: 10.1128/MCB.23.2.708-720.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Piñol-Roma S., Dreyfuss G. Cell cycle-regulated phosphorylation of the pre-mRNA-binding (heterogeneous nuclear ribonucleoprotein) C proteins. Mol. Cell. Biol. 1993;13:5762–5770. doi: 10.1128/mcb.13.9.5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou A., Ou A.C., Cho A., Benz E.J., Huang S.-C. Novel splicing factor RBM25 modulates Bcl-x pre-mRNA 5′ splice site selection. Mol. Cell. Biol. 2008;28:5924–5936. doi: 10.1128/MCB.00560-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shalek A.K., Satija R., Adiconis X., Gertner R.S., Gaublomme J.T., Raychowdhury R., Schwartz S., Yosef N., Malboeuf C., Lu D., et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013;498:236–240. doi: 10.1038/nature12172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N.J., Livak K.J., Mikkelsen T.S., Rinn J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shalek A.K., Satija R., Shuga J., Trombetta J.J., Gennert D., Lu D., Chen P., Gertner R.S., Gaublomme J.T., Yosef N., et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature. 2014;509:363–369. doi: 10.1038/nature13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patel A.P., Tirosh I., Trombetta J.J., Shalek A.K., Gillespie S.M., Wakimoto H., Cahill D.P., Nahed B.V., Curry W.T., Martuza R.L., et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dey S.S., Kester L., Spanjaard B., Bienko M., van Oudenaarden A. Integrated genome and transcriptome sequencing of the same cell. Nat. Biotechnol. 2015;33:285–289. doi: 10.1038/nbt.3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagano T., Lubling Y., Stevens T.J., Schoenfelder S., Yaffe E., Dean W., Laue E.D., Tanay A., Fraser P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 2013;502:59–64. doi: 10.1038/nature12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buenrostro J.D., Wu B., Litzenburger U.M., Ruff D., Gonzales M.L., Snyder M.P., Chang H.Y., Greenleaf W.J. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523:486–490. doi: 10.1038/nature14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rotem A., Ram O., Shoresh N., Sperling R.A., Goren A., Weitz D.A., Bernstein B.E. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol. 2015;33:1165–1172. doi: 10.1038/nbt.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY DATA