Type III Interferons Produced by Human Placental Trophoblasts Confer Protection Against Zika Virus Infection (original) (raw)
. Author manuscript; available in PMC: 2017 May 11.
Published in final edited form as: Cell Host Microbe. 2016 Apr 5;19(5):705–712. doi: 10.1016/j.chom.2016.03.008
Summary
During mammalian pregnancy, the placenta acts as a barrier between the maternal and fetal compartments. The recently observed association between Zika virus (ZIKV) infection during human pregnancy and fetal microcephaly and other anomalies suggests that ZIKV may bypass the placenta to reach the fetus. This led us to investigate ZIKV infection of primary human trophoblasts (PHT), which are the barrier cells of the placenta. We discovered that PHT cells from full-term placentas are refractory to ZIKV infection. In addition, medium from uninfected PHT cells protects non-placental cells from ZIKV infection. PHT cells constitutively release the type III interferon (IFN) IFNλ1, which functions in both a paracrine and autocrine manner to protect trophoblast and non-trophoblast cells from ZIKV infection. Our data suggests that for ZIKV to access the fetal compartment, it must evade restriction by trophoblast-derived IFNλ1 and other trophoblast-specific antiviral factors and/or use alternative strategies to cross the placental barrier.
Keywords: Virus, Zika virus, Placenta, Trophoblasts, type III interferon, IFNλ
Graphical Abstract
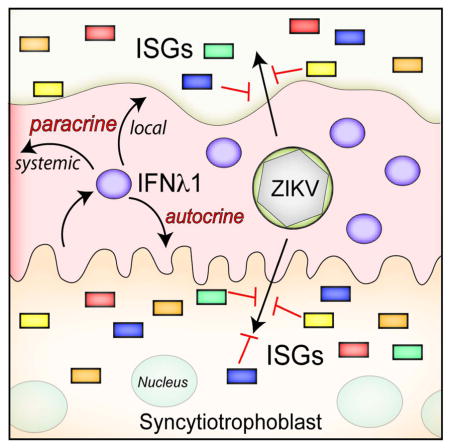
Introduction
In eutherian organisms, the placenta acts as a physical and immunological barrier between the maternal and fetal compartments, and protects the developing fetus from the vertical transmission of viruses. In the human hemochorial placenta, the frontline of fetal protection are the syncytiotrophoblasts, which cover the surfaces of the human placental villous tree and are directly bathed in maternal blood following the establishment of the maternal circulatory system during the later stages of the first trimester.
The mechanisms by which viruses can be transmitted vertically are multifaceted, and can involve entry into the gestational sac via direct hematogenous spread, trophoblastic transcellular or paracellular pathways, transport within immune cells or infected sperm, pre-pregnancy uterine colonization, introduction during invasive procedures during pregnancy, and/or transvaginal ascending infection. The emerging ZIKV pandemic poses a new threat to the developing fetus. While usually causing relatively mild symptoms in non-pregnant individuals, ZIKV infection in Brazil has been associated with increased incidence of microcephaly (Cauchemez et al., 2016; Oliveira Melo et al., 2016; Schuler-Faccini et al., 2016; Ventura et al., 2016a). In addition, ZIKV infections have also been associated with other disorders such as placental insufficiency and fetal growth restriction, ocular disorders, other CNS anomalies, and even fetal death (Brasil et al., 2016; Ventura et al., 2016b).
While direct evidence for a causal relationship between ZIKV infections and the development of abnormal pregnancy outcomes is still emerging, recent reports have directly identified the presence of viral RNA and infectious virus in the placentas, amniotic cavity and brains of fetuses that had developed fetal anomalies (Calvet et al., 2016; Martines et al., 2016; Mlakar et al., 2016). Interestingly, other flaviviruses, such as dengue virus (DENV), which is endemic in the regions of Brazil most impacted by the recent ZIKV outbreak, have not been associated with microcephaly or other congenital disorders, suggesting that ZIKV may exhibit unique mechanism(s) to directly infect and/or bypass the placental barrier, and access the fetal compartment and cause organ-specific damage.
The innate immune system is a primary host defense strategy to suppress viral infections and converges on the induction of interferons (IFNs), which function in autocrine and paracrine manners to upregulate a cadre of other genes, known as interferon stimulated genes (ISGs). The effects of IFNs and ISGs are potent and wide-ranging; they are pro-inflammatory, enhance adaptive immunity, and are directly antiviral (Schneider et al., 2014). In most cell types, type I IFNs, which include IFNα and IFNβ, are the primary IFNs that are generated in response to viral infections. In contrast, cells of epithelial origin mount antiviral responses primarily mediated by type III IFNs, which include IFNλ1-4 (also known as IL-29, IL-28A–C) (Lazear et al., 2015b). The role of IFN signaling in the protection of placental trophoblasts from viral infections is unclear. Previous work has pointed to unidentified IFN(s) present in first trimester human placentas (Lefevre and Boulay, 1993). Ruminants express IFNτ at various stages of gestation (Bazer et al., 1996), and the mouse placenta can produce IFNλs in response to Listeria monocytogenes infection (Bierne et al., 2012).
Here we show that primary human trophoblast (PHT) cells, isolated from full-term placentas, are refractory to infection by two strains of ZIKV, one derived from an African lineage, and one derived from an Asian lineage that exhibits >99% amino acid sequence similarity to strains currently circulating in Brazil (Haddow et al., 2012). We also found that conditioned medium isolated from PHT cells protected non-trophoblast cells from ZIKV infection through the constitutive release of the type III IFN IFNλ1. Our findings thus suggest that for ZIKV to infect syncytiotrophoblasts, it must overcome the restriction imparted by IFNλ1 and other syncytiotrophoblast-specific antiviral factors, and/or gain access to the fetal compartment by a mechanism that does not involve syncytiotrophoblast infection, at least in the latter stages of pregnancy.
Results
PHT cells resist ZIKV infection
To assess the ability of ZIKV to replicate in human placental trophoblasts, we measured the replication of two strains of ZIKV, one of African lineage (Haddow et al., 2012) (MR766, termed ZIKVM hereafter) and one of Asian lineage (Haddow et al., 2012) (FSS13025, termed ZIKVC hereafter) in PHT cells and a panel of trophoblast-derived cell lines including BeWo, JEG-3, and JAR choriocarcinoma cells, and the extravillous trophoblast cell line HTR8/SVneo (Graham et al., 1993). In addition, we compared the level of infection of these cell types by DENV. We also compared the infectivity of these cell types with that of human brain microvascular endothelial cells (HBMEC), which is a cell-based model of the blood-brain barrier (Stins et al., 2001) and is permissive to DENV and both strains of ZIKV (Figure 1A, Figure S1A). We found that BeWo, JEG-3, JAR, and HTR8/SVneo cells supported infection by both ZIKVM and ZIKVC, although BeWo cells were less susceptible to infection by both DENV and ZIKV than the other trophoblast-derived cells lines (Figure 1A and Figure S1A). In contrast, we were unable to detect any evidence of ZIKV or DENV replication in PHT cells by immunofluorescence microscopy (not shown). Consistent with this, we found that PHT cells resisted infection by ZIKVM, ZIKVC, and DENV, as evidenced by very low levels of total viral RNA (vRNA) (Figure S1B and S1C) and the lack of production of the negative strand of vRNA, which is only produced during viral replication (Figure 1B). These results are consistent with our previous observations that PHT cells resist infection by diverse RNA and DNA viruses (Delorme-Axford et al., 2013) and show that ZIKV is unable to replicate efficiently in primary trophoblasts.
Figure 1. ZIKV infects placental trophoblast cell lines, but not PHT cells.

(A), The indicated cell lines were infected with DENV, ZIKVM, or ZIKVC for ~24hrs, fixed, and then stained with anti-dsRNA (J2) antibody. Data are shown as the percent of vRNA positive cells relative to the total number of nuclei (as assessed by DAPI). (B), Levels of DENV, ZIKVM, or ZIKVC negative strand vRNA were assessed by RT-qPCR in HBMEC or PHT cells infected for ~48hrs. (C), HBMEC were exposed to non-conditioned (NCM) PHT medium or conditioned PHT medium (CM, two independent preparations) for ~24hrs and then infected with DENV, DENV, ZIKVM, or ZIKVC. The level of infection was assessed for fluorescence microscopy for dsRNA. Data are shown as the percent of vRNA positive cells relative to the total number of nuclei (as assessed by DAPI). (D), Control HeLa cells, or HeLa cells constitutively expressing a DENV replicon were exposed to NCM or three independent preparations of PHT CM and then the levels of DENV vRNA assessed by RT-qPCR ~24hrs after exposure. In all, data are shown as mean ± standard deviation (*p<0.05, **p<0.01, ***p<0.001).
Conditioned medium isolated from PHT cells protects nonplacental cells from ZIKV infection
In addition to the resistance of PHT cells to ZIKV infection, we found that conditioned medium (CM) isolated from uninfected PHT cells protected non-placental recipient cells from infection by both isolates of ZIKV and DENV (Figure 1C). Interestingly, we found that this protection was lost when CM was added after the establishment of viral replication, as PHT CM exhibited no inhibitory effects on the production of vRNA in cells stably propagating a DENV subgenomic replicon (Figure 1D, Figure S1D–E).
Using microarrays, we found that exposure of human fibrosarcoma HT1080 (2fTGH) cells to PHT CM induced a subset of previously characterized ISGs (Schoggins et al., 2011), which did not occur in HT1080 cells with defective signal transducer and activator of transcription 1 (STAT1; 2fTGH-U3A cells) signaling (McKendry et al., 1991) (Figure 2A and Table S1). We obtained similar results when cells were treated with IFNβ (Figure 2A), as previously described (Shu et al., 2015). We confirmed these results by RT-qPCR in human osteosarcoma U2OS cells that were exposed to PHT CM, which led to the robust induction of two known ISGs, interferon induced protein 44 like (IFI44L) and interferon-induced protein with tetratricopeptide repeats 1 (IFIT1) (Figure 2B) and in human monocyte THP-1 cells as determined by an interferon regulatory factor (IRF)-inducible SEAP reporter assay (Figure S2A). In addition, RNASeq revealed that PHT cells express high levels of ISGs (Figure 2C and Table S2). In contrast, the trophoblast cell line JEG-3 did not endogenously express ISGs (Figure 2C and Tables S2) and CM isolated from these cells did not induce ISGs in non-placental recipient cells (Figure S2B).
Figure 2. Conditioned medium from PHT cells induces ISGs.

(A), A heat map of interferon stimulated genes (ISGs) differentially expressed between control (TGH) or STAT1 signaling deficient (U3A) HT1080 cells exposed to purified IFNβ or PHT CM for 24hrs. (B), RT-qPCR analysis for IFI44L or IFIT1 in U2OS cells exposed to control PHT non-conditioned medium (NCM) or five independent preparations of PHT CM. Data are shown as a fold change from NCM. (C), Heat map of differentially expressed interferon stimulated genes (ISGs) between two cultures of JEG-3 cells and a two preparations of PHT cells (samples 2 and 3 are biological replicates of the same PHT preparation) as assessed by RNASeq (p<0.05). (D), Two preparations of PHT cells were exposed to dimethyl sulfoxide (DMSO) to inhibit cell fusion, CM collected, and then IFI44L induction assessed by RT-qPCR (left y axis). In parallel, the levels of human chorionic gonadotropin (hCG) were determined by ELISA (right y axis). (E), Two preparations of PHT cells were exposed to epidermal growth factor (EGF) to enhance cell fusion, CM collected, and then IFI44L induction assessed by RT-qPCR. (F), BeWo cells were exposed to forskolin to induce fusion, CM collected, and ISG induction in CM-exposed cells assessed by RT-qPCR (for IFI44L, left y axis). In parallel, the levels of hCG were assessed by ELISA (right y axis). In (B), (D–F), data are shown as mean ± standard deviation (*p<0.05, **p<0.01, ***p<0.001, ns not significant). The color intensity in (A) and (C) indicates the level of gene expression (yellow for up-regulation and blue for down-regulation), and grey indicates that no transcripts were detected in that sample.
During culturing in vitro, PHT cells undergo fusion to form syncytiotrophoblasts (Figure S2C) similar to their natural differentiation process in vivo, which can be inhibited by exposing the cultures to dimethyl sulfoxide (DMSO) (Thirkill and Douglas, 1997). We found that attenuation of PHT differentiation by DMSO reduced the ability of PHT CM to induce IFI44L in recipient cells (Figure 2D). Consistent with a role for syncytiotrophoblast fusion in the induction of ISGs, we found that exposure of PHT cells to epidermal growth factor (EGF), which promotes cell-cell fusion of trophoblasts (Morrish et al., 1987), enhanced the ISG inducing properties of PHT CM (Figure 2E). Importantly, ISG induction in recipient cells was specific for PHT CM and did not occur when cells were exposed to CM from the trophoblast-derived cell lines BeWo, JEG-3, JAR, or HTR8 cells, suggesting that this induction is specific for CM derived from primary trophoblasts (Figure 2E and Figure S2B). Furthermore, although BeWo cell fusion can be stimulated by forskolin treatment (Wice et al., 1990), this treatment did not confer ISG-inducing properties to BeWo CM (Figure 2F), suggesting that cell-cell fusion alone is not sufficient to confer ISG inducing properties to trophoblasts. Lastly, we previously showed that PHT-derived exosomes released into PHT CM mediated some of the antiviral properties of PHT CM (Delorme-Axford et al., 2013). We found that CM depleted of vesicles was still capable of inducing ISGs in recipient cells (Figure S2D), indicating that an ISG-inducing pathway is present in PHT CM and bestows antiviral properties independently from PHT-derived exosomes.
PHT cells release the type III IFN IFNλ1
We found by ELISAs that PHT CM contained negligible levels of IFNβ that were comparable to those in control non-CM, but contained IFNλ1, and to a lesser extent, IFNλ2, which was detected in one PHT preparation (Figure 3A). In addition, PHT cells expressed high levels of IFNλ1 mRNA (Figure 3B), which were consistent with the levels induced in non-PHT cells (HBMEC) transfected with the synthetic ligand polyinosinic-polycytidylic acid (poly I:C) to induce IFN production (Figure 3B). In addition, we found that anti-IFNλ /2 neutralizing antibodies partially inhibited the induction of the ISG IFI44L by PHT CM (Figure 3C). Furthermore, although CM isolated from uninfected trophoblast-derived cell lines did not contain detectable levels of IFNλ1 (Figure S3A), we found that these cells potently induced type III IFNs, primarily IFNλ1, in response to infection by Sendai Virus (SeV, Figure 3D) and by both DENV and ZIKV (Figure 3E). In contrast, PHT cells did not induce IFNλ1 or the ISG 2′-5′-Oligoadenylate Synthetase 1 (OAS1) in response to ZIKV, or DENV, infection, yet were highly resistant to infection when compared to JEG-3 cells (Figure 3F and Figure S3B). However, PHT cells do induce both IFNλ1 and ISGs in response to toll like receptor 3 (TLR3) stimulation by poly I:C (Figure S3C). Finally, we found that RNAi-mediated silencing of a subunit of the type III IFN receptor (IL28RA) partially restored ZIKV infection in recipient cells exposed to PHT CM depleted of vesicles (Figure 3G). Collectively, these data point to a direct role for type III IFNs, particularly IFNλ1, in the antiviral signaling of placental syncytiotrophoblasts to restrict viral infections, including ZIKV.
Figure 3. Conditioned medium from PHT cells contains IFNλ1, which is required for ISG induction.

(A), ELISA for IFNβ, IFNλ1, and IFNλ2 in four independent PHT CM preparations (left y axis). In parallel, the extent of ISG induction in each sample was determined by RT-qPCR for the levels of IFI44L induced in U2OS cells exposed to the sample (right y axis). (B), The levels of IFNβ and IFNλ1 mRNA in three preparations of PHT cells was assessed by RT-qPCR. In parallel, IFNβ and IFNλ1 mRNA levels were determined in mock-treated HBMEC, or in HBMEC exposed to 10μg poly I:C (‘floated’ in the medium) for ~24hrs. Data are shown as a fold change from mock-treated HBMEC cells. (C), Level of ISG induction (as assessed by IFI44L RT-qPCR) in U2OS cells exposed to purified IFNλ1, or to three preparations of PHT CM incubated with a non-neutralizing monoclonal antibody (MOPC21) or anti-IFNλ-1-3 neutralizing antibodies. (D), RT-qPCR for IFNβ, IFNλ1, or IFNλ2 in indicated trophoblast cell lines infected with Sendai virus (SeV) for ~24hrs. (E), RT-qPCR for IFNλ1 or IFNλ2 in the indicated trophoblast cell lines infected with DENV or ZIKVM for ~24hrs. (F), RT-qPCR for IFNλ1 or OAS1 in JEG-3 or PHT cells infected with DENV, ZIKVM, or ZIKVC for ~24hrs. (G), ZIKVC infection in HBMEC transfected with control siRNA (CONsi) or IL28RA siRNAs and exposed to PHT conditioned medium depleted of vesicles for ~24hrs prior to infection. In all panels, data are shown as mean ± standard deviation (*p<0.05, **p<0.01, ***p<0.001, ns not significant).
Discussion
The strong association between ZIKV infection in pregnant women with the development of fetal growth restriction and/or CNS and other fetal congenital abnormalities, in addition to the positive culture of ZIKV from feto-placental tissues of affected pregnancies, suggests that ZIKV is capable of gaining access into the intrauterine cavity to directly affect fetal development. Our work presented here suggests that ZIKV is unlikely to access the fetal compartment by its direct replication in placental syncytiotrophoblasts, at least in the later stages of pregnancy, unless ZIKV can bypass the antiviral properties of type III IFNs and other syncytiotrophoblast-derived antiviral pathways during in vivo infection of pregnant women. Because we observed potent protection from ZIKV infection by type III IFNs, specifically IFNλ1, which is constitutively produced by syncytiotrophoblasts, it is likely functioning in an autocrine manner to protect these cells from viral infections. In addition, we show that trophoblast-derived IFNλ1 protects non-placental cells from ZIKV infection in a paracrine manner. A schematic of the human placenta and the mechanisms by which IFNλ 1 protects syncytiotrophoblasts from ZIKV infection is shown in Figure 4. Our work thus provides evidence that ZIKV may not directly infect placental villous syncytiotrophoblasts during later stages of pregnancy, suggesting instead that the virus must either evade the potent type III IFN antiviral signaling pathways generated by these cells and/or bypass these cells through an as-yet-unknown pathway to gain access to the fetal compartment.
Figure 4. Schematic depicting the structure of the human placenta and the role of IFNλ1 in protecting against ZIKV infection.

(A), The intrauterine environment during human pregnancy. Embryonic structures include the villous tree of the human hemochorial placenta and the umbilical cord, which transfers blood between the placenta and the fetus. (B), An overview of a single placental villus. Extravillous trophoblasts invade and anchor the placenta to the maternal decidua and to the inner third of the myometrium. The villous tree consists of both floating and anchoring villi. Multinucleated syncytiotrophoblasts overlie the surfaces of the villous tree and are in direct contact with maternal blood, which fills the intervillous space (IVS) once the placenta is fully formed. Mononuclear cytotrophoblasts are subjacent to the syncytiotrophoblasts and the basement membrane of the villous tree, and serve to replenish the syncytiotrophoblast layer throughout pregnancy. (C), In the work presented here, we show that syncytiotrophoblasts release IFNλ1 that can act in both autocrine and paracrine manners to induce ISGs, which protect against ZIKV, and other viral infections. The paracrine function of IFNλ could work locally within the direct maternal-fetal compartment, or might circulate more systemically to act on other maternal target cells.
Our previous studies implicated a role for trophoblast-specific miRNAs associated with the placental-specific chromosome 19 miRNA cluster (C19MC), contained within PHT-derived exosomes, as part of the antiviral arsenal secreted by PHT cells (Bayer et al., 2015; Delorme-Axford et al., 2013). Indeed, our work presented here demonstrates another facet of the antiviral mechanisms engaged by PHTs to protect the developing fetus. These potent antiviral pathways likely function in parallel to provide multiple mechanisms to protect syncytiotrophoblasts, and other cell types at the maternal-fetal interface, from ZIKV, and other viruses. It is also possible that other as-yet-undiscovered pathways intrinsic to placental trophoblasts provide additional mechanisms to protect these cells from viral infections. While we have not been able to reliably measure IFNλ in the plasma of pregnant women, this may be because IFNλ is below the limits of detection in the expanded plasma volume of pregnant women and/or that the effects of IFNλ are local, affecting trophoblastic and non-trophoblastic placental cells (such as villous fibroblasts) in the immediate vicinity of the feto-placental unit.
Type III IFNs share significant structural homology with members of the IL-10 cytokine family (Gad et al., 2009), but induce ISGs similar to type I IFNs (Kotenko et al., 2003) through a distinct receptor (Sheppard et al., 2003). We found that PHT cells expressed high levels of IFNλ1. Remarkably, IFNλ1 was constitutively released from PHT cells and did not require the activation of antiviral innate immune signaling pathways to become induced. Thus, in addition to studies that implicate an important role for type III IFNs in antiviral signaling in the respiratory and gastrointestinal tracts and the blood-brain barrier (Lazear et al., 2015b), our work directly points to a role for type III IFNs, specifically IFNλ1, in antiviral signaling at the maternal-fetal interface. Although type I IFNs are conserved between mice and humans, there is significant divergence in the type III IFN pathway, where humans express IFNλ1-4 and mice express only IFNλ2 and IFNλ3. PHT cells expressed IFNλ2 at significantly lower levels than IFNλ1 and did not express mRNA for either IFNλ3 or IFNλ4. Thus, in addition to the morphological differences between the human and mouse placentas (Maltepe et al., 2010), these data suggest that the IFNλ1-mediated antiviral properties of placental syncytiotrophoblasts may be distinct between humans and mice, which may complicate the use of the mouse placenta as a model for viral infections of the placenta during human pregnancy.
Another important implication of our work is that cells that do not express the type III IFN receptor, or do not respond robustly to type III IFNs, may be more susceptible to ZIKV infection, particularly at the maternal-fetal interface. In mice, the expression of the alpha subunit of the IFN-lambda receptor (IL-28RA) is restricted to epithelial-derived cells, which respond most robustly to type III IFNs (Sommereyns et al., 2008). Recent evidence also supports a role for type III IFNs in the microvascular endothelium comprising the blood brain barrier (Lazear et al., 2015a). Because syncytiotrophoblasts, and other trophoblasts that are epithelial, are likely protected by the potent stimulation of ISGs in response to their constitutive production of IFNλ1, our data suggest that ZIKV may invade the intrauterine cavity by mechanisms that are independent of direct trophoblast infection. In addition to the trophoblast cell layers, the human placenta is also composed of mesenchymal cells, placental-specific macrophages (termed Hofbauer cells), and fibroblasts located within the villous core between trophoblasts and fetal vessels, that may exhibit differences in their responsiveness to IFNλs. In addition, it is also possible that less differentiated, first trimester trophoblasts as well as extravillous trophoblasts are more permissive than late pregnancy villous trophoblasts to ZIKV infection and/or the antiviral effects of IFNλs. Finally, it is possible that the levels of IFNλ1 vary throughout pregnancy, or between individuals, which could markedly impact the ability of the virus to infect the syncytiotrophoblast cell layer at specific times during pregnancy, or in specific individuals
The rapidly emerging human health crisis associated with the ZIKV epidemic highlights the growing need to identify mechanisms by which ZIKV accesses the fetal compartment. These data will be instrumental in order to design therapeutic measures to limit ZIKV replication and/or spread. Our experimental cell system is directly relevant to the study of congenital ZIKV infections, by defining unique antiviral mechanisms at play in this specialized environment. We provide evidence that ZIKV is unlikely to access the fetal compartment by its direct infection of late pregnancy villous syncytiotrophoblasts, and potentially neighboring cells that express IL28RA, due to the role of type III IFNs in the antiviral defense produced by human trophoblasts and suggests that the virus may circumnavigate these cells or overcome this restriction in vivo in order to bypass the placental barrier.
Experimental Procedures
Culture of primary human trophoblasts
PHT cells were isolated from healthy singleton term placentas using the trypsin-DNAse-dispase/Percoll method as described (Kliman et al., 1986), with previously published modifications under an exempt protocol approved by the institutional review board at the University of Pittsburgh. Patients provided written consent for the use of de-identified and discarded tissues for research purposes upon admission to the hospital. Cells were maintained in DMEM (Sigma) containing 10% FBS (HyClone) and antibiotics at 37°C in a 5% CO2 air atmosphere. Cells were then maintained for 72 h after plating, with cell quality ensured by microscopy and production of human chorionic gonadotropin (hCG), determined by ELISA (DRG international). The cells exhibited a characteristic increase in medium hCG levels as the cytotrophoblasts differentiated into syncytiotrophoblasts.
Cells and viruses
Human osteosarcoma U2OS cells, Vero cells, 2fTGH (STAT1 wild-type) and U3A (STAT1 mutant) fibrosarcoma cells (previously described (McKendry et al., 1991)) were cultured in DMEM supplemented with 10% FBS and antibiotics. BeWo cells were maintained in F12K Kaighn’s modified medium supplemented with 10% FBS and antibiotics. JAR cells and immortalized, human, first-trimester, extravillous trophoblast cells (HTR8/SVneo) were maintained in RPMI 1640 medium supplemented with 10% FBS with antibiotics. Human choriocarcinoma JEG-3 cells were maintained in EMEM medium, supplemented with 10% FBS with antibiotics. Human brain microvascular endothelial cells (HBMEC) were maintained in RPMI 1640 medium supplemented with 10% FBS, 10% NuSerum, MEM vitamins, non-essential amino acids, sodium pyruvate, and antibiotics. HeLa CCL-2 cells were maintained in MEM supplemented with 10% FBS, non-essential amino acids, sodium pyruvate, and antibiotics. Development of HeLa cells stably propagating a DENV subgenomic replicon has been previously described (Ansarah-Sobrinho et al., 2008). Plasmids used to generate stable replicon cells were provided by Theodore Pierson (Viral Pathogenesis Section Laboratory of Viral Diseases, NIH/NIAID). Aedes albopictus midgut C6/36 cells were maintained in DMEM supplemented with 10% FBS and antibiotics at 28°C in a 5% CO2 air atmosphere.
DENV2 16681 and ZIKV FSS13025 (Cambodian origin) were propagated in C6/36 cells, as previously described (Vasilakis et al., 2008). ZIKV MR766 (Ugandan origin) was propagated in Vero cells. Viral titers were determined by fluorescent focus assay, as previously described (Payne et al., 2006), using anti-DENV envelope protein monoclonal antibody 4G2 (provided by Margaret Kielian, Albert Einstein College of Medicine) for DENV and anti-double stranded RNA monoclonal antibody J2 (provided by Saumendra Sarkar, University of Pittsburgh) for ZIKV (specificity of the J2 antibody is shown in Figure S1G). SeV was purchased from Charles River Laboratories. Experiments measuring productive DENV and ZIKV infection were performed with 1 to 3 focus forming units/cell for 24hr, unless otherwise stated, and SeV was used at 100 hemagglutination units/cell for 24hr. Infection was determined by either RT-qPCR or immunofluorescence microscopy, as stated in the figure legends.
Preparation and characterization of conditioned medium
CM samples from PHT cells or other cells were harvested at 72h after plating followed by centrifugation at 800 xg for 5 min. Non-conditioned medium (NCM) was complete PHT medium (described above) that had not been exposed to PHT cells. Recipient cells were exposed to conditioned medium for ~24 h before assays. Vesicle depleted conditioned medium was generated by three centrifugation steps: 2,500 xg for 5 min at room temperature, followed by 12,000 xg for 20min at room temperature, and 100,000 xg for 2 h at 4°C. Antiviral activity of CM preparations was determined in HBMEC exposed to CM for 24 h prior to infection with DENV, ZIKVM, or ZIKVC.
RNA extraction, cDNA synthesis, and Real-Time quantitative PCR
For cellular mRNA analysis, total RNA was extracted using TRI reagent (MRC) or GenElute total RNA miniprep kit (Sigma) according to the manufacturer protocol. RNA samples were treated with RNase-free DNAse (Qiagen or Sigma). Total RNA was reverse transcribed using HiCapacity cDNA synthesis kit (Applied Biosystems) or iScript cDNA synthesis kit (Bio-Rad) according to the manufacturer protocol. Strand specific cDNA was produced with primers targeting the negative RNA strand DENV or ZIKV using iScript Select cDNA Synthesis kit (Bio-Rad). RT-qPCR was performed using SYBR select or iQ SYBR green supermix (BioRad) in a StepOnePlus real-time PCR system (Applied Biosystems), ViiA 7 system (Applied Biosystems), or CFX96 Real-Time system (Bio-Rad). Gene expression was calculated using the 2-delta delta CT method normalized to GAPDH or actin. Primer sequences are located in the Supplemental Experimental Procedures. The specificity of ZIKV and DENV primers were confirmed by RT-qPCR analysis (Figure S1F).
RNASeq and microarray analyses
RNASeq from JEG-3 and PHT cells was performed as previously described (McConkey CA, 2016). Briefly, libraries were prepared with the NEB Ultra Library Preparation kit and library quality was determined using the Qubit assay and the Agilent 2100 Bioanalyzer. Sequencing was performed with the Illumina HiSeq2500 rapid-run mode on one flow cell (two lanes). CLC Genomics Workbench 8 (Qiagen) was used to process, normalize, and map sequence data to the human reference genome (hg19). Differentially expressed genes were identified using DESeq2 (Love et al., 2014) with a significance cut-off of 0.05, and heat maps were generated using MeViewer software. Files associated with RNASeq studies have been deposited into Sequence Read Archive (SRA), accession SRP067137.
We used high-throughput microarray analysis as previously described (Shu et al., 2015), to screen for transcriptional changes in control (2fTGH) vs. STAT1 signaling deficient (U3A) HT1080 cells, both exposed to 100U of purified IFNβ (PBL) or PHT CM. for 24 h. In parallel, mock-treated 2fTGH and U3A were also included and were used to identify differentially expressed genes in IFNβ- and CM-treated cells. Datasets related to these arrays have been deposited in GEO (dataset accession GSE72342).
Statistics
Experiments were performed at least three times as indicated in the figure legends or as detailed. Data are presented as mean ± standard deviation. Except were specified, a Student’s t-test was used to determine statistical significance for virus infection assays when two sets were compared, and one-way ANOVA with Bonferroni’s correction used for post hoc multiple comparisons was used to determine statistical significance. Specific p-values are detailed in the figure legends.
Supplementary Material
1
2
3
Acknowledgments
We thank Saumendra Sarkar and Donald Burke (University of Pittsburgh) for reagents and/or advice, Ted Pierson (NIH/NIAID) and Margaret Kielian (Albert Einstein College of Medicine) for DENV reagents, and Kwang Sik Kim (Johns Hopkins University) for the HBMEC used in the study. This project was supported by NIH R01-AI081759 [C.B.C.], NIH R01-HD075665 [C.B.C. and Y.S.], and T32-AI049820 [N.J.L.]. In addition, C.B.C. and S.C. are supported by Burroughs Wellcome Investigators in the Pathogenesis of Infectious Disease Awards.
Footnotes
Author Contributions
A.B., N.J.L., Y.O., J.C.B, S.M., and C.B.C. performed experiments, A.B., N.J.L., Y.S., and C.B.C. analyzed data, E.M. and S.C. contributed essential reagents, and A.B., N.J.L, S.C., Y.S., and C.B.C. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature cited
- Ansarah-Sobrinho C, Nelson S, Jost CA, Whitehead SS, Pierson TC. Temperature-dependent production of pseudoinfectious dengue reporter virus particles by complementation. Virology. 2008;381:67–74. doi: 10.1016/j.virol.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer A, Delorme-Axford E, Sleigher C, Frey TK, Trobaugh DW, Klimstra WB, Emert-Sedlak LA, Smithgall TE, Kinchington PR, Vadia S, et al. Human trophoblasts confer resistance to viruses implicated in perinatal infection. Am J Obstet Gynecol. 2015;212:71, e71–78. doi: 10.1016/j.ajog.2014.07.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazer FW, Spencer TE, Ott TL. Placental interferons. Am J Reprod Immunol. 1996;35:297–308. doi: 10.1111/j.1600-0897.1996.tb00485.x. [DOI] [PubMed] [Google Scholar]
- Bierne H, Travier L, Mahlakoiv T, Tailleux L, Subtil A, Lebreton A, Paliwal A, Gicquel B, Staeheli P, Lecuit M, et al. Activation of type III interferon genes by pathogenic bacteria in infected epithelial cells and mouse placenta. PLoS One. 2012;7:e39080. doi: 10.1371/journal.pone.0039080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasil P, Pereira JP, Jr, Raja Gabaglia C, Damasceno L, Wakimoto M, Ribeiro Nogueira RM, Carvalho de Sequeira P, Machado Siqueira A, Abreu de Carvalho LM, Cotrim da Cunha D, et al. Zika Virus Infection in Pregnant Women in Rio de Janeiro - Preliminary Report. N Engl J Med. 2016 doi: 10.1056/NEJMoa1602412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvet G, Aguiar RS, Melo AS, Sampaio SA, de Filippis I, Fabri A, Araujo ES, de Sequeira PC, de Mendonca MC, de Oliveira L, et al. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: a case study. Lancet Infect Dis. 2016 doi: 10.1016/S1473-3099(16)00095-5. [DOI] [PubMed] [Google Scholar]
- Cauchemez S, Besnard M, Bompard P, Dub T, Guillemette-Artur P, Eyrolle-Guignot D, Salje H, Van Kerkhove MD, Abadie V, Garel C, et al. Association between Zika virus and microcephaly in French Polynesia, 2013–15: a retrospective study. Lancet. 2016 doi: 10.1016/S0140-6736(16)00651-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorme-Axford E, Donker RB, Mouillet JF, Chu T, Bayer A, Ouyang Y, Wang T, Stolz DB, Sarkar SN, Morelli AE, et al. Human placental trophoblasts confer viral resistance to recipient cells. Proc Natl Acad Sci U S A. 2013;110:12048–12053. doi: 10.1073/pnas.1304718110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gad HH, Dellgren C, Hamming OJ, Vends S, Paludan SR, Hartmann R. Interferon-lambda is functionally an interferon but structurally related to the interleukin-10 family. J Biol Chem. 2009;284:20869–20875. doi: 10.1074/jbc.M109.002923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham CH, Hawley TS, Hawley RG, MacDougall JR, Kerbel RS, Khoo N, Lala PK. Establishment and characterization of first trimester human trophoblast cells with extended lifespan. Experimental cell research. 1993;206:204–211. doi: 10.1006/excr.1993.1139. [DOI] [PubMed] [Google Scholar]
- Haddow AD, Schuh AJ, Yasuda CY, Kasper MR, Heang V, Huy R, Guzman H, Tesh RB, Weaver SC. Genetic characterization of Zika virus strains: geographic expansion of the Asian lineage. PLoS Negl Trop Dis. 2012;6:e1477. doi: 10.1371/journal.pntd.0001477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliman HJ, Nestler JE, Sermasi E, Sanger JM, Strauss JF., 3rd Purification, characterization, and in vitro differentiation of cytotrophoblasts from human term placentae. Endocrinology. 1986;118:1567–1582. doi: 10.1210/endo-118-4-1567. [DOI] [PubMed] [Google Scholar]
- Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, Langer JA, Sheikh F, Dickensheets H, Donnelly RP. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- Lazear HM, Daniels BP, Pinto AK, Huang AC, Vick SC, Doyle SE, Gale M, Jr, Klein RS, Diamond MS. Interferon-lambda restricts West Nile virus neuroinvasion by tightening the blood-brain barrier. Sci Transl Med. 2015a;7:284ra259. doi: 10.1126/scitranslmed.aaa4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazear HM, Nice TJ, Diamond MS. Interferon-lambda: Immune Functions at Barrier Surfaces and Beyond. Immunity. 2015b;43:15–28. doi: 10.1016/j.immuni.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefevre F, Boulay V. A novel and atypical type one interferon gene expressed by trophoblast during early pregnancy. J Biol Chem. 1993;268:19760–19768. [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltepe E, Bakardjiev AI, Fisher SJ. The placenta: transcriptional, epigenetic, and physiological integration during development. J Clin Invest. 2010;120:1016–1025. doi: 10.1172/JCI41211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martines RB, Bhatnagar J, Keating MK, Silva-Flannery L, Muehlenbachs A, Gary J, Goldsmith C, Hale G, Ritter J, Rollin D, et al. Notes from the Field: Evidence of Zika Virus Infection in Brain and Placental Tissues from Two Congenitally Infected Newborns and Two Fetal Losses - Brazil, 2015. MMWR Morb Mortal Wkly Rep. 2016;65:159–160. doi: 10.15585/mmwr.mm6506e1. [DOI] [PubMed] [Google Scholar]
- McConkey CA, D-AE, Nickerson CA, Kim KS, Sadovsky Y, Boyle JP, Coyne CB. A three-dimensional culture system recapitulates placental syncytiotrophoblast development and microbial resistance. Science Advances. 2016;2 doi: 10.1126/sciadv.1501462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKendry R, John J, Flavell D, Muller M, Kerr IM, Stark GR. High-frequency mutagenesis of human cells and characterization of a mutant unresponsive to both alpha and gamma interferons. Proc Natl Acad Sci U S A. 1991;88:11455–11459. doi: 10.1073/pnas.88.24.11455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlakar J, Korva M, Tul N, Popovic M, Poljsak-Prijatelj M, Mraz J, Kolenc M, Resman Rus K, Vesnaver Vipotnik T, Fabjan Vodusek V, et al. Zika Virus Associated with Microcephaly. N Engl J Med. 2016 doi: 10.1056/NEJMoa1600651. [DOI] [PubMed] [Google Scholar]
- Morrish DW, Bhardwaj D, Dabbagh LK, Marusyk H, Siy O. Epidermal growth factor induces differentiation and secretion of human chorionic gonadotropin and placental lactogen in normal human placenta. J Clin Endocrinol Metab. 1987;65:1282–1290. doi: 10.1210/jcem-65-6-1282. [DOI] [PubMed] [Google Scholar]
- Oliveira Melo AS, Malinger G, Ximenes R, Szejnfeld PO, Alves Sampaio S, Bispo de Filippis AM. Zika virus intrauterine infection causes fetal brain abnormality and microcephaly: tip of the iceberg? Ultrasound Obstet Gynecol. 2016;47:6–7. doi: 10.1002/uog.15831. [DOI] [PubMed] [Google Scholar]
- Payne AF, Binduga-Gajewska I, Kauffman EB, Kramer LD. Quantitation of flaviviruses by fluorescent focus assay. J Virol Methods. 2006;134:183–189. doi: 10.1016/j.jviromet.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler-Faccini L, Ribeiro EM, Feitosa IM, Horovitz DD, Cavalcanti DP, Pessoa A, Doriqui MJ, Neri JI, Neto JM, Wanderley HY, et al. Possible Association Between Zika Virus Infection and Microcephaly - Brazil, 2015. MMWR Morb Mortal Wkly Rep. 2016;65:59–62. doi: 10.15585/mmwr.mm6503e2. [DOI] [PubMed] [Google Scholar]
- Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, Kuestner R, Garrigues U, Birks C, Roraback J, et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol. 2003;4:63–68. doi: 10.1038/ni873. [DOI] [PubMed] [Google Scholar]
- Shu Q, Lennemann NJ, Sarkar SN, Sadovsky Y, Coyne CB. ADAP2 Is an Interferon Stimulated Gene That Restricts RNA Virus Entry. PLoS Pathog. 2015;11:e1005150. doi: 10.1371/journal.ppat.1005150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommereyns C, Paul S, Staeheli P, Michiels T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008;4:e1000017. doi: 10.1371/journal.ppat.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stins MF, Badger J, Sik Kim K. Bacterial invasion and transcytosis in transfected human brain microvascular endothelial cells. Microb Pathog. 2001;30:19–28. doi: 10.1006/mpat.2000.0406. [DOI] [PubMed] [Google Scholar]
- Thirkill TL, Douglas GC. Differentiation of human trophoblast cells in vitro is inhibited by dimethylsulfoxide. J Cell Biochem. 1997;65:460–468. doi: 10.1002/(sici)1097-4644(19970615)65:4<460::aid-jcb2>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Vasilakis N, Tesh RB, Weaver SC. Sylvatic dengue virus type 2 activity in humans, Nigeria, 1966. Emerg Infect Dis. 2008;14:502–504. doi: 10.3201/eid1403.070843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura CV, Maia M, Bravo-Filho V, Gois AL, Belfort R., Jr Zika virus in Brazil and macular atrophy in a child with microcephaly. Lancet. 2016a;387:228. doi: 10.1016/S0140-6736(16)00006-4. [DOI] [PubMed] [Google Scholar]
- Ventura CV, Maia M, Ventura BV, Linden VV, Araujo EB, Ramos RC, Rocha MA, Carvalho MD, Belfort R, Jr, Ventura LO. Ophthalmological findings in infants with microcephaly and presumable intra-uterus Zika virus infection. Arq Bras Oftalmol. 2016b;79:1–3. doi: 10.5935/0004-2749.20160002. [DOI] [PubMed] [Google Scholar]
- Wice B, Menton D, Geuze H, Schwartz AL. Modulators of cyclic AMP metabolism induce syncytiotrophoblast formation in vitro. Experimental cell research. 1990;186:306–316. doi: 10.1016/0014-4827(90)90310-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1
2
3