DNA Sequence Alignment during Homologous Recombination (original) (raw)
Abstract
Homologous recombination allows for the regulated exchange of genetic information between two different DNA molecules of identical or nearly identical sequence composition, and is a major pathway for the repair of double-stranded DNA breaks. A key facet of homologous recombination is the ability of recombination proteins to perfectly align the damaged DNA with homologous sequence located elsewhere in the genome. This reaction is referred to as the homology search and is akin to the target searches conducted by many different DNA-binding proteins. Here I briefly highlight early investigations into the homology search mechanism, and then describe more recent research. Based on these studies, I summarize a model that includes a combination of intersegmental transfer, short-distance one-dimensional sliding, and length-specific microhomology recognition to efficiently align DNA sequences during the homology search. I also suggest some future directions to help further our understanding of the homology search. Where appropriate, I direct the reader to other recent reviews describing various issues related to homologous recombination.
Keywords: biophysics, DNA recombination, DNA repair, homologous recombination, single-molecule biophysics, Dmc1, Rad51, RecA, biochemistry, homology search
Homologous Recombination
Homologous recombination enables the exchange of genetic information between different DNA molecules and is a major driving force in evolution. Homologous recombination contributes to double-strand DNA break (DSB)2 repair, the rescue of stalled or collapsed replication forks, programmed and aberrant chromosomal rearrangements, horizontal gene transfer, and meiosis (1–5). The importance of homologous recombination is underscored by the findings that defects in key recombination proteins can result in a loss of genome integrity and lead to gross chromosomal rearrangements that are a hallmark of cancer. During recombination, a presynaptic ssDNA is paired with the complementary strand of a homologous dsDNA, resulting in the displacement of the noncomplementary strand from the duplex to generate a D-loop intermediate (Fig. 1) (6–8). This intermediate can be channeled through a number of alternative pathways, any of which can allow for the repair of the originally broken DNA molecule using information derived from the template (1, 9, 10).
FIGURE 1.

Overview of homologous recombination. A, simplified schematic highlighting the early stages of recombination. DSBs are resected to yield long 3′ ssDNA overhangs that serve as the binding sites for Rad51/RecA DNA recombinases. The recombinases form long filaments on the ssDNA that are referred to as the presynaptic complex. The presynaptic complex then searches for a homologous DNA and pairs the processed ssDNA overhang with its homologous partner to generate a D-loop intermediate. B, RecA-ssDNA crystal structure highlighting the base triplet organization of the presynaptic ssDNA (34). Figure adapted with permission from Ref. 57.
Key reactions in homologous recombination are catalyzed by the Rad51/RecA family DNA recombinases, which are ATP-dependent proteins that form helical filaments on DNA (6–8, 11). These recombinases are broadly conserved, and prominent family members include bacterial RecA, the archaeal protein RadA, and the eukaryotic recombinases Rad51 and Dmc1. RecA is the archetypal recombinase originally identified in genetic screens for Escherichia coli mutants defective in recombination by Clark and Margulies in 1965 (12). This discovery set the stage for years of investigation into the RecA protein, including its biochemical purification and characterization by the Radding, Howard-Flanders, Lehman, and Roberts laboratories (13–20).
The importance of this early work on bacterial recombination was underscored by studies in 1992 from the Ogawa and Kleckner groups (21, 22), which identified the first eukaryotic Rad51 and Dmc1 recombinases, respectively, with amino acid sequence homology to RecA. The identification of these eukaryotic recombinases provided the crucial confirmation that recombination proteins were conserved throughout biology, and enabled biochemical study of eukaryotic recombination. Subsequent work from the Sung and West laboratories (23, 24) demonstrated that yeast and human Rad51 catalyze the same types of strand exchange reactions as RecA, providing a clear indication that the prokaryotic and eukaryotic recombinases promoted recombination through conserved mechanisms. Although Dmc1 proved more recalcitrant to biochemical analysis, this meiosis-specific recombinase was eventually shown to catalyze strand exchange (25). Interestingly, Dmc1 is thought to have diverged from Rad51 shortly after the emergence of eukaryotes (26, 27), although the reason why most eukaryotes require a specialized meiotic recombinase for meiosis remains unclear (28, 29).
Electron microscopy studies showed that RecA, Rad51, and Dmc1 all form extended polymers on DNA and stretch the bound DNA by ∼50% relative to the contour length of normal B-form DNA (23, 30–33). It was often assumed that the DNA was uniformly extended, but no one could obtain diffraction quality crystals of any recombinase bound to DNA. A major breakthrough came from the Pavletich group (34), which in 2008 reported crystal structures of RecA-ssDNA and RecA-dsDNA pre- and post-synaptic complexes, revealing that the DNA was organized into near B-form base triplets separated by ∼8 Å between adjacent triplets (Fig. 1B). We refer to this unique DNA architecture as RS-DNA (Rad51/RecA stretched-DNA) to help distinguish it from other forms of mechanically stretched DNA (35).
We have learned much about the basic biochemical and biophysical features of homologous recombination since the identification of the first recA mutants in 1965. We know now what recombinases are involved, what types of reactions they catalyze, and the genetic consequence of defects in these proteins, and we even have atomic level details for some proteins and complexes. Despite our depth of understanding, many questions remain, and of central importance are questions regarding reaction dynamics and transient intermediates that are essential to any biochemical pathway.
The Homology Search
In 1979, Charles Radding wrote, “Nothing is more intriguing about homologous recombination than its beginning” (13). For our purposes, we will consider the “beginning” as what takes place during the alignment of DNA strands during the homology search. The remainder of this minireview will attempt to describe our current understanding of the homology search, and I also direct the reader to excellent reviews, each providing a different perspective on the topic (36–39).
The homology search underlies all homologous recombination reactions, and its importance can be understood by recognizing that misalignment by even a single base pair can potentially render crucial genetic information inaccessible. The presynaptic complex can be considered a large site-specific DNA binding entity, whose specificity is defined by the sequence of the bound ssDNA. Therefore the principles derived from studies of the biological target search processes of simpler site-specific DNA-binding proteins also apply to the homology search. Our understanding of these types of search problems goes back to early experimental work of Riggs et al. (40), who in 1970 demonstrated that the lac repressor could locate target sites more quickly than expected based on random diffusion. This surprising observation prompted Peter von Hippel, Otto Berg, and colleagues (41, 42) to establish three models to explain how DNA-binding proteins might locate target sites. These hypothetical mechanisms include (i) hopping, where the protein moves via a series of microscopic dissociation and rebinding events; (ii) one-dimensional diffusion or sliding, which involves a random walk along the DNA without dissociation; and (iii) intersegmental transfer, involving movement from one site to another via a looped intermediate. Collectively referred to as facilitated diffusion, these models laid the groundwork for most subsequent target search studies.
Early Biochemical Insights
The availability of purified RecA led to rapid advances in our understanding of what reactions the protein could catalyze and insights into how these activities might be related to genetic recombination. As highlighted below, several early efforts were directed toward understanding the homology search.
Many of the earliest attempts to study the homology search were made by Radding and colleagues (43–46). Through a series of studies using plasmid-length substrates, they reported that strand exchange proceeded through an intermediate referred to as a “coaggregate,” composed of a stable intermeshed network of RecA presynaptic complexes and dsDNA, and that the search itself involved facilitated diffusion within these coaggregates. The Lehman and Kowalczykowski laboratories (18, 47, 48) revealed that RecA does not require ATP hydrolysis to promote strand exchange, demonstrating that the search process must be driven by thermal energy. Camerini-Otero and colleagues (49) later found that RecA can form stable pairing interactions with homologous substrates as short as 8 bp, providing perhaps the first hint that RecA might employ a length-based recognition mechanism to help align homologous sequences (see below). Finally, the Adzuma laboratory (50) employed a novel approach based on tandem arrays of homologous bindings to ask whether one-dimensional sliding might contribute to the homology search. The premise of these assays was straightforward: if sliding contributed to the search, then there should be preferential recognition of the outermost targets within a tandem array, whereas a search based upon random three-dimensional diffusion would allow efficient recognition for any site regardless of its position within the array. These experiments convincingly ruled out the involvement of extensive one-dimensional sliding during the homology search.
In sum, these early investigations supported a search mechanism involving: (i) facilitated diffusion within large nucleoprotein networks; (ii) no need for ATP hydrolysis; (iii) the possibility of length-based sequence recognition; and (iv) the absence of long-distance one-dimensional sliding. Given these observations, and considering the polyvalent nature of the presynaptic complex, it seemed plausible that the homology search might take place via the original intersegmental transfer model of Berg and von Hippel (41, 42). However, definite proof of this mechanism would need to await development of new technologies capable of watching the search take place in real time.
Single-molecule Studies
Following the early biochemical work, further advances in our understanding of the homology search came much more slowly. This can perhaps be ascribed to two reasons: first, there were (and remain) many other exciting aspects of recombination that warranted investigation; and second, target search problems are very difficult to study because the relevant intermediates are both transient and heterogeneous. However, the advent of single-molecule approaches, which have the unique capacity to probe transient and heterogeneous intermediates, led to resurgent interest in the homology search, and the common theme among the studies described below is that they sought to observe search intermediates using optical microscopy.
One of the first single molecule studies of the homology search came from Forget and Kowalczykowski (51). These studies first sought to visualize fluorescently labeled RecA-ssDNA presynaptic complexes (430 or 1,762 nt in length) interacting with dsDNA (λ phage DNA; 48,502 bp) tethered by either one or two ends to the surface of a sample chamber. Initial experiments revealed that homologous pairing interactions could only be detected with dsDNA molecules that were tethered by one end to the sample chamber surface (no homologous pairing interactions were found for extended dsDNAs tethered by both ends), suggesting that a successful homology search required randomly coiled dsDNA. This hypothesis was verified by elegant microscopy experiments that visualized RecA-ssDNA presynaptic complexes as they searched for homology within a dsDNA whose ends were held within a dual-beam optical trap, which was used to manipulate the extension of the dsDNA (Fig. 21_A_) (51, 52). In addition, this study identified the existence of intermediates involving multiple, simultaneous contacts between the presynaptic complex and the dsDNA via along-distance looping interactions. Taken together, these experiments indicated that the homology search required a collapsed dsDNA, allowing interactions to take place within three-dimensional space through a mechanism identical with the original intersegmental transfer model from von Hippel and colleagues (41, 42). This crucial finding remains perhaps the clearest experimental example of any biological target search based upon intersegmental transfer.
FIGURE 2.
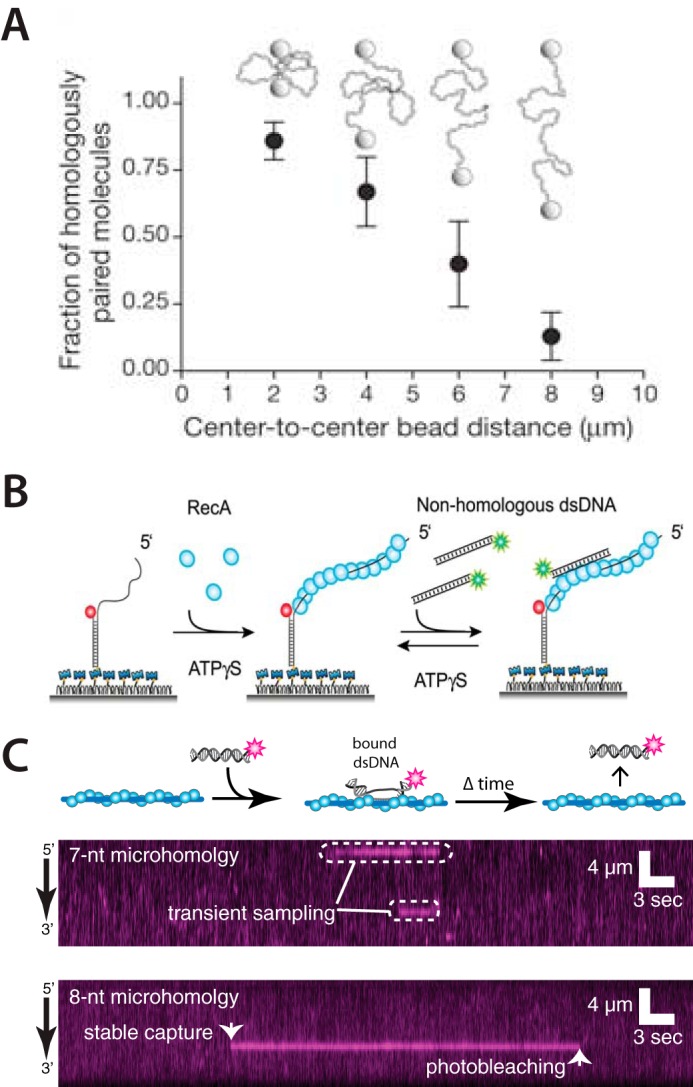
Single-molecule optical microscopy studies of the homology search. A, example illustrating how modulating dsDNA extension in a dual-beam optical trap can affect the efficiency of DNA pairing interactions during homologous recombination. Adapted with permission from Ref. 51. Error bars represent the standard error of the mean from multiple experiments. B, schematic illustration of a singe-molecule FRET measurement of the homology search. Presynaptic complexes labeled with an acceptor fluorophore (red) are anchored to a surface. Duplex DNA labeled with a donor fluorophore (green) is then injected into the sample chamber. Fluorescence from the acceptor is detected when the donor-labeled dsDNA binds to the presynaptic complex. Dynamic behaviors of the resulting intermediates are revealed as distance-dependent changes in the FRET signal. ATPγS, adenosine 5′-_O_-(thiotriphosphate). Adapted with permission from Ref. 53. C, examples of kymographs from a DNA curtain assay using fluorescently tagged dsDNA fragments. Transient binding events are detected with DNA fragments containing ≤7 nt of microhomology, and more stable events are observed for fragments with ≥8 nt of microhomology. Adapted with permission from Ref. 55.
Shortly thereafter, the laboratory of Taekjip Ha (53) demonstrated that one-dimensional sliding over short distances may contribute to the alignment of DNA sequences by RecA. These experiments used single molecule fluorescence resonance energy transfer (smFRET) to show that short nonhomologous dsDNA fragments could slide along a RecA presynaptic filament (Fig. 2B) (53, 54). The resulting data revealed a diffusion coefficient of ∼0.9 × 10−3 μm2/s, corresponding to a sliding distance of only ∼60–300 bp, which was in good agreement with prior bulk biochemical results (50). These authors also used a clever assay with two short homologous sequences embedded within the same presynaptic ssDNA, allowing the dsDNA to be aligned into either of two positions. Remarkably, they detected two distinct FRET signals, and when these homologous sequences were between 5 and 7 nt, the dsDNA could rapidly oscillate between the two sites. Interestingly, when the length of homology was increased to 8 nt, the dsDNA could again align at either of the two sites, but could no longer oscillate between the sites. This finding was consistent with earlier bulk biochemical assays from the Camerini-Otero group (49) showing that 8 nt of homology was sufficient for stable dsDNA capture by the RecA presynaptic complex.
Most recently, my colleagues and I have used DNA curtains to visualize Saccharomyces cerevisiae Rad51, human Rad51, S. cerevisiae Dmc1, human Dmc1, and E. coli RecA presynaptic filaments as they sampled short (70-bp) fluorescently tagged dsDNA molecules with short tracts of sequence microhomology complementary to the presynaptic ssDNA (Fig. 2C) (55–57). These studies provided further evidence that stable dsDNA binding required an 8-nt tract of microhomology, suggesting that this 8-nt requirement is a fundamental feature of the Rad51/RecA family of recombinases. This work also revealed that all five recombinases rapidly sample and reject sequences bearing fewer than 8 nt of microhomology. These intermediates exhibited power-law kinetics, implying the existence of a diverse ensemble of nonspecific binding states, rather than a single nonspecifically bound species, as the presynaptic complex interrogates different nonhomologous sequences. A key conclusion from this work, also predicted from several theoretical studies (see below), was that the Rad51/RecA family of recombinases reduces search complexity by confining stable search intermediates to defined length tracts of microhomology that have a high probability of being the “correct” homologous target (Fig. 3A).
FIGURE 3.

Homology search model. A, graphical representation of microhomology length versus abundance as calculated for the S. cerevisiae genome. Sequences ≤7 nt in length can be ignored during the homology search, whereas longer sequences, which are far less abundant, are more carefully scrutinized. Adapted with permission from Ref. 55. B, the search process involves multiple contact points between the presynaptic complex and the dsDNA molecule that is being interrogated for homology. The most stable contacts bear ≥8 nt of microhomology, allowing the presynaptic complex to probe flanking sequences for additional homology. Contacts with ≤7 nt of microhomology are rapidly released, ensuring that the search is focused only on sequences with a high probability of being the correct target. Each individual contact can slide short distances, and release of a single contact allows the presynaptic complex to sample a different region of the dsDNA for sequence homology. For clarity, the proteins and flanking dsDNA are omitted from the presynaptic complex.
These single molecule studies, together with the preceding bulk biochemical studies, help present a unified picture of the basic principles that guide sequence alignment during the early phases of genetic recombination involving a combination of (i) intersegmental transfer, (ii) short-distance one-dimensional sliding, and (iii) a reduction in search complexity allowing the presynaptic complex to kinetically ignore tracts of microhomology less than 8 nt in length (Fig. 3B).
These studies also point toward new avenues of investigation that should help expand our understanding of the homology. For instance, does ATP hydrolysis play any role at all in the homology search? What are the orientation and footprint of the dsDNA bound during each sliding event? How does the stiffness of the presynaptic complex, which has a persistence length of ∼800 nm, impact its interactions with dsDNA during intersegmental transfer? How does chromosome structure impact the search, and how might the search be augmented by the presence of other accessory proteins that are necessary for recombination? What is the temporal relationship between DNA end processing, presynaptic filament assembly, and the homology search? Can the search take place concurrently with end processing (i.e. as the recombinase is being loaded onto the ssDNA), or are these truly separate stages of the reaction, with the search only taking place once end processing is completed? All of these questions might be accessible to single molecule approaches, or other emerging technologies.
Theoretical Considerations
Despite recent experimental advances, we still do not have a full grasp of the molecular details that make genetic recombination possible. For instance, we do not understand how recombinases access sequence information within a bound dsDNA molecule and then compare this information to the presynaptic ssDNA. Although base flipping is likely to be involved (58, 59), exactly how this might take place remains poorly understood. Again, our lack of understanding of this process can be traced to the transient and heterogeneous nature of the underlying intermediates, and the relevant intermediates cannot be accessed by typical experimental methodologies. For this reason, it will be crucial to integrate experimental findings with detailed theoretical models that can help reveal facets of genetic recombination that are otherwise inaccessible. Here I very briefly touch upon a few examples of how theory and computational modeling have impacted our understanding of the homology search.
Several experimental studies have reported an 8-nt minimum length requirement for stable dsDNA binding by the presynaptic complex (49, 53, 55), raising the question of “Why eight?” The importance of length-dependent sequence recognition as a means for rapidly rejecting incorrect sequences was first recognized by Charles Thomas, Jr. (60), who in 1966 suggested that it would be beneficial for recombination to take place between nonrepetitive minimal recognition lengths composed of “words” that can be uniquely identified within the genome, and these original concepts have been further elaborated by several groups (55, 61–63). Simply put, longer sequences are less common than shorter sequences, so there is a benefit to search for these longer sequences, which will also have a higher probability of being the correct homologous target (Fig. 3A). These simple mathematical considerations illustrate why minimal recognition lengths can be beneficial, but they do not address why 8 nt seems to be preferred.
A compelling physical explanation for this length-dependent recognition mechanism comes from the recent work of Prentiss and colleagues (39, 64). Using molecular dynamics simulations, these researchers identified an initial binding intermediate in which two adjacent bases, each from three successive triplets (i.e. eight contiguous bases in total) within a dsDNA molecule, had flipped open and paired with the presynaptic ssDNA (Fig. 4A). The third base of each triplet remained unpaired because a conserved DNA-binding loop within the RecA presynaptic complex (Loop 2) blocked formation of a complete triplet. This result provides crucial insights into the structure of the initial binding intermediate and also suggests an explanation for why 8 nt is the preferred minimal recognition length. In addition, this work indicates that the L2 loop must reorient to allow triplet formation. It is tempting to speculate that initial pairing with the correct 8-nt site will result in the reorientation of L2 and that this conformation transition may propagate along the interior of the presynaptic complex, ensuring that subsequent base triplet interactions can form unimpeded as longer regions of DNA are paired by the recombinases.
FIGURE 4.
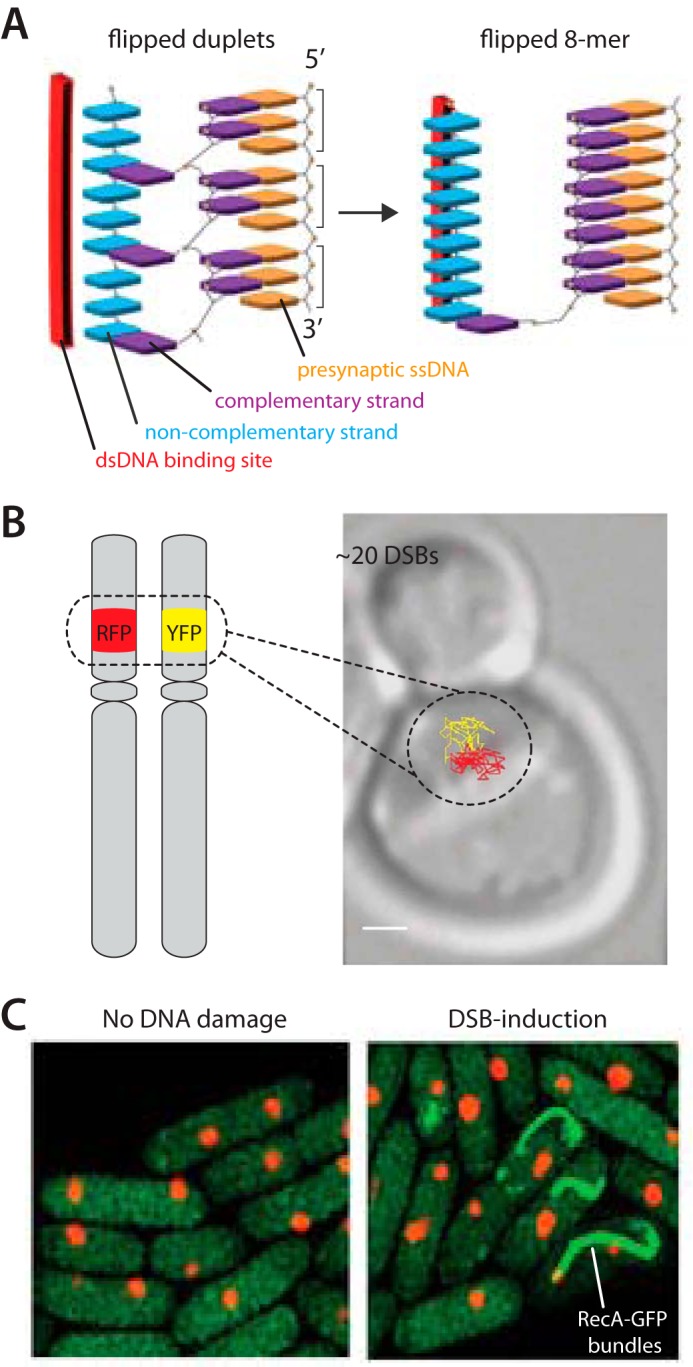
Theoretical studies and in vivo observations of the homology search. A, schematic of the duplet pairing interactions observed in molecular dynamics simulations and proposed transition leading to a more stable binding configuration. Adapted with permission from Ref. 64. B, live cell microscopy assay to visualize DSB-dependent motion of RFP- and YFP-tagged chromosomal loci. The yellow and red trajectories superimposed upon a differential interference contrast image of the cell reveal diffusive motions of the DNA after γ irradiation. Adapted with permission from Ref. 85. C, live cell microscopy revealing the existence of DSB-dependent RecA bundles (green). Adapted with permission from Ref. 94.
Important insights also arise from considering how the extended structure of RS-DNA may impact the mechanism by which the presynaptic complex will interrogate sequence information buried within dsDNA. For instance, the modeling and experimental studies of Prentiss and colleagues (65, 66) have revealed that upon binding to the presynaptic complex, the two strands of the incoming dsDNA will be stretched to differing extents: the noncomplementary strand will be stretched a bit more than the complementary strand. This unequal extension would help open the dsDNA to promote pairing interactions between the complementary strand and the presynaptic ssDNA.
Savir and Tlusty (67) have also examined the potential relationship between RS-DNA extension and pairing fidelity. Their work suggests that the deformation necessary to extend B-DNA to match the extension of RS-DNA may enhance the ability of the presynaptic complex to reject nonhomologous sequences during the homology search. They propose that the energetic penalty associated with dsDNA extension reduces the binding free energies of successive sets of base triplets, and that this deformation-dependent reduction in binding free energy is optimized against the gain in binding free energy for a correct triplet. In contrast, mispaired triplets are not sufficiently stable to overcome the energetic penalty associated with dsDNA extension, allowing the presynaptic complex to quickly discriminate against nonhomologous triplets.
These few simple examples help illustrate how theory and modeling can provide crucial insights into recombination that cannot necessarily be accessed by experiment, and the continued combination of theory and experiment will be necessary for developing a deeper understanding of genetic recombination.
Homology Searches in Living Cells
Recent studies have begun probing how recombination takes place in vivo, and these experiments serve to underscore that regulatory cofactors, accessory proteins, chromatin, and chromosome organization must impact the homology search in ways that we do not yet fully understand. Several excellent reviews are available describing issues related to the in vivo homology search (36–38), and here I briefly describe a few interesting observations that perhaps warrant further exploration.
Although nearly all in vitro studies of the homology search use just a single protein (i.e. RecA, Rad51, or Dmc1), it is almost certain that the presynaptic complex present in living cells will be bound by other recombination proteins. Therefore it will be essential to more fully define the length and protein composition of the “search entity” that exists within living cells. For instance, the eukaryotic protein Rad54, which is an ATP-dependent DNA translocase, is just one example of a protein known to interact with the presynaptic complex (68–70). Rad54 and the closely related protein Rdh54 perform multiple functions during the different stages of recombination (68, 71, 72). Using time-resolved ChIP assays, the Jentsch laboratory (73) has recently provided evidence that Rad54 and Rdh54 are necessary for the homology search in S. cerevisiae. In the presence of Rad54/Rdh54, the Rad51 presynaptic complex sampled sites throughout a broken chromosome, but the homology search failed in the absence of the two translocases. One interpretation of these findings is that the motor activities of Rad54 and Rdh54 may promote ATP-driven unidirectional motion of the presynaptic complex along duplex regions of the genome while searching for homology (73). However, there may be alternative role(s) for Rad54/Rdh54 in these early stages of recombination: for instance, studies have implicated these proteins as contributing to the stability of the presynaptic complex (74), promoting nucleosome remodeling during recombination (75–77), and altering DNA topology to enhance strand unwinding (78–81). Future work will be essential to fully understand how all the activities of these proteins contribute to recombination.
Interestingly, Rad54/Rad51-mediated search is accompanied by phosphorylation of histone H2AX (73, 82). The simplest interpretation of these data is that the corresponding kinase might be an integral component of the presynaptic complex, but why it would phosphorylate sites of the chromosome that have already been searched (and rejected) remains unclear.
Recent studies have revealed that fluorescently tagged DSBs within S. cerevisiae can undergo highly dynamic diffusive motion, and this increased mobility is thought to reflect enhanced movement of the processed DNA ends as they explore the nuclear volume while looking for the homologous template (Fig. 4B) (83–87). The local movement of the DSB is also accompanied by increased mobility of all the chromosomes throughout the nucleus. Interestingly, there is some evidence suggesting that the two ends generated by a single DSB appear to remain associated with one another during the search process, which may help coordinate the repair process by ensuring that both DSB ends locate the homologous target at the same time (86). DSB mobility is dependent upon both Rad51 and Rad54, suggesting that it reflects the movement of a fully assembled and actively searching presynaptic complex.
In addition to diffusive motion, the Greenberg group (88) has reported evidence for unidirectional motion of the presynaptic complex during Rad51-dependent recombination at telomeres in mammalian cells. Surprisingly, this movement occurred over distances ranging from ∼1.3–5 μm and was dependent upon the Hop2-Mnd1 protein, which is not normally considered to be involved in mitotic recombination, but is instead required for recombination during meiosis. All of these studies raise crucial and interesting questions with respect to what factors influence DSB mobility in living cells and how changes in mobility might be related to the homology search.
Eukaryotic chromosomes are not randomly organized, but instead appear to have preferred positions within the nucleus, and the existence of this higher-order spatial organization has important implications in replication, transcription, and recombination (89–92). The Haber laboratory (93) has recently begun examining how the spatial organization of the S. cerevisiae genome might impact homologous recombination. For these experiments, they generated yeast strains bearing a single DSB that could be repaired using a template that was positioned at one of 24 distinct locations throughout the genome. This work revealed that repair efficiency was strongly influenced by spatial proximity: DSBs were more readily repaired when in close spatial proximity to the repair template, but more difficult to repair when further away from the template in three-dimensional space. These findings suggest that the homology search itself may be the rate-limiting step in homologous recombination and also raises interesting questions regarding how the physiological state of the cell might impact both chromosome organization and recombination, and whether cell-to-cell variability in chromosome organization could influence recombination, and if so, whether such differences might be heritable.
As a final note, the Sherratt laboratory (94) has shown that DSBs trigger formation of large RecA bundles in E. coli (Fig. 4D). The constituent proteins are not all bound to ssDNA, but instead form a scaffold along the inner membrane that guides the DSB ends toward the homologous locus through an ATP hydrolysis-dependent mechanism (94, 95). These findings raise crucial questions for how the homology search might be taking place within the context of RecA bundles and to what extent the in vitro homology search reflects the in vivo search process. This finding also raises the question of whether similar protein scaffolds may be involved in eukaryotic mitotic recombination, meiotic recombination, or both. It will be crucial to recapitulate these fascinating RecA bundles in an in vitro setting to determine how they affect the homology search and subsequent strand exchange.
Conclusion
We are beginning to grasp the basic principles of the homology search, but the problem is far from solved, and future advances will likely require contributions from a number of distinct disciplines, including cell biology, biochemistry, biophysics, and molecular modeling approaches. Emergent technologies such as real-time super-resolution in vivo imaging (96) and high-resolution cryo-EM (97) also offer the potential for many new insights. How is pairing fidelity controlled, to what extent are degenerate sequences tolerated, and how might partner proteins affect these features? How does recombination intersect with replication and other repair pathways? What are the fundamental differences between mitotic and meiotic recombination, and how do these differences contribute to identification of the correct homologous template? Answers to some of these questions may be forthcoming, whereas others may prove more challenging. Of course, the other stages of recombination (e.g. end processing, strand invasion, choice of pathway, etc.) remain just as interesting as the homology search and are in many ways still just as mystifying. This abundance of open questions will help ensure that homologous recombination remains a fruitful area of scientific inquiry.
Acknowledgments
I thank Daniel Duzdevich and Fabian Erdel for their comments on this manuscript. I apologize to colleagues whose work may not have been cited due to length limitations.
*
This work was supported by National Institutes of Health Grant GM074739 and National Science Foundation (NSF) Grant MCB-1154511 (to E. C. G.) The author declares that he has no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
2
The abbreviations used are:
DSB
double-strand DNA break
nt
nucleotide(s)
RS-DNA
Rad51/RecA stretched-DNA.
References
- 1.San Filippo J., Sung P., and Klein H. (2008) Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 77, 229–257 [DOI] [PubMed] [Google Scholar]
- 2.Cromie G. A., Connelly J. C, and Leach D. R. (2001) Recombination at double-strand breaks and DNA ends: conserved mechanisms from phage to humans. Mol. Cell 8, 1163–1174 [DOI] [PubMed] [Google Scholar]
- 3.Hunter N. (2015) Meiotic recombination: the essence of heredity. Cold Spring Harb Perspect Biol. 7, a016618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cox M. M. (2001) Historical overview: searching for replication help in all of the rec places. Proc. Natl. Acad. Sci. U.S.A. 98, 8173–8180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cox M. M., Goodman M. F., Kreuzer K. N., Sherratt D. J., Sandler S. J., and Marians K. J. (2000) The importance of repairing stalled replication forks. Nature 404, 37–41 [DOI] [PubMed] [Google Scholar]
- 6.Bianco P. R., Tracy R. B., and Kowalczykowski S. C. (1998) DNA strand exchange proteins: a biochemical and physical comparison. Front Biosci. 3, D570–D603 [DOI] [PubMed] [Google Scholar]
- 7.Morrical S. W. (2015) DNA-pairing and annealing processes in homologous recombination and homology-directed repair. Cold Spring Harb. Perspect Biol. 7, a016444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kowalczykowski S. C. (2015) An overview of the molecular mechanisms of recombinational DNA repair. Cold Spring Harb Perspect Biol. 7, a016410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pâques F., and Haber J. E. (1999) Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol. Biol. Rev. 63, 349–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Symington L. S., Rothstein R., and Lisby M. (2014) Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae. Genetics 198, 795–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conway A. B., Lynch T. W., Zhang Y., Fortin G. S., Fung C. W., Symington L. S., and Rice P. A. (2004) Crystal structure of a Rad51 filament. Nat. Struct. Mol. Biol. 11, 791–796 [DOI] [PubMed] [Google Scholar]
- 12.Clark A. J., and Margulies A. D. (1965) Isolation and characterization of recombination-deficient mutants of Escherichia coli K12. Proc. Natl. Acad. Sci. U.S.A. 53, 451–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shibata T., Cunningham R. P., DasGupta C., and Radding C. M. (1979) Homologous pairing in genetic recombination: complexes of recA protein and DNA. Proc. Natl. Acad. Sci. U.S.A. 76, 5100–5104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shibata T., DasGupta C., Cunningham R. P., and Radding C. M. (1979) Purified Escherichia coli recA protein catalyzes homologous pairing of superhelical DNA and single-stranded fragments. Proc. Natl. Acad. Sci. U.S.A. 76, 1638–1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cunningham R. P., Shibata T., DasGupta C., and Radding C. M. (1979) Single strands induce recA protein to unwind duplex DNA for homologous pairing. Nature 281, 191–195 [DOI] [PubMed] [Google Scholar]
- 16.Howard-Flanders P., West S. C., and Stasiak A. (1984) Role of RecA protein spiral filaments in genetic recombination. Nature 309, 215–219 [DOI] [PubMed] [Google Scholar]
- 17.West S. C., Cassuto E., and Howard-Flanders P. (1981) Mechanism of E. coli RecA protein directed strand exchanges in post-replication repair of DNA. Nature 294, 659–662 [DOI] [PubMed] [Google Scholar]
- 18.McEntee K., Weinstock G. M., and Lehman I. R. (1979) Initiation of general recombination catalyzed in vitro by the recA protein of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 76, 2615–2619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Craig N. L., and Roberts J. W. (1980) E. coli recA protein-directed cleavage of phage λ repressor requires polynucleotide. Nature 283, 26–30 [DOI] [PubMed] [Google Scholar]
- 20.Roberts J. W., Roberts C. W., and Craig N. L. (1978) Escherichia coli recA gene product inactivates phage λ repressor. Proc. Natl. Acad. Sci. U.S.A. 75, 4714–4718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bishop D. K., Park D., Xu L., and Kleckner N. (1992) DMC1: a meiosis-specific yeast homolog of E. coli recA required for recombination, synaptonemal complex formation, and cell cycle progression. Cell 69, 439–456 [DOI] [PubMed] [Google Scholar]
- 22.Shinohara A., Ogawa H., and Ogawa T. (1992) Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell 69, 457–470 [DOI] [PubMed] [Google Scholar]
- 23.Sung P. (1994) Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science 265, 1241–1243 [DOI] [PubMed] [Google Scholar]
- 24.Baumann P., Benson F. E., and West S. C. (1996) Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell 87, 757–766 [DOI] [PubMed] [Google Scholar]
- 25.Sehorn M. G., Sigurdsson S., Bussen W., Unger V. M., and Sung P. (2004) Human meiotic recombinase Dmc1 promotes ATP-dependent homologous DNA strand exchange. Nature 429, 433–437 [DOI] [PubMed] [Google Scholar]
- 26.Chintapalli S. V., Bhardwaj G., Babu J., Hadjiyianni L., Hong Y., Todd G. K., Boosalis C. A., Zhang Z., Zhou X., Ma H., Anishkin A., van Rossum D. B., and Patterson R. L. (2013) Reevaluation of the evolutionary events within recA/RAD51 phylogeny. BMC Genomics 14, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin Z., Kong H., Nei M., and Ma H. (2006) Origins and evolution of the recA/RAD51 gene family: evidence for ancient gene duplication and endosymbiotic gene transfer. Proc. Natl. Acad. Sci. U.S.A. 103, 10328–10333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown M. S., and Bishop D. K. (2015) DNA strand exchange and RecA homologs in meiosis. Cold Spring Harb. Perspect. Biol. 7, a016659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neale M. J., and Keeney S. (2006) Clarifying the mechanics of DNA strand exchange in meiotic recombination. Nature 442, 153–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Egelman E. H., and Stasiak A. (1986) Structure of helical RecA-DNA complexes. Complexes formed in the presence of ATP-γ-S or ATP. J. Mol. Biol. 191, 677–697 [DOI] [PubMed] [Google Scholar]
- 31.Ogawa T., Yu X., Shinohara A., and Egelman E. H. (1993) Similarity of the yeast RAD51 filament to the bacterial RecA filament. Science 259, 1896–1899 [DOI] [PubMed] [Google Scholar]
- 32.Sheridan S. D., Yu X., Roth R., Heuser J. E., Sehorn M. G., Sung P., Egelman E. H., and Bishop D. K. (2008) A comparative analysis of Dmc1 and Rad51 nucleoprotein filaments. Nucleic Acids Res. 36, 4057–4066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu X., Jacobs S. A., West S. C., Ogawa T., and Egelman E. H. (2001) Domain structure and dynamics in the helical filaments formed by RecA and Rad51 on DNA. Proc. Natl. Acad. Sci. U.S.A. 98, 8419–8424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Z., Yang H., and Pavletich N. P. (2008) Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures. Nature 453, 489–494 [DOI] [PubMed] [Google Scholar]
- 35.King G. A., Gross P., Bockelmann U., Modesti M., Wuite G. J., and Peterman E. J. (2013) Revealing the competition between peeled ssDNA, melting bubbles, and S-DNA during DNA overstretching using fluorescence microscopy. Proc. Natl. Acad. Sci. U.S.A. 110, 3859–3864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Renkawitz J., Lademann C. A., and Jentsch S. (2014) Mechanisms and principles of homology search during recombination. Nat. Rev. Mol. Cell Biol. 15, 369–383 [DOI] [PubMed] [Google Scholar]
- 37.Barzel A., and Kupiec M.(2008) Finding a match: how do homologous sequences get together for recombination? Nat. Rev. Genet. 9, 27–37 [DOI] [PubMed] [Google Scholar]
- 38.Weiner A., Zauberman N., and Minsky A. (2009) Recombinational DNA repair in a cellular context: a search for the homology search. Nat. Rev. Microbiol. 7, 748–755 [DOI] [PubMed] [Google Scholar]
- 39.Prentiss M., Prévost C., and Danilowicz C. (2015) Structure/function relationships in RecA protein-mediated homology recognition and strand exchange. Crit. Rev. Biochem. Mol. Biol. 50, 453–476 [DOI] [PubMed] [Google Scholar]
- 40.Riggs A. D., Bourgeois S., and Cohn M. (1970) The lac repressor-operator interaction. 3. Kinetic studies. J. Mol. Biol. 53, 401–417 [DOI] [PubMed] [Google Scholar]
- 41.Berg O. G., Winter R. B., and von Hippel P. H. (1981) Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry 20, 6929–6948 [DOI] [PubMed] [Google Scholar]
- 42.von Hippel P. H., and Berg O. G. (1989) Facilitated target location in biological systems. J. Biol. Chem. 264, 675–678 [PubMed] [Google Scholar]
- 43.Tsang S. S., Chow S. A., and Radding C. M. (1985) Networks of DNA and RecA protein are intermediates in homologous pairing. Biochemistry 24, 3226–3232 [DOI] [PubMed] [Google Scholar]
- 44.Gonda D. K., and Radding C. M. (1986) The mechanism of the search for homology promoted by recA protein: facilitated diffusion within nucleoprotein networks. J. Biol. Chem. 261, 13087–13096 [PubMed] [Google Scholar]
- 45.Gonda D. K., and Radding C. M. (1983) By searching processively RecA protein pairs DNA molecules that share a limited stretch of homology. Cell 34, 647–654 [DOI] [PubMed] [Google Scholar]
- 46.Chow S. A., and Radding C. M. (1985) Ionic inhibition of formation of RecA nucleoprotein networks blocks homologous pairing. Proc. Natl. Acad. Sci. U.S.A. 82, 5646–5650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Menetski J. P., Bear D. G., and Kowalczykowski S. C. (1990) Stable DNA heteroduplex formation catalyzed by the Escherichia coli RecA protein in the absence of ATP hydrolysis. Proc. Natl. Acad. Sci. U.S.A. 87, 21–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rehrauer W. M., and Kowalczykowski S. C. (1993) Alteration of the nucleoside triphosphate (NTP) catalytic domain within Escherichia coli recA protein attenuates NTP hydrolysis but not joint molecule formation. J. Biol. Chem. 268, 1292–1297 [PubMed] [Google Scholar]
- 49.Hsieh P., Camerini-Otero C. S., and Camerini-Otero R. D. (1992) The synapsis event in the homologous pairing of DNAs: RecA recognizes and pairs less than one helical repeat of DNA. Proc. Natl. Acad. Sci. U.S.A. 89, 6492–6496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adzuma K. (1998) No sliding during homology search by RecA protein. J. Biol. Chem. 273, 31565–31573 [DOI] [PubMed] [Google Scholar]
- 51.Forget A. L., and Kowalczykowski S. C. (2012) Single-molecule imaging of DNA pairing by RecA reveals a three-dimensional homology search. Nature 482, 423–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Forget A. L., Dombrowski C. C., Amitani I., and Kowalczykowski S. C. (2013) Exploring protein-DNA interactions in 3D using in situ construction, manipulation and visualization of individual DNA dumbbells with optical traps, microfluidics and fluorescence microscopy. Nat. Protoc 8, 525–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ragunathan K., Liu C., and Ha T. (2012) RecA filament sliding on DNA facilitates homology search. Elife 1, e00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roy R., Hohng S., and Ha T. (2008) A practical guide to single-molecule FRET. Nat. Methods 5, 507–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Qi Z., Redding S., Lee J. Y., Gibb B., Kwon Y., Niu H., Gaines W. A., Sung P., and Greene E. C. (2015) DNA sequence alignment by microhomology sampling during homologous recombination. Cell 160, 856–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Collins B. E., Ye L. F., Duzdevich D., and Greene E. C. (2014) DNA curtains: novel tools for imaging protein-nucleic acid interactions at the single-molecule level. Methods Cell Biol. 123, 217–234 [DOI] [PubMed] [Google Scholar]
- 57.Lee J. Y., Terakawa T., Qi Z., Steinfeld J. B., Redding S., Kwon Y., Gaines W. A., Zhao W., Sung P., and Greene E. C. (2015) Base triplet stepping by the Rad51/RecA family of recombinases. Science 349, 977–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Folta-Stogniew E., O'Malley S., Gupta R., Anderson K. S., and Radding C. M. (2004) Exchange of DNA base pairs that coincides with recognition of homology promoted by E. coli RecA protein. Mol. Cell 15, 965–975 [DOI] [PubMed] [Google Scholar]
- 59.Gupta R. C., Folta-Stogniew E., O'Malley S., Takahashi M., and Radding C. M. (1999) Rapid exchange of A:T base pairs is essential for recognition of DNA homology by human Rad51 recombination protein. Mol. Cell 4, 705–714 [DOI] [PubMed] [Google Scholar]
- 60.Thomas C. A., Jr. (1966) Recombination of DNA molecules. Prog. Nucleic Acid Res. Mol. Biol. 5, 315–337 [DOI] [PubMed] [Google Scholar]
- 61.Jiang L., and Prentiss M. (2014) RecA-mediated sequence homology recognition as an example of how searching speed in self-assembly systems can be optimized by balancing entropic and enthalpic barriers. Phys. Rev. E. Stat. Nonlin. Soft Matter Phys. 90, 022704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kates-Harbeck J., Tilloy A., and Prentiss M. (2013) Simplified biased random walk model for RecA-protein-mediated homology recognition offers rapid and accurate self-assembly of long linear arrays of binding sites. Phys. Rev. E. Sta.t Nonlin. Soft Matter Phys. 88, 012702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.De Vlaminck I., van Loenhout M. T., Zweifel L., den Blanken J., Hooning K., Hage S., Kerssemakers J., and Dekker C. (2012) Mechanism of homology recognition in DNA recombination from dual-molecule experiments. Mol. Cell 46, 616–624 [DOI] [PubMed] [Google Scholar]
- 64.Yang D., Boyer B., Prévost C., Danilowicz C., and Prentiss M. (2015) Integrating multi-scale data on homologous recombination into a new recognition mechanism based on simulations of the RecA-ssDNA/dsDNA structure. Nucleic Acids Res. 43, 10251–10263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vlassakis J., Feinstein E., Yang D., Tilloy A., Weiller D., Kates-Harbeck J., Coljee V. W., and Prentiss M. (2013) Tension on dsDNA bound to ssDNA-RecA filaments may play an important role in driving efficient and accurate homology recognition and strand exchange. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 87, 032702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Danilowicz C., Feinstein E., Conover A., Coljee V. W., Vlassakis J., Chan Y. L., Bishop D. K., and Prentiss M. (2012) RecA homology search is promoted by mechanical stress along the scanned duplex DNA. Nucleic Acids Res. 40, 1717–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Savir Y., and Tlusty T. (2010) RecA-mediated homology search as a nearly optimal signal detection system. Mol. Cell 40, 388–396 [DOI] [PubMed] [Google Scholar]
- 68.Heyer W. D., Li X., Rolfsmeier M., and Zhang X. P. (2006) Rad54: the Swiss Army knife of homologous recombination? Nucleic Acids Res. 34, 4115–4125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mazin A. V., Bornarth C. J., Solinger J. A., Heyer W. D., and Kowalczykowski S. C. (2000) Rad54 protein is targeted to pairing loci by the Rad51 nucleoprotein filament. Mol. Cell 6, 583–592 [DOI] [PubMed] [Google Scholar]
- 70.Amitani I., Baskin R. J., and Kowalczykowski S. C. (2006) Visualization of Rad54, a chromatin remodeling protein, translocating on single DNA molecules. Mol. Cell 23, 143–148 [DOI] [PubMed] [Google Scholar]
- 71.Mazin A. V., Mazina O. M., Bugreev D. V., and Rossi M. J. (2010) Rad54, the motor of homologous recombination. DNA Repair (Amst.) 9, 286–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tan T. L., Kanaar R., and Wyman C. (2003) Rad54, a Jack of all trades in homologous recombination. DNA Repair (Amst.) 2, 787–794; Correction (2003) DNA Repair (Amst.)2, 1293 [DOI] [PubMed] [Google Scholar]
- 73.Renkawitz J., Lademann C. A., Kalocsay M., and Jentsch S. (2013) Monitoring homology search during DNA double-strand break repair in vivo. Mol. Cell 50, 261–272 [DOI] [PubMed] [Google Scholar]
- 74.Mazin A. V., Alexeev A. A., and Kowalczykowski S. C. (2003) A novel function of Rad54 protein: stabilization of the Rad51 nucleoprotein filament. J. Biol. Chem. 278, 14029–14036 [DOI] [PubMed] [Google Scholar]
- 75.Alexeev A., Mazin A., and Kowalczykowski S. C. (2003) Rad54 protein possesses chromatin-remodeling activity stimulated by the Rad51-ssDNA nucleoprotein filament. Nat. Struct. Biol. 10, 182–186 [DOI] [PubMed] [Google Scholar]
- 76.Wolner B., and Peterson C. L. (2005) ATP-dependent and ATP-independent roles for the Rad54 chromatin remodeling enzyme during recombinational repair of a DNA double strand break. J. Biol. Chem. 280, 10855–10860 [DOI] [PubMed] [Google Scholar]
- 77.Zhang Z., Fan H. Y., Goldman J. A., and Kingston R. E. (2007) Homology-driven chromatin remodeling by human RAD54. Nat. Struct. Mol. Biol. 14, 397–405 [DOI] [PubMed] [Google Scholar]
- 78.Petukhova G., Sung P., and Klein H. (2000) Promotion of Rad51-dependent D-loop formation by yeast recombination factor Rdh54/Tid1. Genes Dev. 14, 2206–2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Petukhova G., Van Komen S., Vergano S., Klein H., and Sung P. (1999) Yeast Rad54 promotes Rad51-dependent homologous DNA pairing via ATP hydrolysis-driven change in DNA double helix conformation. J. Biol. Chem. 274, 29453–29462 [DOI] [PubMed] [Google Scholar]
- 80.Ristic D., Wyman C., Paulusma C., and Kanaar R. (2001) The architecture of the human Rad54-DNA complex provides evidence for protein translocation along DNA. Proc. Natl. Acad. Sci. U.S.A. 98, 8454–8460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Van Komen S., Petukhova G., Sigurdsson S., Stratton S., and Sung P. (2000) Superhelicity-driven homologous DNA pairing by yeast recombination factors Rad51 and Rad54. Mol. Cell 6, 563–572 [DOI] [PubMed] [Google Scholar]
- 82.Renkawitz J., Lademann C. A., and Jentsch S. (2013) γH2AX spreading linked to homology search. Cell Cycle 12, 2526–2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dion V., and Gasser S. M. (2013) Chromatin movement in the maintenance of genome stability. Cell 152, 1355–1364 [DOI] [PubMed] [Google Scholar]
- 84.Dion V., Kalck V., Horigome C., Towbin B. D., and Gasser S. M. (2012) Increased mobility of double-strand breaks requires Mec1, Rad9 and the homologous recombination machinery. Nat. Cell Biol. 14, 502–509 [DOI] [PubMed] [Google Scholar]
- 85.Miné-Hattab J., and Rothstein R. (2012)Increased chromosome mobility facilitates homology search during recombination. Nat. Cell Biol. 14, 510–517 [DOI] [PubMed] [Google Scholar]
- 86.Miné-Hattab J., and Rothstein R. (2013)DNA in motion during double-strand break repair. Trends Cell Biol. 23, 529–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Strecker J., Gupta G. D., Zhang W., Bashkurov M., Landry M. C., Pelletier L., and Durocher D. (2016) DNA damage signalling targets the kinetochore to promote chromatin mobility. Nat. Cell Biol. 18, 281–290 [DOI] [PubMed] [Google Scholar]
- 88.Cho N. W., Dilley R. L., Lampson M. A., and Greenberg R. A. (2014) Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell 159, 108–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aparicio O. M. (2013) Location, location, location: it's all in the timing for replication origins. Genes Dev. 27, 117–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dekker J., and Mirny L. (2016) The 3D genome as moderator of chromosomal communication. Cell 164, 1110–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dekker J., and Misteli T. (2015) Long-range chromatin interactions. Cold Spring Harb. Perspect. Biol. 7, a019356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dekker J., Rippe K., Dekker M., and Kleckner N. (2002) Capturing chromosome conformation. Science 295, 1306–1311 [DOI] [PubMed] [Google Scholar]
- 93.Lee C. S., Wang R. W., Chang H. H., Capurso D., Segal M. R., and Haber J. E. (2016) Chromosome position determines the success of double-strand break repair. Proc. Natl. Acad. Sci. U.S.A. 113, E146–E54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lesterlin C., Ball G., Schermelleh L., and Sherratt D. J. (2014) RecA bundles mediate homology pairing between distant sisters during DNA break repair. Nature 506, 249–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rajendram M., Zhang L., Reynolds B. J., Auer G. K., Tuson H. H., Ngo K. V., Cox M. M., Yethiraj A., Cui Q., and Weibel D. B. (2015) Anionic phospholipids stabilize RecA filament bundles in Escherichia coli. Mol. Cell 60, 374–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Uphoff S., and Kapanidis A. N.(2014) Studying the organization of DNA repair by single-cell and single-molecule imaging. DNA Repair (Amst.) 20, 32–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bai X. C., McMullan G., and Scheres S. H. (2015) How cryo-EM is revolutionizing structural biology. Trends Biochem. Sci. 40, 49–57 [DOI] [PubMed] [Google Scholar]