Sirtuin 1 Deacetylase: A Key Regulator of Hepatic Lipid Metabolism (original) (raw)
. Author manuscript; available in PMC: 2016 Aug 8.
Summary
Obesity is a serious medical problem worldwide and disruption of metabolic/energy homeostasis plays a pivotal role in this global epidemic. In obese people, fatty liver (steatosis) develops, which increases the risk for diabetes, cardiovascular disease, and even, liver cancer. Sirtuin 1 (SIRT1) is a NAD+-dependent deacetylase that functions as a key metabolic/energy sensor and mediates homeostatic responses to nutrient availability. Accumulating evidence indicates that SIRT1 is a master regulator of the transcriptional networks that control hepatic lipid metabolism. During energy-deprived conditions, SIRT1 deacetylates and alters the expression and activities of key transcriptional regulators involved in hepatic lipogenesis, fatty acid β-oxidation, and cholesterol/bile acid metabolism. This review will discuss the latest advances in this field, focusing on beneficial roles of SIRT1 in hepatic lipid metabolism including its potential as a therapeutic target for treatment of steatosis and other obesity-related metabolic diseases.
Keywords: SIRT1, NAD+, fatty liver, obesity, lipogenesis, β-oxidation, SREBP-1c
I. Introduction
Obesity is a serious medical problem worldwide and dysregulation of metabolic pathways, caused by imbalances in energy metabolism, plays a pivotal role in this global epidemic (Grundy, et al. 2004; Russell and Proctor. 2006). In nearly 80% of obese individuals, fatty liver (hepatic steatosis) develops, which substantially increases the risk for type 2 diabetes, cardiovascular disease, and even, liver cancer (Stefan, et al. 2008; Cohen, et al. 2011). Despite its clinical importance, what causes hepatic steatosis and possible therapeutic options to treat steatosis remain poorly understood.
The liver is the chief organ that is responsible for maintaining lipid and glucose levels in the body. In response to nutritional and hormonal signals, the liver controls lipid and carbohydrate metabolic pathways to maintain homeostasis. A growing body of evidence indicates that SIRT1 is a master regulator of hepatic lipid (fatty acid, triglyceride, cholesterol, bile acid) metabolism. SIRT1 is a NAD+-dependent deacetylase that links cellular energy and metabolic status to transcriptional outcomes to meet energy deficits under energy-deprived conditions, such as fasting or exercise (Guarente. 2007; Canto and Auwerx. 2009; Chalkiadaki and Guarente. 2012; Houtkooper, et al. 2012). SIRT1 regulates transcriptional programs of hepatic metabolism by deacetylating transcriptional regulatory proteins, as well as, histones at its metabolic target genes (Chalkiadaki and Guarente. 2012; Houtkooper, et al. 2012). In this review, we will provide an update of the latest advances in the SIRT1 field, with a focus on the role of SIRT1 in hepatic lipid metabolism. We will discuss how the expression and activity of hepatic SIRT1 are regulated under normal physiological conditions and how they are dysregulated in fatty liver in obesity. We will also discuss how SIRT1 modulates transcriptional regulators involved in hepatic lipid metabolism and further discuss the underlying mechanisms by which SIRT1 regulates their expression levels and activities by protein deacetylation. Finally, we will discuss the therapeutic potential of SIRT1 as a novel target for treating hepatic steatosis and other obesity-related disease, such as type 2 diabetes, cardiovascular disease, and certain cancers.
II. SIRT1 as a master regulator of metabolism
A. SIRT1 as a link between metabolic/energy status and transcriptional outcomes
In mammals, there are seven sirtuin proteins (SIRT1–7) (Guarente. 2007; Yamamoto, et al. 2007). They are ubiquitously expressed and share a common catalytic core domain but also have distinct features in their subcellular localization, enzymatic activities, and target proteins. Of the sirtuins, SIRT1 is the best-characterized protein. SIRT1 is a mammalian ortholog of the yeast silent information regulator 2 (sir2) (Klar, et al. 1979; Sinclair and Guarente. 1997). Yeast sir2 was originally discovered as one of several genes that encode transcriptional regulators that silence gene expression and was shown to be a longevity protein that increased life span (Gottlieb and Esposito. 1989; Sinclair and Guarente. 1997). SIRT1 was then shown to be a NAD+-dependent enzyme that catalyzes deacetylation of histones (Imai, et al. 2000). Intensive studies during the last decade have demonstrated that SIRT1 also deacetylates a wide range of non-histone regulatory proteins including transcriptional nuclear receptors involved in diverse biological pathways, such as metabolism, stress response, and possibly the aging process (Finkel. 2003; Sinclair, et al. 2006; Canto and Auwerx. 2009). Well-known transcriptional regulators involved in the cellular stress response and metabolism that are modulated by SIRT1-mediated deacetylation include p53, FOXOs, PGC-1α, NF-κB, CRTC2, LXRs, FXR, and SREBPs.
NAD+-dependent SIRT1 activity is particularly important in the regulation of metabolic/energy homeostasis because it can sense nutrient availability and connect cellular energy/metabolic status to transcriptional outcomes. A large body of evidence indicates that cellular NAD+ levels in mammalian tissues are increased in response to metabolic stress conditions, such as fasting, calorie restriction, and vigorous exercise, which leads to increased SIRT1 deacetylase activity (Guarente. 2007; Chalkiadaki and Guarente. 2012; Houtkooper, et al. 2012). Further, recent studies demonstrated that NAD+ levels also fluctuate in response to circadian rhythm and that the clock genes are regulated by SIRT1. In addition to SIRT1, AMP-activated kinase (AMPK) is another major cellular energy sensor that maintains metabolic/energy homeostasis in the body (Kahn, et al. 2005). AMPK activator agonists like AICAR were shown to increase NAD+ levels and activate the SIRT1activity (Canto, et al. 2009). All these studies demonstrate that SIRT1 senses changes in cellular NAD+ levels that reflect intracellular energy levels and adjusts energy outputs by protein deacetylation, which modulates levels and activities of transcriptional metabolic regulators and alters usually, but not always, transcriptional programs of cellular metabolic pathways.
B. Regulation of SIRT1 in physiology and disease
Fluctuating nutrient and hormonal signals during fasting and feeding cycles dynamically regulate the expression of SIRT1 (Fig. 1A). Recent studies have shown that cAMP-responsive element binding protein (CREB) positively activates SIRT1 gene transcription during fasting, while carbohydrate response element binding protein (ChREBP) negatively regulates SIRT1 expression after feeding (Chalkiadaki and Guarente. 2011; Noriega, et al. 2011). After short-term fasting (5–6 h in mice), increased glucagon levels lead to the dephosphorylation of CREB-regulated transcriptional coactivator 2 (CRTC2), which results in transport of CRTC2 into the nucleus (Liu, et al. 2008). The CREB/CRTC2 transcriptional complex then activates transcription of the SIRT1 gene as well as gluconeogenic genes. Under more prolonged fasting conditions (at least 12 h–18 h in mice), SIRT1 deacetylates CRTC2, which results in its ubiquitination and proteasomal degradation (Noriega, et al. 2011). Furthermore after prolonged fasting, SIRT1 deacetylates key regulators of gluconeogenesis and fatty acid β-oxidation, PGC-1α and Foxo-1, thus, increasing their transcriptional activities (Brunet, et al. 2004; Rodgers, et al. 2005). These coordinated transcriptional events lead to fatty acid β-oxidation and continued gluconeogenesis to meet energy demands during prolonged fasting. Conversely, under feeding conditions, the expression of SIRT1 is transiently decreased by ChREBP, a transcriptional factor that couples hepatic glucose utilization and lipid synthesis (Uyeda and Repa. 2006). ChREBP is transported into the nucleus and binds to a site in the SIRT1 promoter that overlaps the CREB binding site resulting in repression of SIRT1 gene transcription (Noriega, et al. 2011).
Figure 1.
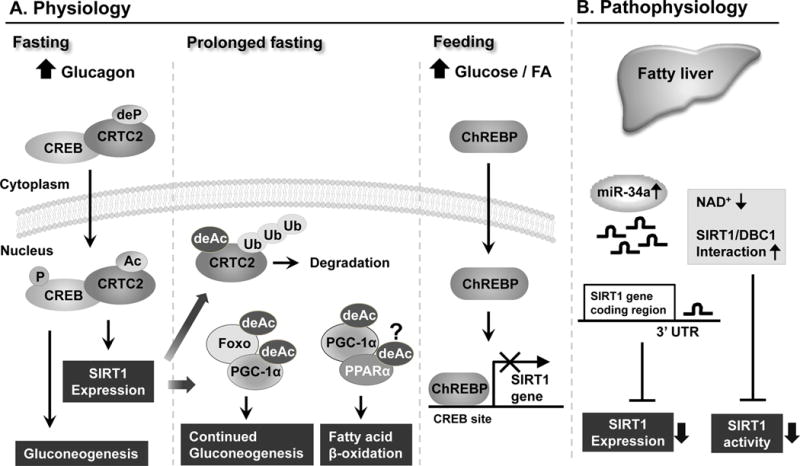
Recent studies have suggested that the expression levels and deacetylase activity of SIRT1 are constitutively decreased under pathological conditions, such as in fatty livers of obese mice (Fig. 1B) and that small non-coding microRNAs (miRs) play a key role in this abnormal regulation (Coste, et al. 2008; Lee, et al. 2010). MiRs have been recently demonstrated as important cellular regulators that function as negative gene regulators by directly binding to the 3′untranslated region (3′ UTR) of target genes (Bartel. 2004; Neilson and Sharp. 2008). Consistent with their critical biological functions, miRs are aberrantly expressed in human diseases (Esau, et al. 2006; Cheung, et al. 2008; Cermelli, et al. 2011). MiR-34a is the most highly elevated hepatic miR in fatty liver disease patients and obese mice (Cheung, et al. 2008; Lee, et al. 2010; Cermelli, et al. 2011; Trajkovski, et al. 2011). In recent studies, our group has shown that SIRT1 expression is post-transcriptionally inhibited by abnormally elevated levels of miR-34a in fatty livers of dietary or genetic obese mice (Lee, et al. 2010). SIRT1 deacetylase activity was also shown be inhibited by decreased levels of intracellular NAD+ levels in fatty liver of obese mice (Yoshino, et al. 2011). In obesity, SIRT1 activity is also inhibited by an unusual protein interaction between SIRT1 and a negative regulator of SIRT1, DBC1. Metabolic stress induced by a high fat diet increases the interaction between SIRT1 and DBC1, resulting in decreased SIRT1 deacetylase activity (Escande, et al. 2010). Unlike normal physiological regulation of SIRT1, transcriptional regulation of SIRT1 under pathological conditions has not been well-studied. Further, it remains largely unknown whether aberrant post-translational modifications of SIRT1 negatively regulate the expression levels and activities of SIRT1 under pathological conditions.
C. Metabolic functions of SIRT1
The role of SIRT1 in cellular metabolism has been demonstrated in a number of studies using small molecule SIRT1 activators or transgenic mice that are deficient for SIRT1 expression or that overexpress SIRT1. The small molecule compounds activating SIRT1 include resveratrol, a polyphenolic compound abundant in red-grape skin, and synthetic compounds, such as SRT1720 (Baur, et al. 2006; Lagouge, et al. 2006; Feige, et al. 2008). In vivo animal studies using these natural or synthetic compounds have revealed that activation of SIRT1 ameliorates insulin resistance, increases mitochondrial content, improves metabolic profiles, and increases survival in mice challenged with a high fat diet (Baur, et al. 2006; Lagouge, et al. 2006; Feige, et al. 2008). Remarkably, a recent human study has shown that calorie restriction-like beneficial effects on energy metabolism and metabolic profiles were observed in obese humans when treated with a resveratrol-supplemented diet for 30 days (Timmers, et al. 2011).
In transgenic studies, SIRT1-overexpressing mice were resistant to body weight gain and also, insulin resistance and glucose intolerance was ameliorated when these mice were fed a high fat diet compared to wild-type control mice (Banks, et al. 2008; Rodgers and Puigserver. 2007; Pfluger, et al. 2008). Consistent with these SIRT1 gain-of-function studies, detrimental metabolic effects were observed in loss-of-function studies. In SIRT1 liver-specific knockout (SIRT1 LKO) mice fed a high fat diet, fatty acid metabolism was altered and the fat (triglyceride) was accumulated abnormally in the liver, resulting in hepatic steatosis and elevated inflammatory responses (Purushotham, et al. 2009). Further, in SIRT1 LKO mice generated by a different group, mTorc2/Akt signaling was impaired, which led to hyperglycemia, oxidative stress, and insulin resistance (Wang, et al. 2011).
All told, these recent studies demonstrate that SIRT1 is a master regulator of cellular metabolism and mediates beneficial metabolic effects in various metabolic tissues. In the following sections, we will discuss beneficial metabolic functions of SIRT1, with a focus on hepatic lipid metabolism.
III. Regulation of hepatic fat (triglyceride, TG) metabolism by SIRT1
A. Regulation of hepatic TG levels
The liver is the central organ in the body that ensures metabolic homeostasis. The liver controls nearly all aspects of lipid (fatty acid and TG, cholesterol/bile acids) and carbohydrate (glucose, glycogen) metabolic pathways. In these sections, we will focus on the regulation of hepatic fatty acid and TG metabolic pathways in response to metabolic signals. Hepatic fat (TG) metabolism largely involves the uptake, synthesis, storage, and utilization of fatty acids and TG in response to nutrient and hormonal metabolic signals under fasting and feeding conditions (Cohen, et al. 2011). During fasting, the liver converts TG into usable energy through the process of fatty acid β-oxidation to meet energy deficits. Conversely, under feeding conditions, lipid metabolic programs in the liver are reversed in order to store extra energy in the form of lipid TG droplets. Under normal physiological conditions, these processes are tightly controlled, but in disease states, such as non-alcoholic fatty liver disease (NAFLD), regulation of these processes is impaired, resulting in an imbalance between hepatic storage and removal of TG (fat) and abnormal accumulations of fat in the liver. In NAFLD, hepatic insulin resistance, ER stress, and chronic inflammation are increased (Meshkani and Adeli. 2009; Baker, et al. 2011), which substantially increases the risk for type 2 diabetes, cardiovascular disease, and hepatobiliary liver diseases, such as gallstones, steatohepatitis, cirrhosis, and liver cancer.
As shown in Figure 2, there are four major pathways controlling hepatic TG levels: 1) TG Import through uptake of dietary TG, 2) TG synthesis (lipogenesis), 3) utilization of TG through fatty acid β-oxidation, and 4) TG export through VLDL lipoproteins. First, after a meal, dietary lipids taken up in the small intestine are packaged as TG-rich chylomicrons and released into the systemic circulation. About 20% of chylomicrons are taken up by the liver to provide a source of lipid for hepatic TG (Cohen, et al. 2011). Second, hepatic TG levels are also increased by lipogenesis by conversion of fatty acids into TG. Two major transcription factors, sterol response element binding protein-1c (SREBP-1c) and ChREBP, are largely responsible for the activation of the hepatic lipogenic program of fatty acid synthesis and subsequent TG production (Brown and Goldstein. 1997; Uyeda and Repa. 2006). Third, during prolonged fasting, TG levels are decreased by release of fatty acids from TG followed by fatty acid β-oxidation in mitochondria that provides energy fuel sources for the body, especially ketone bodies for brain. In addition, fatty acids produced by β-oxidation can also be re-esterified and converted back to TG in the liver, which partially restores hepatic TG levels. Fourth, hepatic TG levels are decreased by export into the blood as the very low density lipoprotein (VLDL) lipoprotein particle. Human subjects with a heterozygous mutation in the gene encoding apolipoprotein B (Apo B), a structural protein of VLDL, showed 3-fold increase in hepatic TG levels compared to normal individuals (Tanoli, et al. 2004).
Figure 2.
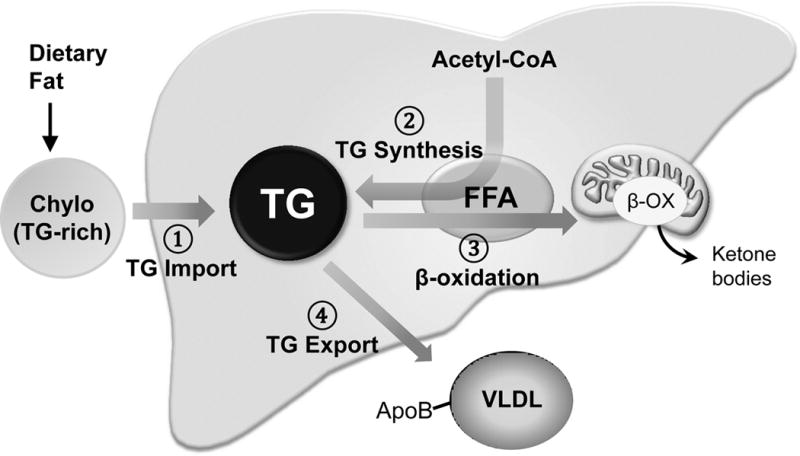
Recent studies have shown that SIRT1 decreases hepatic TG levels by inhibiting lipogenesis and stimulating fatty acid β-oxidation (Purushotham, et al. 2009; Ponugoti, et al. 2010; Walker, et al. 2010). SIRT1 inhibits hepatic fatty acid and TG synthesis by targeting SREBP-1c and stimulates β-oxidation by targeting PPARα and its coactivator, PGC-1α, which will be discussed further in the following sections.
B. Inhibition of hepatic lipogenesis by SIRT1
Recent studies from our group and A. Narr and his colleagues have shown that SIRT1 inhibits hepatic lipogenesis by deacetylation of SREBP-1c, a key lipogenic activator, during fasting (Ponugoti, et al. 2010; Walker, et al. 2010). SREBPs are DNA binding transcription factors that are critically involved in the regulation of cellular lipid and cholesterol levels (Brown and Goldstein. 1997). There are three isoforms of SREBPs (SREBP-1c, -1a, and 2) encoded by two genes (Horton. 2002). SREBP-1a and -1c are synthesized from the same gene and SREBP-2 from a separate gene. SREBP-1c is more abundantly expressed in adult liver, whereas SREBP-1a is the predominant isoform in cultured hepatic cells. SREBP-1a and -1c regulate fatty acid and TG synthesis and SREBP-2 is a critical regulator of cholesterol synthesis (Shimano, et al. 1997; Horton, et al. 1998). In response to increased insulin levels in the fed state, SREBP-1c binds to and activates its lipogenic target genes, such as fatty acid synthase, acetyl coA carboxylase, stearyl coA desaturase, and also its own gene, SREBP-1c, which results in increased fatty acid synthesis (Repa, et al. 2000; Chen, et al. 2004). In contrast, SREBP-2 is not transcriptionally regulated upon feeding (Shimano, et al. 1997; Horton, et al. 1998).
Recent studies independently carried out by two groups, including us, support the conclusion that SIRT1 deacetylates and thus, downregulates hepatic SREBP-1c lipogenic activity during fasting (Fig. 3) (Ponugoti, et al. 2010; Walker, et al. 2010). Biochemical proteomic analysis along with mechanistic studies have revealed that SREBP-1c is acetylated by p300 and deacetylated by SIRT1 at Lys-289 and Lys-309 and that deacetylation of SREBP-1c by SIRT1 downregulates SREBP-1c transcriptional activity by promoting ubiquitination and possibly, proteasomal degradation (Ponugoti, et al. 2010; Walker, et al. 2010). Consistently, treatment with small molecule activators of SIRT1 decreased nuclear SREBP-1c levels, whereas SIRT1 inhibitors increased the levels (Walker, et al. 2010). Further, deacetylation by SIRT1 inhibited binding of SREBP-1c to its target lipogenic genes. Remarkably, while SREBP-1c acetylation levels were dynamically regulated in response to fasting and feeding under normal physiological conditions, SREBP-1c acetylation levels were constitutively elevated in fatty livers of obese mice (Ponugoti, et al. 2010). Consistent with the role of SIRT1 in downregulating SREBP-1c, adenoviral-mediated hepatic overexpression of SIRT1 or treatment with resveratrol daily for 1 week decreased SREBP-1c acetylation levels in these mice with hepatic gene expression profiles consistent with beneficial metabolic effects (Ponugoti, et al. 2010).
Figure 3.
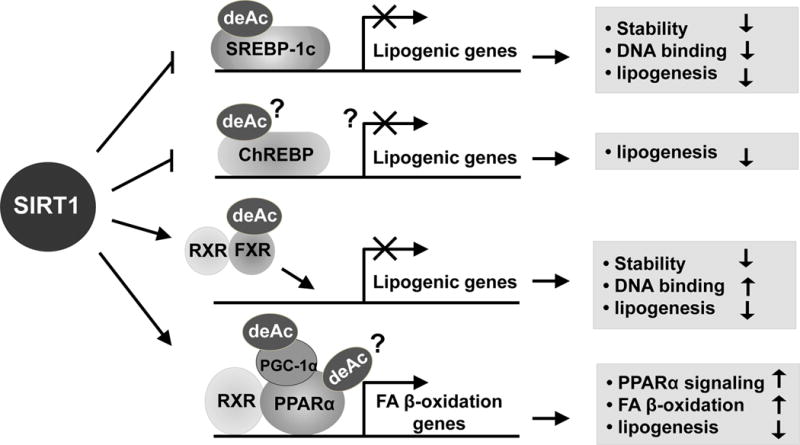
In addition to SREBP-1c, ChREBP is also an important lipogenic activator (Fig. 3). ChREBP is a transcription factor that couples hepatic glucose utilization and fatty acid and TG synthesis and is translocated into the nucleus when glucose and fatty acid levels are elevated during feeding (Uyeda and Repa. 2006). Increased ChREBP expression was observed in SIRT1 LKO mice and further, hepatic steatosis was evident even under normal feeding conditions (Wang, et al. 2010). However, it is not known whether the expression of ChREBP-1c is a direct SIRT1 target for deacetylation or is indirectly regulated by SIRT1.
The nuclear bile acid receptor, Farnesoid X receptor (FXR), is also a direct target of SIRT1 in the regulation of hepatic lipid metabolism (Kemper, et al. 2009; Kemper. 2011). FXR is the primary biosensor for bile acids and plays a central role in maintaining bile acid homeostasis (Lee, et al. 2006; Lefebvre, et al. 2009). In addition to controlling bile acid levels, FXR also indirectly regulates transcription of genes involved in TG synthesis and clearance, leading to decreased hepatic TG levels (Watanabe, et al. 2004; Zhang, et al. 2004). In recent studies, our group has shown that FXR acetylation levels were dynamically regulated during fasting and feeding in normal mice, but constitutively elevated in fatty liver of obese mice (Fig. 3) (Kemper, et al. 2009). SIRT1 deacetylates FXR during fasting and enhances FXR transactivation potential by increasing occupancy of FXR/RXRα heterodimers at their target genes. Notably, downregulation of hepatic SIRT1 increases FXR acetylation levels, resulting in gene expression patterns expected to be detrimental, including increased expression of the lipogenic SREBP-1c and FAS genes. Further, treatment of obese mice with resveratrol daily for 1 week substantially decreased FXR acetylation levels and beneficial metabolic effects (Kemper, et al. 2009). These findings indicate an intriguing functional link between elevated FXR acetylation levels and decreased hepatic SIRT1 activity with deleterious effects, further providing evidence that SIRT1 is a potential therapeutic target for treating metabolic disease.
C. Stimulation of hepatic fatty acid β-oxidation by SIRT1
Fatty acid β-oxidation is a major means of TG utilization in liver under energy-limited conditions, and the transcriptional regulators, PPARα and PGC-1α, play key roles in this process (Evans, et al. 2004; Rodgers, et al. 2008). While SIRT1 increases transcriptional activity of the coactivator PGC-1α by deacetylation, it is not known whether PPARα is also a direct target of SIRT1 deacetylation during fasting (Fig. 3). Studies utilizing liver-specific SIRT1 LKO mice or adenoviral-mediated liver-specific acute downregulation of SIRT1 in mice have shown that SIRT1 stimulates β-oxidation and decreases hepatic TG levels in response to fasting (Purushotham, et al. 2009). Acute downregulation of hepatic SIRT1 by adenoviral shRNA resulted in reduced expression of β-oxidation genes in livers of fasted mice. Consistent with these findings, a recent SIRT1 LKO mouse study showed that hepatic deletion of SIRT1 results in impaired PPARα signaling and decreased expression of genes involved in β-oxidation, whereas overexpression of SIRT1 leads to increased PPARα transcriptional signaling. Further, SIRT1 LKO mice were more susceptible to high fat diet-induced gain weight and hepatic steatosis (Purushotham, et al. 2009).
However, contradictory results were observed in another study using similarly generated SIRT1 LKO mice. In these SIRT1 LKO mice, a reduction in weight gain and reduced liver TG accumulation were observed when the mice were fed a high fat diet (Chen, et al. 2008). Because these mice are the same mouse strain CV57BL and similarly generated, the differences in dietary composition (different carbohydrate and cholesterol levels in the high fat diet) and/or the ages of the mice may account for the discrepancies in these two studies.
IV. Regulation of hepatic cholesterol and bile acid metabolism by SIRT1
The liver is the chief metabolic organ that maintains whole body cholesterol and bile acid homeostasis. Cholesterol and bile acids are steroid molecules that play essential roles in biological functions. Cholesterol serves as a component of the cell membrane and also is a precursor for biosynthesis of steroid hormones, lipid-soluble vitamins, and bile acids (Russell and Setchell. 1992; Russell. 2003). Bile acids are detergent-like amphipathic molecules that facilitate digestion of lipid-soluble nutrients. In addition to this traditional role, bile acids have been recently recognized as key signaling molecules that control integrative metabolism and energy balance in the body (Chiang. 2009; Hylemon, et al. 2009). Consistent with their critical functions in the body, excesses of cholesterol and bile acids lead to human diseases, such as cardiovascular disease, hepatobiliary disease, and liver/colon cancer. Therefore, the levels of cholesterol and bile acids are tightly regulated under normal physiological conditions. Liver plays the key role in accomplishing these important functions because first, cholesterol conversion to bile acids exclusively occurs in the liver and second, this process is the major route for elimination of cholesterol from the body because a minor fraction (about 5 %) of cholesterol (as bile acids) is excreted daily through feces (Russell. 1999; Chiang. 2009). In this section, we will discuss how SIRT1 mediates beneficial metabolic effects to maintain cholesterol and bile acid homeostasis by modulating key regulators of hepatic cholesterol and bile acid metabolism.
A. The role of SIRT1 in hepatic cholesterol metabolism
SIRT1 plays a beneficial metabolic role in cholesterol metabolism by deacetylating and thus, increasing the activity of the oxysterol receptors, liver X receptors (LXRs). LXRs (LXRα and LXRβ) are nuclear receptors that function as sterol sensors and are key regulators of body lipid homeostasis (Tontonoz and Mangelsdorf. 2003; Kalaany and Mangelsdorf. 2006). LXRs promote reverse hepatic cholesterol transport by promoting efflux of HDL cholesterol-containing lipoproteins from peripheral tissues and uptake in the liver, where lipoproteins can be degraded and the released cholesterol can then be removed from the body through its conversion into bile acids (Tontonoz and Mangelsdorf. 2003). Therefore, this LXR-mediated reverse cholesterol transport prevents the formation of foam cells in arterial walls, which reduces atherosclerosis and the incidence of cardiovascular diseases.
Recently, Li et al. have shown that LXRs are acetylated at Lys-432 in LXRα and Lys-433 in LXRβ adjacent to the ligand-regulated activation function 2 (AF2) domain of the receptor (Li, et al. 2007). Upon activation by ligands, LXRs interact with SIRT1, which promotes deacetylation and consequently ubiquitination/degradation of LXRs. Mutation of Lys-432 eliminates transcriptional activation of LXRs mediated by the SIRT1 demonstrating that deacetylation of LXRs by SIRT1 is important for their activity. Further, in SIRT1 KO mice, decreased expression of LXR downstream target genes involved in cholesterol metabolism including the ATP binding cassette transporter, impaired cholesterol homeostasis, and hepatic cholesterol accumulation were observed. The role of SIRT1 in cholesterol metabolism was also shown by other studies in which adenoviral-mediated acute hepatic down regulation of SIRT1 reduced expression of CYP7A1, the rate-limiting hepatic enzyme for bile acid biosynthesis from cholesterol (Rodgers and Puigserver. 2007). These results are consistent with the abnormal cholesterol accumulation in the liver observed in the SIRT1 LKO mice describe above (Li, et al. 2007).
There is a major puzzling issue in the activation of LXRs by SIRT1 deacetylation. In addition to reversed cholesterol transport, LXRs also promote hepatic lipogenesis by activating the transcriptional program of fatty acid synthesis, especially by increasing transcription of SREBP-1c, a key lipogenic activator (Repa, et al. 2000). SIRT1-mediated deacetylation of LXRs, therefore, can lead to both beneficial (promoting reverse cholesterol transport) and deleterious (promoting hepatic lipogenesis) effects in hepatic lipid metabolism. However, SIRT1 deacetylates not only LXRs but also SREBP-1c (Walker, et al. 2010). Under fasting, SIRT1 deacetylates SREBP-1c and thus, reduces its transcriptional activity. It is known that both LXRs and SREBP-1c contribute to transcriptional activation of the SREBP-1c gene (Yoshikawa, et al. 2001; Chen, et al. 2004) and therefore, abrogation of SREBP-1c activity by SIRT1-mediated deacetylation may allow SIRT1 to show beneficial effects on hepatic cholesterol metabolism. It will be important to elucidate precise molecular mechanisms by which deacetylation of LXRs by SIRT1 selectively regulates hepatic cholesterol and fatty acid metabolism in gene- and metabolic pathway-specific manners.
B. The role of SIRT1 in hepatic bile acid metabolism
SIRT1 also plays beneficial roles in maintaining cholesterol and bile acid homeostasis by modulating hepatic FXR signaling. FXR belongs to the nuclear receptor superfamily and functions as the primary biosensor for endogenous bile acids (Mangelsdorf and Evans. 1995; Kalaany and Mangelsdorf. 2006). Upon activation by elevated hepatic bile acid levels, FXR mediates transcriptional programs for every aspect of hepatic bile acid metabolism, including biosynthesis from cholesterol, transport (import and export), and bile acid metabolism (Lee, et al. 2006; Lefebvre, et al. 2009; Kemper. 2011). Since bile acids are detergent-like molecules and toxic to hepatocytes, dysregulation resulting in excess bile acids can cause hepatobiliary disease and liver cancer (Russell and Setchell. 1992; Lefebvre, et al. 2009). FXR protects the liver from elevated toxic bile acid levels by mediating these processes and SIRT1 directly targets hepatic FXR signaling in hepatocytes.
Recent studies from several laboratories including ours have shown that SIRT1 regulates hepatic transcriptional programs of FXR signaling at multiple levels (Fig. 4). SIRT1 and FXR can form interactive regulatory loops in the liver during fasting. First, SIRT1 increases FXR gene transcription by upregulating the transcriptional complex, HNF-1α and PGC-1 α (Purushotham, et al. 2012), although the underlying mechanisms remain unclear. Occupancy of HNF-1α at the FXR gene was decreased in SIRT1 LKO mice, suggesting that SIRT1 may increase HNF-1α DNA binding activity. Notably, SIRT1 LKO mice had deranged bile acid metabolism including impaired transport of biliary bile acids and phospholipids, which predisposed the mice to the development of cholesterol gallstones when fed a lithogenic diet containing high fat, cholesterol, and bile acid (Purushotham, et al. 2012). Second, SIRT1 deacetylates FXR and enhances its transactivation potential by increasing heterodimerization with RXRα and binding to FXR target genes (Kemper, et al. 2009). Well-known direct FXR target genes involved in hepatic lipid metabolism include small heterodimer partner (SHP), a key negative regulator of bile acid synthesis, and hepatic bile acid export transporters, like BSEP, as well as PLTP and Apo CII, stimulators of TG clearance (Lee, et al. 2006; Lefebvre, et al. 2009; Kemper. 2011). Notably, FXR acetylation levels are constitutively elevated in fatty liver of obese mice, which results in abnormal transcriptional programs with deleterious metabolic effects (Kemper, et al. 2009). Third, SIRT1 further activates transcription of FXR target genes by deacetylating and increasing PGC-1α activity. It was shown that PGC-1α enhances hepatic FXR activity during fasting by both increasing transcription of FXR and also by acting as a coactivator of FXR (Zhang, et al. 2004). Fourth, FXR, in turns, reciprocally increase the expression of SIRT1 by inhibiting miR-34a, which directly binds to the 3′UTR of the SIRT1 mRNA (Yamakuchi, et al. 2008; Lee and Kemper. 2010; Lee, et al. 2010). These interactive regulatory loops are critical for maintaining bile acid homeostasis and dysregulation of these networks can lead to metabolic abnormalities, resulting in hepatobiliary disease, such as gallstones, cholestasis, steatosis, and hepatocellular carcinoma (liver cancer).
Figure 4.
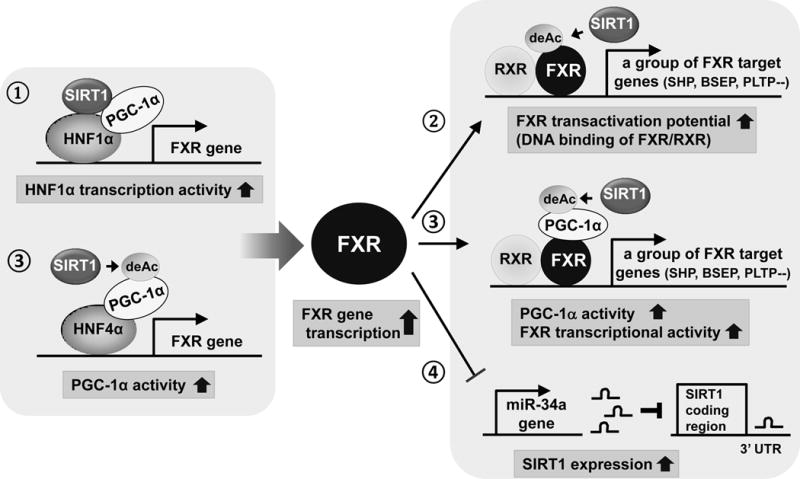
Combined, SIRT1 plays beneficial roles in maintaining cholesterol and bile acid levels by assisting the key bile acid regulator FXR.
V. Conclusion and Future Perspectives
SIRT1 was originally discovered as a longevity protein in yeast and soon after, also identified in multicellular organisms like C. elegans and drosophila (Kaeberlein, et al. 1999; Tissenbaum and Guarente. 2001). Over a decade ago, SIRT1 was further identified as a NAD+-dependent deacetylase that mediates mammalian metabolic responses to nutrient availability (Imai, et al. 2000). Since then, SIRT1 has been intensively studied because of its great potential for human health benefits. As discussed in this review, SIRT1 plays beneficial roles in hepatic lipid metabolism by inhibiting hepatic lipogenesis, stimulating fatty acid β-oxidation, and maintaining cholesterol and bile acid levels. Whether or not SIRT1 is a life-span extension protein in human has not yet been established, but, it is evident that SIRT1 mediates beneficial effects on mammalian physiology and, therefore, activation of SIRT1 should enhance the quality of life by affecting human health in many ways. Not surprisingly, there have been a number of studies done to understand how SIRT1 levels and activity are regulated at multiple levels: transcriptionally, post-transcriptionally through microRNAs, and post-translationally through its covalent modifications (Kwon and Ott. 2008; Chalkiadaki and Guarente. 2011; Houtkooper, et al. 2012). Also, numerous studies have been done to understand how the SIRT1 activity can be enhanced by small molecule compounds, SIRT1-interacting regulatory proteins, and by increasing cellular NAD+ levels (Canto and Auwerx. 2012). All these SIRT1-based studies should be extremely valuable for the development of therapeutic drugs for the treatment of age-related human diseases, such as obesity-related steatosis discussed in this review, diabetes, cardiovascular disease, neurodegenerative disease, and cancer.
Acknowledgments
We apologize to those authors whose original works were not discussed in this review due to space limitations. We thank Byron Kemper for critical reading of the manuscript. Research discussed from the authors’ laboratory was supported by grants, NIH DK062777, NIH DK095842, and a Basic Science Award from the American Diabetes Association.
References
- Baker RG, Hayden MS, Ghosh S. NF-kappaB, Inflammation, and Metabolic Disease. Cell Metab. 2011;13:11–22. doi: 10.1016/j.cmet.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks AS, Kon N, Knight C, Matsumoto M, Gutierrez-Juarez R, Rossetti L, Gu W, Accili D. SirT1 Gain of Function Increases Energy Efficiency and Prevents Diabetes in Mice. Cell Metab. 2008;8:333–341. doi: 10.1016/j.cmet.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, Ramaswamy S, Fishbein KW, Spencer RG, Lakatta EG, Le Couteur D, Shaw RJ, Navas P, Puigserver P, Ingram DK, de Cabo R, Sinclair DA. Resveratrol Improves Health and Survival of Mice on a High-Calorie Diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. The SREBP Pathway: Regulation of Cholesterol Metabolism by Proteolysis of a Membrane-Bound Transcription Factor. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-Dependent Regulation of FOXO Transcription Factors by the SIRT1 Deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Canto C, Auwerx J. Targeting Sirtuin 1 to Improve Metabolism: All You Need is NAD(+)? Pharmacol Rev. 2012;64:166–187. doi: 10.1124/pr.110.003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Auwerx J. Caloric Restriction, SIRT1 and Longevity. Trends Endocrinol Metab. 2009;20:325–331. doi: 10.1016/j.tem.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK Regulates Energy Expenditure by Modulating NAD+ Metabolism and SIRT1 Activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cermelli S, Ruggieri A, Marrero JA, Ioannou GN, Beretta L. Circulating microRNAs in Patients with Chronic Hepatitis C and Non-Alcoholic Fatty Liver Disease. PLoS One. 2011;6:e23937. doi: 10.1371/journal.pone.0023937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkiadaki A, Guarente L. Sirtuins Mediate Mammalian Metabolic Responses to Nutrient Availability. Nat Rev Endocrinol. 2012;8:287–296. doi: 10.1038/nrendo.2011.225. [DOI] [PubMed] [Google Scholar]
- Chalkiadaki A, Guarente L. Metabolic Signals Regulate SIRT1 Expression. EMBO Rep. 2011;12:985–986. doi: 10.1038/embor.2011.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Bruno J, Easlon E, Lin SJ, Cheng HL, Alt FW, Guarente L. Tissue-Specific Regulation of SIRT1 by Calorie Restriction. Genes Dev. 2008;22:1753–1757. doi: 10.1101/gad.1650608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Liang G, Ou J, Goldstein JL, Brown MS. Central Role for Liver X Receptor in Insulin-Mediated Activation of Srebp-1c Transcription and Stimulation of Fatty Acid Synthesis in Liver. Proc Natl Acad Sci U S A. 2004;101:11245–11250. doi: 10.1073/pnas.0404297101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung O, Puri P, Eicken C, Contos MJ, Mirshahi F, Maher JW, Kellum JM, Min H, Luketic VA, Sanyal AJ. Nonalcoholic Steatohepatitis is Associated with Altered Hepatic MicroRNA Expression. Hepatology. 2008;48:1810–1820. doi: 10.1002/hep.22569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang JY. Bile Acids: Regulation of Synthesis. J Lipid Res. 2009;50:1955–1966. doi: 10.1194/jlr.R900010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JC, Horton JD, Hobbs HH. Human Fatty Liver Disease: Old Questions and New Insights. Science. 2011;332:1519–1523. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coste A, Louet JF, Lagouge M, Lerin C, Antal MC, Meziane H, Schoonjans K, Puigserver P, O’Malley BW, Auwerx J. The Genetic Ablation of SRC-3 Protects Against Obesity and Improves Insulin Sensitivity by Reducing the Acetylation of PGC-1{Alpha} Proc Natl Acad Sci U S A. 2008;105:17187–17192. doi: 10.1073/pnas.0808207105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, Watts L, Booten SL, Graham M, McKay R, Subramaniam A, Propp S, Lollo BA, Freier S, Bennett CF, Bhanot S, Monia BP. MiR-122 Regulation of Lipid Metabolism Revealed by in Vivo Antisense Targeting. Cell Metab. 2006;3:87–98. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Escande C, Chini CC, Nin V, Dykhouse KM, Novak CM, Levine J, van Deursen J, Gores GJ, Chen J, Lou Z, Chini EN. Deleted in Breast Cancer-1 Regulates SIRT1 Activity and Contributes to High-Fat Diet-Induced Liver Steatosis in Mice. J Clin Invest. 2010;120:545–558. doi: 10.1172/JCI39319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RM, Barish GD, Wang YX. PPARs and the Complex Journey to Obesity. Nat Med. 2004;10:355–361. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- Feige JN, Lagouge M, Canto C, Strehle A, Houten SM, Milne JC, Lambert PD, Mataki C, Elliott PJ, Auwerx J. Specific SIRT1 Activation Mimics Low Energy Levels and Protects Against Diet-Induced Metabolic Disorders by Enhancing Fat Oxidation. Cell Metab. 2008;8:347–358. doi: 10.1016/j.cmet.2008.08.017. [DOI] [PubMed] [Google Scholar]
- Finkel T. Ageing: A Toast to Long Life. Nature. 2003;425:132–133. doi: 10.1038/425132a. [DOI] [PubMed] [Google Scholar]
- Gottlieb S, Esposito RE. A New Role for a Yeast Transcriptional Silencer Gene, SIR2, in Regulation of Recombination in Ribosomal DNA. Cell. 1989;56:771–776. doi: 10.1016/0092-8674(89)90681-8. [DOI] [PubMed] [Google Scholar]
- Grundy SM, Brewer HB, Jr, Cleeman JI, Smith SC, Jr, Lenfant C. Definition of Metabolic Syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association Conference on Scientific Issues Related to Definition. Circulation. 2004;109:433–438. doi: 10.1161/01.CIR.0000111245.75752.C6. [DOI] [PubMed] [Google Scholar]
- Guarente L. Sirtuins in Aging and Disease. Cold Spring Harb Symp Quant Biol. 2007;72:483–488. doi: 10.1101/sqb.2007.72.024. [DOI] [PubMed] [Google Scholar]
- Haigis MC, Sinclair DA. Mammalian Sirtuins: Biological Insights and Disease Relevance. Annu Rev Pathol. 5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD. Sterol Regulatory Element-Binding Proteins: Transcriptional Activators of Lipid Synthesis. Biochem Soc Trans. 2002;30:1091–1095. doi: 10.1042/bst0301091. [DOI] [PubMed] [Google Scholar]
- Horton JD, Bashmakov Y, Shimomura I, Shimano H. Regulation of Sterol Regulatory Element Binding Proteins in Livers of Fasted and Refed Mice. Proc Natl Acad Sci U S A. 1998;95:5987–5992. doi: 10.1073/pnas.95.11.5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as Regulators of Metabolism and Healthspan. Nat Rev Mol Cell Biol. 2012;13:225–238. doi: 10.1038/nrm3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hylemon PB, Zhou H, Pandak WM, Ren S, Gil G, Dent P. Bile Acids as Regulatory Molecules. J Lipid Res. 2009;50:1509–1520. doi: 10.1194/jlr.R900007-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional Silencing and Longevity Protein Sir2 is an NAD-Dependent Histone Deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 Complex and SIR2 Alone Promote Longevity in Saccharomyces Cerevisiae by Two Different Mechanisms. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn BB, Alquier T, Carling D, Hardie DG. AMP-Activated Protein Kinase: Ancient Energy Gauge Provides Clues to Modern Understanding of Metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Kalaany NY, Mangelsdorf DJ. LXRS and FXR: The Yin and Yang of Cholesterol and Fat Metabolism. Annu Rev Physiol. 2006;68:159–191. doi: 10.1146/annurev.physiol.68.033104.152158. [DOI] [PubMed] [Google Scholar]
- Kemper JK, Xiao Z, Ponugoti B, Miao J, Fang S, Kanamaluru D, Tsang S, Wu S, Chiang CM, Veenstra TD. FXR Acetylation is Normally Dynamically Regulated by p300 and SIRT1 but Constitutively Elevated in Metabolic Disease States. Cell Metabolism. 2009;10:392–404. doi: 10.1016/j.cmet.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemper JK. Regulation of FXR Transcriptional Activity in Health and Disease: Emerging Roles of FXR Cofactors and Post-Translational Modifications. Biochim Biophys Acta. 2011;1812:842–850. doi: 10.1016/j.bbadis.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klar AJ, Fogel S, Macleod K. MAR1-a Regulator of the HMa and HMalpha Loci in SACCHAROMYCES CEREVISIAE. Genetics. 1979;93:37–50. doi: 10.1093/genetics/93.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HS, Ott M. The Ups and Downs of SIRT1. Trends Biochem Sci. 2008;33:517–525. doi: 10.1016/j.tibs.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J. Resveratrol Improves Mitochondrial Function and Protects Against Metabolic Disease by Activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Lee FY, Lee H, Hubbert ML, Edwards PA, Zhang Y. FXR, a Multipurpose Nuclear Receptor. Trends Biochem Sci. 2006;31:572–580. doi: 10.1016/j.tibs.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Lee J, Kemper JK. Controlling SIRT1 Expression by microRNAs in Health and Metabolic Disease. Aging (Albany NY) 2010;2:527–534. doi: 10.18632/aging.100184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Padhye A, Sharma A, Song G, Miao J, Mo YY, Wang L, Kemper JK. A Pathway Involving Farnesoid X Receptor and Small Heterodimer Partner Positively Regulates Hepatic Sirtuin 1 Levels Via microRNA-34a Inhibition. J Biol Chem. 2010;285:12604–12611. doi: 10.1074/jbc.M109.094524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of Bile Acids and Bile Acid Receptors in Metabolic Regulation. Physiol Rev. 2009;89:147–191. doi: 10.1152/physrev.00010.2008. [DOI] [PubMed] [Google Scholar]
- Li X, Zhang S, Blander G, Tse JG, Krieger M, Guarente L. SIRT1 Deacetylates and Positively Regulates the Nuclear Receptor LXR. Mol Cell. 2007;28:91–106. doi: 10.1016/j.molcel.2007.07.032. [DOI] [PubMed] [Google Scholar]
- Liu Y, Dentin R, Chen D, Hedrick S, Ravnskjaer K, Schenk S, Milne J, Meyers DJ, Cole P, Yates J, 3rd, Olefsky J, Guarente L, Montminy M. A Fasting Inducible Switch Modulates Gluconeogenesis Via activator/coactivator Exchange. Nature. 2008;456:269–273. doi: 10.1038/nature07349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Evans RM. The RXR Heterodimers and Orphan Receptors. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- Meshkani R, Adeli K. Hepatic Insulin Resistance, Metabolic Syndrome and Cardiovascular Disease. Clin Biochem. 2009;42:1331–1346. doi: 10.1016/j.clinbiochem.2009.05.018. [DOI] [PubMed] [Google Scholar]
- Neilson JR, Sharp PA. Small RNA Regulators of Gene Expression. Cell. 2008;134:899–902. doi: 10.1016/j.cell.2008.09.006. [DOI] [PubMed] [Google Scholar]
- Noriega LG, Feige JN, Canto C, Yamamoto H, Yu J, Herman MA, Mataki C, Kahn BB, Auwerx J. CREB and ChREBP Oppositely Regulate SIRT1 Expression in Response to Energy Availability. EMBO Rep. 2011;12:1069–1076. doi: 10.1038/embor.2011.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschop MH. Sirt1 Protects Against High-Fat Diet-Induced Metabolic Damage. Proc Natl Acad Sci U S A. 2008;105:9793–9798. doi: 10.1073/pnas.0802917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponugoti B, Kim DH, Xiao Z, Smith Z, Miao J, Zang M, Wu SY, Chiang CM, Veenstra TD, Kemper JK. SIRT1 Deacetylates and Inhibits SREBP-1C Activity in Regulation of Hepatic Lipid Metabolism. J Biol Chem. 2010;285:33959–33970. doi: 10.1074/jbc.M110.122978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purushotham A, Xu Q, Lu J, Foley JF, Yan X, Kim DH, Kemper JK, Li X. Hepatic Deletion of SIRT1 Decreases Hepatocyte Nuclear Factor 1alpha/farnesoid X Receptor Signaling and Induces Formation of Cholesterol Gallstones in Mice. Mol Cell Biol. 2012;32:1226–1236. doi: 10.1128/MCB.05988-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X. Hepatocyte-Specific Deletion of SIRT1 Alters Fatty Acid Metabolism and Results in Hepatic Steatosis and Inflammation. Cell Metab. 2009;9:327–338. doi: 10.1016/j.cmet.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL, Mangelsdorf DJ. Regulation of Mouse Sterol Regulatory Element-Binding Protein-1c Gene (SREBP-1c) by Oxysterol Receptors, LXRalpha and LXRbeta. Genes Dev. 2000;14:2819–2830. doi: 10.1101/gad.844900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Gerhart-Hines Z, Puigserver P. Metabolic Adaptations through the PGC-1 Alpha and SIRT1 Pathways. FEBS Lett. 2008;582:46–53. doi: 10.1016/j.febslet.2007.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Puigserver P. Fasting-Dependent Glucose and Lipid Metabolic Response through Hepatic Sirtuin 1. Proc Natl Acad Sci U S A. 2007;104:12861–12866. doi: 10.1073/pnas.0702509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient Control of Glucose Homeostasis through a Complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- Russell DW. The Enzymes, Regulation, and Genetics of Bile Acid Synthesis. Annu Rev Biochem. 2003;72:137–174. doi: 10.1146/annurev.biochem.72.121801.161712. [DOI] [PubMed] [Google Scholar]
- Russell DW. Nuclear Orphan Receptors Control Cholesterol Catabolism. Cell. 1999;97:539–542. doi: 10.1016/s0092-8674(00)80763-1. [DOI] [PubMed] [Google Scholar]
- Russell DW, Setchell KDR. Bile Acid Biosynthesis. Biochemistry (NY) 1992;31:4737–4749. doi: 10.1021/bi00135a001. [DOI] [PubMed] [Google Scholar]
- Russell JC, Proctor SD. Small Animal Models of Cardiovascular Disease: Tools for the Study of the Roles of Metabolic Syndrome, Dyslipidemia, and Atherosclerosis. Cardiovasc Pathol. 2006;15:318–330. doi: 10.1016/j.carpath.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Shimano H, Horton JD, Shimomura I, Hammer RE, Brown MS, Goldstein JL. Isoform 1c of Sterol Regulatory Element Binding Protein is Less Active than Isoform 1a in Livers of Transgenic Mice and in Cultured Cells. J Clin Invest. 1997;99:846–854. doi: 10.1172/JCI119248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair DA, Lin SJ, Guarente L. Life-Span Extension in Yeast. Science. 2006;312:195–7. doi: 10.1126/science.312.5771.195d. author reply 195–7. [DOI] [PubMed] [Google Scholar]
- Sinclair DA, Guarente L. Extrachromosomal rDNA Circles–a Cause of Aging in Yeast. Cell. 1997;91:1033–1042. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- Stefan N, Kantartzis K, Haring HU. Causes and Metabolic Consequences of Fatty Liver. Endocr Rev. 2008;29:939–960. doi: 10.1210/er.2008-0009. [DOI] [PubMed] [Google Scholar]
- Tanoli T, Yue P, Yablonskiy D, Schonfeld G. Fatty Liver in Familial Hypobetalipoproteinemia: Roles of the APOB Defects, Intra-Abdominal Adipose Tissue, and Insulin Sensitivity. J Lipid Res. 2004;45:941–947. doi: 10.1194/jlr.M300508-JLR200. [DOI] [PubMed] [Google Scholar]
- Timmers S, Konings E, Bilet L, Houtkooper RH, van de Weijer T, Goossens GH, Hoeks J, van der Krieken S, Ryu D, Kersten S, Moonen-Kornips E, Hesselink MK, Kunz I, Schrauwen-Hinderling VB, Blaak EE, Auwerx J, Schrauwen P. Calorie Restriction-Like Effects of 30 Days of Resveratrol Supplementation on Energy Metabolism and Metabolic Profile in Obese Humans. Cell Metab. 2011;14:612–622. doi: 10.1016/j.cmet.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissenbaum HA, Guarente L. Increased Dosage of a Sir-2 Gene Extends Lifespan in Caenorhabditis Elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Mangelsdorf DJ. Liver X Receptor Signaling Pathways in Cardiovascular Disease. Mol Endocrinol. 2003;17:985–993. doi: 10.1210/me.2003-0061. [DOI] [PubMed] [Google Scholar]
- Trajkovski M, Hausser J, Soutschek J, Bhat B, Akin A, Zavolan M, Heim MH, Stoffel M. MicroRNAs 103 and 107 Regulate Insulin Sensitivity. Nature. 2011;474:649–653. doi: 10.1038/nature10112. [DOI] [PubMed] [Google Scholar]
- Uyeda K, Repa JJ. Carbohydrate Response Element Binding Protein, ChREBP, a Transcription Factor Coupling Hepatic Glucose Utilization and Lipid Synthesis. Cell Metab. 2006;4:107–110. doi: 10.1016/j.cmet.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Walker AK, Yang F, Jiang K, Ji JY, Watts JL, Purushotham A, Boss O, Hirsch ML, Ribich S, Smith JJ, Israelian K, Westphal CH, Rodgers JT, Shioda T, Elson SL, Mulligan P, Najafi-Shoushtari H, Black JC, Thakur JK, Kadyk LC, Whetstine JR, Mostoslavsky R, Puigserver P, Li X, Dyson NJ, Hart AC, Naar AM. Conserved Role of SIRT1 Orthologs in Fasting-Dependent Inhibition of the lipid/cholesterol Regulator SREBP. Genes Dev. 2010;24:1403–1417. doi: 10.1101/gad.1901210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RH, Kim HS, Xiao C, Xu X, Gavrilova O, Deng CX. Hepatic Sirt1 Deficiency in Mice Impairs mTorc2/Akt Signaling and Results in Hyperglycemia, Oxidative Damage, and Insulin Resistance. J Clin Invest. 2011;121:4477–4490. doi: 10.1172/JCI46243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RH, Li C, Deng CX. Liver Steatosis and Increased ChREBP Expression in Mice Carrying a Liver Specific SIRT1 Null Mutation Under a Normal Feeding Condition. Int J Biol Sci. 2010;6:682–690. doi: 10.7150/ijbs.6.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, Moore DD, Auwerx J. Bile Acids Lower Triglyceride Levels Via a Pathway Involving FXR, SHP, and SREBP-1c. J Clin Invest. 2004;113:1408–1418. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakuchi M, Ferlito M, Lowenstein CJ. MiR-34a Repression of SIRT1 Regulates Apoptosis. Proc Natl Acad Sci U S A. 2008;105:13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Schoonjans K, Auwerx J. Sirtuin Functions in Health and Disease. Mol Endocrinol. 2007;21:1745–1755. doi: 10.1210/me.2007-0079. [DOI] [PubMed] [Google Scholar]
- Yoshikawa T, Shimano H, Amemiya-Kudo M, Yahagi N, Hasty AH, Matsuzaka T, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Osuga J, Harada K, Gotoda T, Kimura S, Ishibashi S, Yamada N. Identification of Liver X Receptor-Retinoid X Receptor as an Activator of the Sterol Regulatory Element-Binding Protein 1c Gene Promoter. Mol Cell Biol. 2001;21:2991–3000. doi: 10.1128/MCB.21.9.2991-3000.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide Mononucleotide, a Key NAD(+) Intermediate, Treats the Pathophysiology of Diet- and Age-Induced Diabetes in Mice. Cell Metab. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Castellani LW, Sinal CJ, Gonzalez FJ, Edwards PA. Peroxisome Proliferator-Activated Receptor-Gamma Coactivator 1alpha (PGC-1alpha) Regulates Triglyceride Metabolism by Activation of the Nuclear Receptor FXR. Genes Dev. 2004;18:157–169. doi: 10.1101/gad.1138104. [DOI] [PMC free article] [PubMed] [Google Scholar]