KCNQ2 encephalopathy: Features, mutational hot spots, and ezogabine treatment of 11 patients (original) (raw)
Abstract
Objective:
To advance the understanding of KCNQ2 encephalopathy genotype–phenotype relationships and to begin to assess the potential of selective KCNQ channel openers as targeted treatments.
Methods:
We retrospectively studied 23 patients with KCNQ2 encephalopathy, including 11 treated with ezogabine (EZO). We analyzed the genotype–phenotype relationships in these and 70 previously described patients.
Results:
The mean seizure onset age was 1.8 ± 1.6 (SD) days. Of the 20 EEGs obtained within a week of birth, 11 showed burst suppression. When new seizure types appeared in infancy (15 patients), the most common were epileptic spasms (n = 8). At last follow-up, seizures persisted in 9 patients. Development was delayed in all, severely in 14. The KCNQ2 variants identified introduced amino acid missense changes or, in one instance, a single residue deletion. They were clustered in 4 protein subdomains predicted to poison tetrameric channel functions. EZO use (assessed by the treating physicians and parents) was associated with improvement in seizures and/or development in 3 of the 4 treated before 6 months of age, and 2 of the 7 treated later; no serious side effects were observed.
Conclusions:
KCNQ2 variants cause neonatal-onset epileptic encephalopathy of widely varying severity. Pathogenic variants in epileptic encephalopathy are clustered in “hot spots” known to be critical for channel activity. For variants causing KCNQ2 channel loss of function, EZO appeared well tolerated and potentially beneficial against refractory seizures when started early. Larger, prospective studies are needed to enable better definition of prognostic categories and more robust testing of novel interventions.
Classification of evidence:
This study provides Class IV evidence that EZO is effective for refractory seizures in patients with epilepsy due to KCNQ2 encephalopathy.
In addition to underlying the majority of autosomal dominant cases of benign familial neonatal epilepsy (BFNE), heterozygous variants in KCNQ2 occurring de novo or inherited from a mosaic parent can lead to severe developmental impairment. This was first suggested in the reports of small families with seizures persisting beyond infancy and intellectual disability.1–4 In 2012, a pivotal collaborative study screened 80 patients with early-onset epileptic encephalopathy and found novel KCNQ2 variants in 8.5 Since then, additional examples of “KCNQ2 encephalopathy” have been uncovered in other cohorts.6–12 The distribution of the first few KCNQ2 encephalopathy variants in the structure of the encoded voltage-gated potassium channels implied their potential dominant-negative effects on channel gating, conduction, or surface targeting.13 Indeed, initial studies of those variants in vitro revealed such strong loss of function (60%–90% reduced current).14,15 A majority of known BFNE variants are frameshifts and deletions predicted to reduce the function by half; BFNE missense variants usually exhibit only mild (5%–30%) loss of function in vitro.16 These results suggest that the varying severity of _KCNQ2_-related epilepsy phenotypes may reflect the differences between alleles in the degree of KCNQ2 channel functional deficiency they cause. Recent results indicate that Na+ channel blockers may have some degree of efficacy against KCNQ2 encephalopathy by reducing neuronal hyperactivity arising from KCNQ2 channel deficiency.12 Ezogabine (EZO) (Potiga; GlaxoSmithKline, Philadelphia, PA) acts directly on KCNQ2 channels, increasing their opening; therefore, its use in cases where KCNQ2 variants diminish activity could be beneficial. However, previously reported pediatric use of EZO is limited to a single patient. Here, we describe 23 additional cases of KCNQ2 encephalopathy, including 11 treated with EZO.
METHODS
Patients.
This is a retrospective study of patients followed by pediatric neurologists and geneticists for KCNQ2 encephalopathy. All patients with neonatal-onset epilepsy, developmental delay, and a pathogenic KCNQ2 variant detected during routine clinical care between January 2012 and March 2014 by J.J.M., K.L.P., T.T., B.B.-Z., L.C., R.F., P.M.L., E.M., S.N., V.N., X.R.O.-G., M.C.P., P.L.P., B.P., K.R., E.L.M., M.T., and C.V. were included. Data (demographics; birth, seizure, family, and developmental history; EEG, neuroimaging, and genetic test results; and treatments, drug levels, and treatment responses) were reported by their treating pediatric neurologists (22 patients) and/or a consulting clinical geneticist (1 patient) via an anonymous web-based RedCap survey.17 Seizure classification and magnetic resonance neuroimaging findings were based on the assessment by the treating pediatric neurologist (who directly reviewed the studies) or the geneticist (who reviewed the reports).
EZO treatment.
Ezogabine was prescribed as off-label use of an approved drug, by individual treating neurologists based on their risk-benefit assessment, as part of routine care. Parents were counseled regarding known and potential unknown risks. EZO was given 3 times daily, at milligrams-per-kilogram doses extrapolated from recommendations in adults (table 1). Patients were monitored for known side effects of chromaturia (red-brown urine) and urinary retention18,19 at baseline and during periodic urology visits, by renal/bladder ultrasound and parental diaper counting, as determined by each clinician. No patients were begun on treatment after the U.S. Food and Drug Administration published a warning and revised labeling indicating that EZO can cause retinal pigmentation abnormalities with potential risk of visual loss.20–22 Parents of all patients under treatment at that time were informed of the warning and its detailed content, and the risks and benefits of continued treatment reassessed. Where treatment continued after this warning, the recommended visual monitoring was followed and included dilated funduscopic examinations and assessment of visual fields. Commercial laboratories were used to measure serum levels. Adult mean and 5%–95% range EZO levels were estimated from a published pharmacokinetic model.23
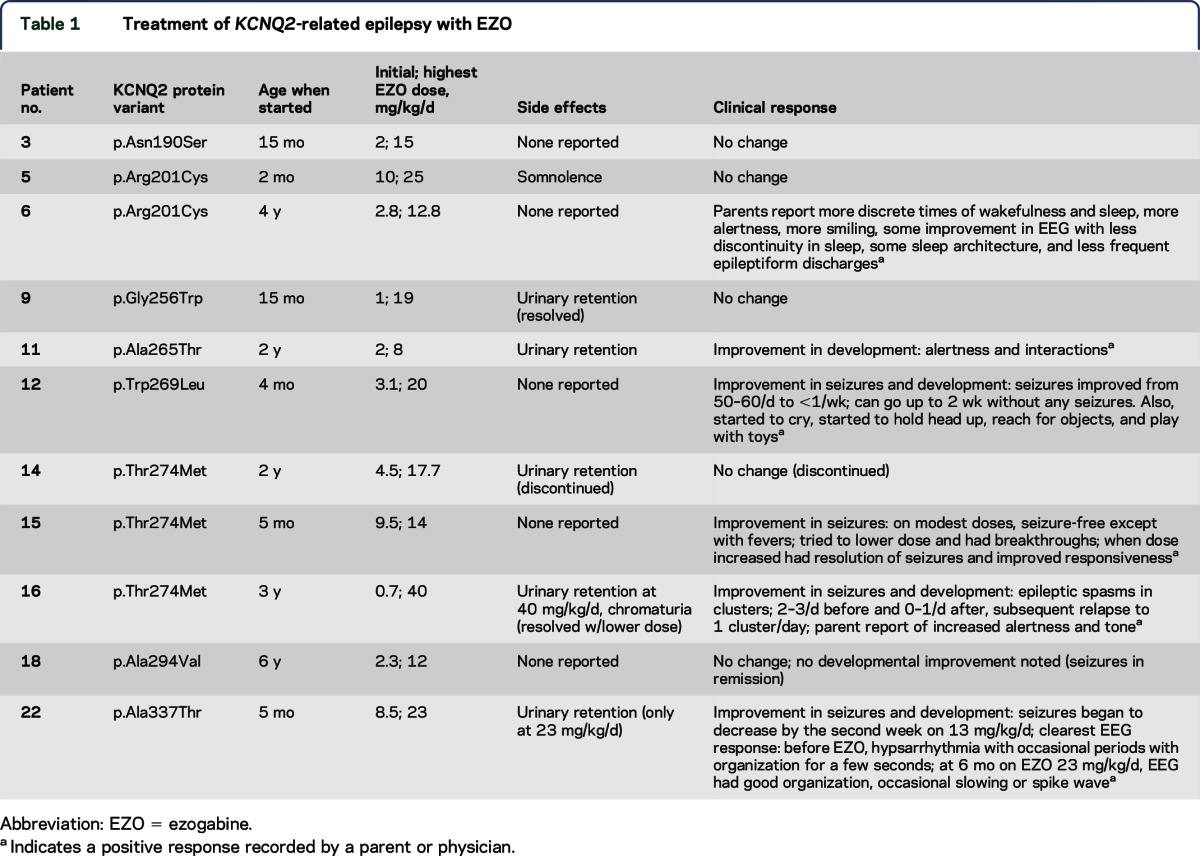
Table 1.
Treatment of _KCNQ2_-related epilepsy with EZO
Standard protocol approvals, registrations, and patient consents.
The retrospective anonymous physician survey was reviewed and exempted by the Colorado Children's Hospital Institutional Review Board. The Rational Intervention for KCNQ2/3 Epileptic Encephalopathy (RIKEE) database and website (www.RIKEE.org) was approved by the Baylor College of Medicine Institutional Review Board.
Research questions and classification of evidence.
This study sought to provide additional information addressing the question, “What are the clinical features of KCNQ2 encephalopathy, including genotype–phenotype relationships and pharmacoresponsiveness?” The study provides Class IV evidence that EZO is effective for the treatment of refractory seizures in patients with epilepsy due to pathogenic KCNQ2 variants.
RESULTS
Patient, family, and epilepsy characteristics.
Of the 23 patients, 12 were female (table e-1 at Neurology.org/ng). Births occurred at a mean gestational age of 38.6 ± 1.16 weeks (SD). A family history of seizures was reported for 5/23 patients. Seizure onset was within the first 3 days of life for 22 patients and on the eighth day of life for 1. Initial seizure frequency was multiple daily for 19 patients. The initial seizure types, as reported by the treating pediatric neurologist, were focal (n = 9), tonic (n = 6), myoclonic (n = 4), mixed focal and generalized (n = 1), generalized tonic-clonic (n = 1), and epileptic spasms (n = 1). Patients developed other seizure types during infancy; the most common were epileptic spasms (n = 8). Seizures were refractory to 3 or more anticonvulsants used alone or in combination in 21 of the 23 patients. Nonetheless, seizures had remitted in 6 of the 12 who were under 2 years of age and in 8 of the 11 who were older than 2 years at last follow-up.
Spectrum of developmental impairment.
Reporting pediatric neurologists made assessments of the development based on milestone achievement at follow-up. For this cohort, mild impairment consisted of appropriate milestones achieved slightly delayed for age, moderate impairment consisted of achievement of most milestones but temporally delayed by >50% (e.g., most walking supported, some speech and/or other communication), and severe impairment consisted of limited or no achievement of developmental milestones in motor, language, and cognitive domains. No patients had normal development. At last follow-up, development was mildly abnormal in 3/23 patients, moderately abnormal in 6/23, and severely abnormal in 14/23. Some mildly to moderately impaired patients were noted to exhibit autistic features. Other neurologic impairments included hypotonia, dysphagia requiring gastrostomy tube, dystonia, spastic quadriplegia, abnormal eye movements, and cortical blindness.
Neonatal EEG and its evolution.
A locally interpreted EEG report from the first week of life was available for 20/23 patients; 11 showed burst suppression, 11 showed dysmaturity and/or focal findings, and 1 was normal. One or more subsequent neonatal or post-neonatal EEGs were available for 21 patients. One EEG continued to show burst suppression with longer bursts and shorter interburst intervals at 3 weeks old. Of the 20 other follow-up EEGs, abnormalities included diffuse/multifocal epileptiform discharges (n = 11), diffuse background slowing (n = 6), background disorganization (n = 6), asymmetric background (n = 2), and hypsarrhythmia (n = 20). The single normal follow-up EEG (at 3 months old) was of patient 1, a child mildly affected in other respects: the neonatal EEG did not show burst suppression, seizures remitted by 1 month old, and developmental impairment was mild.
Neuroimaging.
MRI scans were performed for 22 patients at a mean age of 4 months (range, 1 day to 22 months old); 16 were reported as normal. All abnormal MRI scans except one (at 5 months old) were performed within the first week of life. Abnormalities, as reported by the treating pediatric neurologist, included diffuse atrophy (n = 3), periventricular increased white matter signal (n = 2), thin corpus callosum (n = 3), and increased diffusion in the bilateral globus pallidi (n = 1).
KCNQ2 variant classification.
Sequencing revealed 17 different KCNQ2 variants. We assessed each test result based on a set of criteria: (1) Was the newly sequenced variant a recurring variant identified in another KCNQ2 encephalopathy or BFNE patient or pedigree? (2) Was it a substitution variant resulting in a single amino acid change (i.e., a missense variant)? (3) Was it inherited or de novo? (4) Was the variant located in 1 of the 4 hot spots for the encephalopathy phenotype, namely the S4 voltage-sensor, the pore, the proximal intracellular calmodulin-binding helix, or the distal calmodulin-binding helix?13 Answers to those questions and the clinical features were considered in developing a practical assessment of whether each variant was likely to be pathogenic.
Of the 17 KCNQ2 variants found in our patients, 3 had been reported previously in single epileptic encephalopathy patients, and 4 had been reported in multiple unrelated epileptic encephalopathy patients. Surprisingly, 2 variants (identified in sib pairs 1, 2 and 19, 20) had been identified previously in individuals with BFNE. The 8 remaining variants were novel. One of the novel variants (identified in patient 7) was shown to be inherited from a parent with a BFNE history; 5 of the novel variants occurred de novo; for 2 additional cases, parents had not been tested. Lack of parental testing was typically due to lack of insurance coverage.
Patients 1 and 2, a sibling pair, both exhibiting mild developmental delay, harbored a missense variant, p.Ser122Leu, a position that is outside the KCNQ2 encephalopathy hot zones. The father of patients 1 and 2 had recurrent seizures between 3 months and 2 years of age that remitted but recurred in adolescence; he is now seizure-free without medication and of normal intellect. Parental genetic testing was nevertheless not performed. Notably, p.Ser122Leu was previously reported in a pedigree with 3 affected individuals in 2 generations and normal developmental outcome in all.24 Patient 7, who was mildly delayed developmentally and had an abnormal EEG at 9 months of age, inherited a previously unreported variant (p.Gln204His) from her mother. The mother had been treated for neonatal seizures that remitted and remained seizure-free except for a single seizure at 15 years of age. These 2 families provide examples of variable expressivity at the milder end of the _KCNQ2_-related epilepsy spectrum.
Of the 17 KCNQ2 variants found, 16 resulted in single amino acid substitutions (missense). The exception, a de novo single amino acid deletion (Phe305del), was identified in identical twins (patients 19, 20). Our review of the literature showed no previous examples of single amino acid (or larger) deletions in KCNQ2 encephalopathy patients. Furthermore, Phe305del was previously reported in a de novo case of BFNE,25 while patients 19 and 20 were profoundly delayed. Review of the earlier case report suggested the possibility of mosaicism in the BFNE patient. The 17 KCNQ2 variants were mapped onto a model of the channel's functional domains (figure 1). In 8 cases, the variant was within the voltage-sensor domain (5 within the S4 helix). In 12 cases, the variant was within the pore domain. The 3 remaining cases were within the long C-terminal intracellular domain, within or near the 2 calmodulin-binding helices.
Figure 1. Type and distribution within KCNQ2 protein of variants in self-limiting epilepsy vs epileptic encephalopathy.

Cartoon of KCNQ2 transmembrane topology, including intracellular amino (N) and carboxy (C) terminals, 6 transmembrane segments (S1–S6), and a pore loop between S5 and S6 which partly enters the membrane. The calmodulin-binding segments within the intracellular C-terminal domain are indicated. (A) Mutations found in BFNE/BFNIS/BFIS cases are distributed among all areas of the polypeptide sequence. Each symbol (n = 98) represents a de novo case or a pedigree, placed at the location within the channel polypeptide corresponding to the sequenced genetic variant. Orange symbols (n = 41) are missense variants. Yellow symbols are all other variant types (i.e., loss of start codon, insertions or deletions changing frame, and premature stop codon). Encircled regions, with the exception of the S4 helix, do not appear enriched in missense variants. (B) In patients with KCNQ2 encephalopathy, mutations are nearly always single nucleotide substitutions resulting in a single amino acid change. In one instance (cases 19 and 20 of this series, 2 identical twins), a single amino acid is deleted within an α-helix. Each red sphere represents an unrelated case. Encircled are the 4 hot spots for variants leading to epileptic encephalopathy: the S4 voltage-sensor, the pore, the proximal C-terminal domain that binds phosphatidylinositol 4,5-bisphosphate (PIP2) and calmodulin (CaM A), and the more distal domain which binds calmodulin (CaM B). Variant details for (A) are listed in table e-2; variant details for (B) are listed in table e-3.
Treatment with EZO.
Eleven patients were treated on a TID schedule with EZO, beginning at ages between 2 months and 6 years (table 1). Serum levels were obtained prior to the first or second daily dose in 5 patients (9 measurements). We compared the pediatric levels to a previously reported pharmacokinetic model predicting adult peak (approximately 1 hour after oral dosing) and trough (8 hours after dosing) mean levels and ranges, based on a total of 15,420 adult serum levels obtained during regulatory approval trials.23 Although the number of pediatric measurements was insufficient to allow for detailed analysis, the pediatric levels were all within the range seen in adults (figure 2). Dose-related side effects observed included urinary retention, chromaturia, and somnolence, which all have been reported in adults.18,19 All resolved with dose modification. Serious adverse side effects, including ophthalmologic and skin pigmentary changes included in the FDA black box warning, were not reported during the follow-up period (mean, 11.5 months; range, 2–24 months). These adverse effects were typically observed after 4 or more years of treatment,20,21 which exceeded the amount of follow-up available in our treated patients. Three of the 4 patients treated at less than 6 months of age were reported to exhibit improvement in seizures and development. These patients had failed multiple standard medication trials (at least 4) including several agents with activity on voltage-gated sodium channels (1 tried phenytoin and all 3 responders tried topiramate previously). Detailed dosing information regarding these prior treatment trials was not obtained in our survey. In patient 22, serial neonatal and before and after EZO treatment EEGs were obtained (figure e-1). The neonatal burst-suppression pattern evolved in early infancy to hypsarrhythmia that proved refractory to multiple medications (levetiracetam, topiramate, clobazam, clonazepam, felbamate, phenobarbital, valproic acid, and pyridoxine). Addition of EZO was associated with the emergence of more organization, but persistent intermittent slowing and occasional epileptiform activity were also noted.
Figure 2. EZO levels in infants and young children compared to adults at similar doses.

Adult mean (middle line in bar) and 95%–5% range (top and bottom of bar) EZO levels are shown, along with the levels measured in this study. Adult values at 1 and 8 hours after dosing are taken from a pharmacokinetic model fit to serum EZO levels collected during regulatory trials.23 Adult doses of 300 mg TID (A) and 400 mg TID (B) correspond to 11.8 and 15.8 mg/kg/d, respectively (mean adult study patient weight was 76 ± 16.5 kg, n = 1,219). EZO levels measured in infants and young children, indicated by circles (red, age 6–16 months; blue, age 19 months to 6 years) were within the range observed for comparable doses in adults, and means were similar or somewhat higher than those measured in adults.
Notably, the 3 patients (patients 12, 15, and 22) treated before 6 months of age and who showed improvement had variants in or near the ion-conducting pore domain. By contrast, the patient treated before 6 months of age and who had no response to EZO bore a variant in the S4 helix (patient 5). In 7 patients who started EZO at a later age, where seizures had remitted or were infrequent, our survey tool provided less clear evidence of drug effects. Patient 16, who started treatment when 3 years of age, had an improvement in seizures and development according to the pediatric neurologist and parents. One patient treated at 4 years of age was reported to have improved alertness and EEG background activity (patient 6); 1 patient whose seizures had already remitted when treated at 2 years of age had improvement in development only (patient 11). The association between age groups (<6 months and >6 months) and improved or not improved seizure control with EZO treatment is considered to be statistically significant (Fisher exact test; 2-tailed p value = 0.0242).
DISCUSSION
KCNQ2 variants arising de novo or, more rarely, inherited from a parent have emerged as an important cause of early-onset epileptic encephalopathy. Including the present study, 93 KCNQ2 encephalopathy patients have been reported so far.5–11,14,15,26,27 Considering all cases reported with sufficient phenotypic detail (and omitting a single gain-of-function outlier, discussed below), the most consistent clinical features are very early onset at 2.65 ± 2.26 (SD) days (n = 88) with focal tonic seizures (89% n = 84) and the transient appearance of burst suppression in the EEG (62% n = 86) within the early neonatal period. In most patients, seizures are initially refractory and sometimes evolve through infantile spasms and associated hypsarrhythmia to eventual seizure freedom (73% n = 83). Severe intellectual disability (79% n = 77) emerges in early childhood. In later infancy and childhood, autistic features (language and social impairment, repetitive behaviors, outbursts including potentially self-injurious behavior) are also notable.
Analysis of the first cases suggested a dominant-negative effect as the basis for the severe phenotype in these patients in contrast to the better outcomes in BFNE.13 The clues supporting this hypothesis were that the severe KCNQ2 variants were all missense (unlike BFNE variants, which are diverse and include large deletions) and that they were clustered in putative functional hot spots, unlike BFNE variants, which are randomly distributed throughout the channel polypeptide (figure 1). Moreover, earlier, comprehensive studies of transgenic mice overexpressing a dominant-negative KCNQ2 variant had revealed a severe phenotype.13,28 Because KCNQ2 proteins form tetrameric channels (either homotetramers consisting of KCNQ2 subunits only or heterotetramers consisting of both KCNQ2 and KCNQ3 subunits), variants of one subunit at certain critical locations can poison the entire channel. Of the variants identified in 93 KCNQ2 encephalopathy patients reported with clinical information, 86% are clustered in 4 high-risk zones: the S4 voltage-sensor, the pore, near the C-terminal proximal segment which binds phosphatidylinositol 4,5-bisphosphate and calmodulin, and near the C-terminal B helix, which also binds calmodulin.29 These observed high-risk zones represent less than 20% of the KCNQ2 amino acids. In the context of known potassium channel mechanisms deduced from functional analysis and X-ray crystallography, and initial in vitro studies,14,15 this accumulated clinico-genetic evidence strongly supports dominant-negative suppression as a primary factor underlying severe phenotypes in a large majority of KCNQ2 epileptic encephalopathy cases.13
The exceptions to the usual pattern merit careful analysis. For example, patients 19 and 20, identical twins, were severely affected and bore a single amino acid deletion variant (Phe305del), the first deletion to be reported in KCNQ2 encephalopathy. Notably, a previous report described de novo deletion of the same amino acid in a young female with a classical BFNE phenotype.25 The published DNA sequencing trace data from this earlier case, however, revealed a relatively low abundance of the mutated allele compared to the wild-type allele, raising the possibility of unrecognized mosaicism. Indeed, 2 studies have identified families where a parent was mosaic for a KCNQ2 variant and had experienced BFNE, but their child, a full heterozygote, exhibited severe epileptic encephalopathy.5,30 Thus, sporadic, benign neonatal epilepsy may be caused by a de novo germ line KCNQ2 variant with a mild effect or by post-zygotic mosaicism for a more severe variant. These findings illustrate that screening for mosaicism and accurate variant classification in individuals with apparent de novo KCNQ2 variants are crucial and have important implications for reproductive counseling of such patients.
Three patients (1, 2, and 7), all mildly delayed, had a parent with a history of neonatal seizures. A previous study noted mild developmental delay in 5 of 31 patients in a large KCNQ2 pedigree where other family members exhibited classical BFNE and normal development.31 Such variable expressivity may reflect genetic or nongenetic factors and should be analyzed through broader genetic testing in suitable families.
Previously reported experience with EZO treatment of infants and children with _KCNQ2_-related epilepsy is limited to 1 patient bearing the pore variant p.Gly281Arg, who experienced a marked reduction in seizure severity and frequency when given 40 mg/kg before 22 months of age.11 Although limited by a retrospective survey design and modestly detailed treatment data, the current study describes 11 additional patients treated at ages ranging from 2 months to 6 years. Overall, the patients tolerated these exposures well, with side effects that were dose related, relatively minor, and reversible. The clearest benefit was against refractory seizures, when started before 6 months of age. Including the previously reported case, 4 of 4 patients with pore domain variants treated with EZO as infants experienced what parents and treating doctors described as a clearly positive influence on seizures. Experiments in vitro using Xenopus oocytes have shown that one of these pore domain variants, p.Thr274Met, strongly reduces channel currents and that those effects were reversed by EZO.15 In contrast, for patient 5, EZO at 2 months of age had no apparent effect on seizures. The amino acid variant identified in this patient, p.Arg201Cys, lies within the extracellular portion of the S4 voltage sensor and has been shown to cause gain-of-function by destabilizing the channel closed state.32 The available results suggest that individuals with loss of function may benefit, but those with gain-of-function KCNQ2 variants may not benefit from treatment with KCNQ2 openers. Notably, patient 6, also with p.Arg201Cys, was treated between ages 4 and 6 years, with apparent improvements in behavior and EEG. Studies of larger numbers of patients will be needed to assess such differences.
A recent study analyzed responses of 15 patients with KCNQ2 encephalopathy to several approved antiepileptic drugs, including the sodium-channel inhibitors carbamazepine, phenytoin, and oxcarbazepine.12 Retrospective information was obtained regarding medication dosage and seizure responses to each treatment. In this cohort, 11 patients experienced remission or near remission to either carbamazepine or phenytoin. Three patients had recurrence of seizures following withdrawal of the anticonvulsant with return of control after reintroduction, suggesting that at least several of the responders required ongoing management with the anticonvulsant in question. As in the current study, due to the natural history of this disorder, with frequent resolution of seizures (but persistence of developmental impairment) in infancy, the lack of a second trial in the remaining responders makes the assessment of response more difficult. Also, as in the current series, the heterogeneity in cognitive, language, and motor outcomes associated with different KCNQ2 variants makes it difficult to determine whether early seizure control treatment has affected neurodevelopmental outcome. Further, until clearer KCNQ2 encephalopathy clinical diagnostic criteria emerge or an established pathogenic variant is detected, concerns that carbamazepine can provoke infantile spasms in susceptible patients is another reason for caution.33
The current retrospective study highlights challenges in designing future targeted trials in KCNQ2 encephalopathy. Here, EZO was given after genetic diagnosis, at minimum, months after birth and seizure onset. EZO, though the first KCNQ2 channel opener to obtain regulatory approval, has relatively modest potency and selectivity.34–39 Newer agents, with improved channel subtype-selectivity and greater potency, have been identified, but remain at earlier stages of development.38,39 Optimal testing of KCNQ2 openers as “precision medicines” for KCNQ2 loss-of-function variants will require early diagnosis, clinical advancement of more potent and selective agents, and prospective trials.
Supplementary Material
Data Supplement
Coinvestigators
ACKNOWLEDGMENT
The authors acknowledge participating families for their support of this research.
GLOSSARY
BFNE
benign familial neonatal epilepsy
EZO
ezogabine
Footnotes
AUTHOR AFFILIATIONS
From the Epilepsy Center (J.J.M., S.N.), Ann & Robert H. Lurie Children's Hospital of Chicago, Departments of Pediatrics and Neurology, Northwestern University Feinberg School of Medicine, IL; Division of Neurology, Department of Pediatrics (K.L.P., P.M.L.), University of Colorado School of Medicine, Aurora; Departments of Pediatrics and Neurology (T.T.), Children's National Medical Center, Washington, DC; Department of Pediatrics (B.B.-Z.), Edmond and Lilly Safra Pediatric Hospital, Sheba Medical Center, Ramat-Gan, and Sackler School of Medicine, Tel-Aviv University, Israel; Department of Pediatrics (L.C.), Sainte-Justine Hospital, University of Montreal, Canada; PANDA Neurology (R.F.), Atlanta, GA; Department of Neurology (B.T., N.J., E.C.C.), Baylor College of Medicine, Departments of Neuroscience and Molecular and Human Genetics, Baylor College of Medicine, Houston, TX; Departments of Pediatrics and Neurology (E.M., X.R.O.-G., E.L.M.), Children's Hospital of Philadelphia, Perelman School of Medicine of the University of Pennsylvania; Dorrance Center for Rare Childhood Disorders (V.N., K.R., M.H., R.R., I.S., A.S.), The Translational Genomics Research Institute, Phoenix, AZ; Department of Neurology, Pediatrics, and Medical Genetics (M.C.P.), Mayo Clinic Children's Center, Rochester, MN; Departments of Pediatrics and Neurology (P.L.P.), Boston Children's Hospital, MA; Department of Neurology (B.P.), Stanford University, CA; Department of Medicine and Health Science (M. Taglialatela), University of Molise, Campobasso, Italy; Departments of Pediatrics and Neurology (M. Tracy), Brown University, Providence, RI; Neurology Division (M. Tracy), Department of Pediatrics, Cincinnati Children's Hospital Medical Center (C.V.), OH; Neurogenetics Group (S.W.), Department of Molecular Genetics, VIB, Antwerp, Belgium; Inserm U 1127 (S.W.), CNRS UMR 7225, Sorbonne Universités, UPMC Univ Paris 06 UMR S 1127, Institut du Cerveau et de la Moelle épinière, Service de Neurologie, Paris, France; Virtual Medical Practice, LLC (F.K.), Atlanta, GA; and Department of Pediatrics (M.S.), University of Utah, Salt Lake City.
AUTHOR CONTRIBUTIONS
J.J.M.: design of the study, interpretation of the data, drafting of the manuscript. K.L.P., E.C.C.: design of the study, interpretation of the data, revising of the manuscript for intellectual content. T.T., B.B.-Z., L.C., R.F., N.J., P.M.L., E.M., S.N., V.N., X.R.O.-G., M.C.P., P.L.P., B.P., K.R., E.L.M., M. Taglialatela, M. Tracy, B.T., C.V., and S.W.: interpretation of the data, revising of the manuscript for intellectual content.
STUDY FUNDING
This study was supported by a Research Infrastructure Grant funded jointly by the American Epilepsy Society and the Epilepsy Foundation and by The Jack Pribaz Foundation.
DISCLOSURE
Dr. Millichap serves as an Associate Editor of _Neurology_® and serves on the editorial board of Pediatric Neurology Briefs; volunteers on the medical advisory board of the Jack Pribaz Foundation (KCNQ2.org); received royalties for online monographs from Up-To-Date and BMJ Best Practice; received speaker honoraria from Invitae; served on the scientific advisory board for Mallinkrodt; is principal investigator for a clinical trial funded by UCB Pharma; and is the principal investigator for research grants from Citizens United for Research in Epilepsy (CURE) and Thrasher Research Fund. Dr. Park is funded by a grant from the Epilepsy Study Consortium; received research support from Ovation Pharmaceuticals and Eisai Pharmaceuticals; has received travel funding from Visualase; and was funded by NIH-National Institute of Neurological Disorders and Stroke as a site Principal Investigator for the Epilepsy Phenome Genome Project (U01 NS053998). Dr. Tsuchida has received research support from the Eunice Kennedy Shriver National Institute of Child Health and Human Development, the CURE Foundation, and the Epilepsy Foundation. Dr. Ben-Zeev has served on the editorial board of the European Journal of Pediatric Neurology; and has been a consultant to a startup company developing a drug for Rett syndrome. Dr. Carmant has served on the scientific advisory boards of UCB and Eisai; has received travel funding/speaker honoraria from UCB; and has received research support from Lundbeck US and the CURE Foundation. Dr. Flamini is part of the speaker's bureaus for Lundbeck, Eisai, and Supernus Pharmaceuticals; has received speaker honoraria from Lundbeck and Eisai; and has received research support from GW Pharma. Nishtha Joshi has received travel funding from CURE. Dr. Levisohn reports no disclosures. Dr. Marsh has served on the scientific advisory boards of Stanley Brothers Social Enterprises and the Jefferson University Cannabis Research and Education Center; has served on the editorial board of Epilepsia; has received research support from NIH; is the PI of an RO1 NS082761-02; is currently participating in a GW Pharma study of CBD for Dravet and LGS syndrome; and completed a study with Eisai Pharmaceuticals for rufinamide. Dr. Nangia reports no disclosures. Dr. Narayanan has served on the editorial boards of Pediatric Neurology, the Journal of Child Neurology, and the Journal of Pediatric Neurology; and has received research support from Novartis. Dr. Ortiz-Gonzalez has received research support from National Institute of Neurological Disorders and Stroke and the Robert Wood Johnson Foundation (Harold Amos Award). Dr. Patterson serves as Editor-in-Chief of the Journal of Child Neurology and Child Neurology Open, as an editor of the Journal of Inherited Metabolic Disease, and as a section editor (Pediatric Neurology) for Up-To-Date; has received research support from the NIH (U54NS065768), the National MS Society, Merck Serono, and Actelion Pharmaceuticals; has received travel funding/speaker honoraria from Actelion Pharmaceuticals; has consulted for Actelion Pharmaceuticals, Agios, Amicus, Abbvie, Alexion, Cydan, Genzyme, Orphazyme, Shire HGT, Stem Cells Inc., and Vtesse; has served on a committee for the Institute of Medicine; has served on the WHO International Advisory group for the Revision of ICD-10 Diseases of the Nervous System; and serves on scientific advisory boards of the CDG Family Network, Actelion Pharmaceuticals, Shire Human Genetic Therapies Inc., Working Group, Stem Cells Inc., Amicus Therapeutics, Vtesse, Dana's Angels Research Trust, GLIA, the International Niemann-Pick Disease Foundation, the MLD Foundation, and the National Niemann-Pick Disease Foundation. Dr. Pearl has served on the editorial boards of the Journal of Child Neurology, Annals of Neurology, Neurology, Faculty of 1000, Scientific American Neurology, and Music and Medicine; receives research support from NIH grant HD58553; and receives royalty payments from Up-To-Date and Demos Medical Publishers for the books Inherited Metabolic Epilepsies and Neuro-Logic: A Guide to Localization. Dr. Porter served on the scientific advisory boards of Lundbeck, the Lennox-Gastaut Syndrome Foundation, the CURE Epilepsy Foundation of Northern California, and the Tess Research Foundation; has served on the editorial board of Frontiers in Neurology; has stock and stock options in Johnson and Johnson; and has received research support from Stanford University, Johnson and Johnson, the Simmons Foundation, and NIH NS05622. Keri Ramsey has received research support from TGen. Dr. McGinnis reports no disclosures. Dr. Taglialatela has served on the editorial board of Frontiers in Pharmacology of Ion Channels and Channelopathies; and receives research support from Telethon (GGP15113). Dr. Tracy, Baouyen Tran, Dr. Venkatesan, and Dr. Weckhuysen report no disclosures. Dr. Cooper has received travel funding/speaker honoraria from GlaxoSmithKline, Northwestern University, the University of Montreal, the Gordon Research Conference, and CURE; has served on the editorial board of Epilepsy Currents; receives research support from Baylor College of Medicine, the American Epilepsy Society, the Epilepsy Foundation, KCNQ2 Cure Alliance, NIH/National Institute of Neurological Disorders and Stroke grant R01 NS49119, a research grant from GlaxoSmithKline, a research grant from the Jack Pribaz Foundation, and a research grant from Citizens United for Research in Epilepsy (CURE); is a consultant to SciFluor Biosciences Inc; and serves (no financial compensation) on the scientific advisory boards for the Jack Pribaz Foundation and KCNQ2 Cure Alliance Foundation. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Borgatti R, Zucca C, Cavallini A, et al. . A novel mutation in KCNQ2 associated with BFNC, drug resistant epilepsy, and mental retardation. Neurology 2004;63:57–65. [DOI] [PubMed] [Google Scholar]
- 2.Dedek K, Fusco L, Teloy N, Steinlein OK. Neonatal convulsions and epileptic encephalopathy in an Italian family with a missense mutation in the fifth transmembrane region of KCNQ2. Epilepsy Res 2003;54:21–27. [DOI] [PubMed] [Google Scholar]
- 3.Schmitt B, Wohlrab G, Sander T, Steinlein OK, Hajnal BL. Neonatal seizures with tonic clonic sequences and poor developmental outcome. Epilepsy Res 2005;65:161–168. [DOI] [PubMed] [Google Scholar]
- 4.Steinlein OK, Conrad C, Weidner B. Benign familial neonatal convulsions: always benign?. Epilepsy Res 2007;73:245–249. [DOI] [PubMed] [Google Scholar]
- 5.Weckhuysen S, Mandelstam S, Suls A, et al. . KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann Neurol 2012;71:15–25. [DOI] [PubMed] [Google Scholar]
- 6.Allen NM, Mannion M, Conroy J, et al. . The variable phenotypes of KCNQ-related epilepsy. Epilepsia 2014;55:e99–e105. [DOI] [PubMed] [Google Scholar]
- 7.Kato M, Yamagata T, Kubota M, et al. . Clinical spectrum of early onset epileptic encephalopathies caused by KCNQ2 mutation. Epilepsia 2013;54:1282–1287. [DOI] [PubMed] [Google Scholar]
- 8.Milh M, Boutry-Kryza N, Sutera-Sardo J, et al. . Similar early characteristics but variable neurological outcome of patients with a de novo mutation of KCNQ2. Orphanet J Rare Dis 2013;8:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Numis AL, Angriman M, Sullivan JE, et al. . KCNQ2 encephalopathy: delineation of the electroclinical phenotype and treatment response. Neurology 2014;82:368–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saitsu H, Kato M, Koide A, et al. . Whole exome sequencing identifies KCNQ2 mutations in Ohtahara syndrome. Ann Neurol 2012;72:298–300. [DOI] [PubMed] [Google Scholar]
- 11.Weckhuysen S, Ivanovic V, Hendrickx R, et al. . Extending the KCNQ2 encephalopathy spectrum: clinical and neuroimaging findings in 17 patients. Neurology 2013;81:1697–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pisano T, Numis AL, Heavin SB, et al. . Early and effective treatment of KCNQ2 encephalopathy. Epilepsia 2015;56:685–691. [DOI] [PubMed] [Google Scholar]
- 13.Millichap JJ, Cooper EC. KCNQ2 potassium channel epileptic encephalopathy syndrome: divorce of an electro-mechanical couple? Epilepsy Curr 2012;12:150–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miceli F, Soldovieri MV, Ambrosino P, et al. . Genotype-phenotype correlations in neonatal epilepsies caused by mutations in the voltage sensor of K(v)7.2 potassium channel subunits. Proc Natl Acad Sci USA 2013;110:4386–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orhan G, Bock M, Schepers D, et al. . Dominant-negative effects of KCNQ2 mutations are associated with epileptic encephalopathy. Ann Neurol 2013;75:382–394. [DOI] [PubMed] [Google Scholar]
- 16.Miceli F, Soldovieri MV, Joshi N, Weckhuysen S, Cooper E, Taglialatela M. _KCNQ2_-related disorders. In: Pagon RA, Adam MP, Ardinger HH, et al. editors. GeneReviews [serial online]. Available at: http://www.ncbi.nlm.nih.gov/books/NBK32534/. Accessed June 2, 2016. [Google Scholar]
- 17.Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)–a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform 2009;42:377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brickel N, Gandhi P, VanLandingham K, Hammond J, DeRossett S. The urinary safety profile and secondary renal effects of retigabine (ezogabine): a first-in-class antiepileptic drug that targets KCNQ (K(v)7) potassium channels. Epilepsia 2012;53:606–612. [DOI] [PubMed] [Google Scholar]
- 19.Brodie MJ, Lerche H, Gil-Nagel A, et al. . Efficacy and safety of adjunctive ezogabine (retigabine) in refractory partial epilepsy. Neurology 2010;75:1817–1824. [DOI] [PubMed] [Google Scholar]
- 20.FDA Drug Safety Communication: anti-seizure drug Potiga (ezogabine) linked to retinal abnormalities and blue skin discoloration (4-26-2013) [online]. Available at: http://www.fda.gov/Drugs/DrugSafety/ucm349538.htm. Accessed December 15, 2014.
- 21.FDA Drug Safety Communication: anti-seizure drug Potiga (ezogabine) linked to retinal abnormalities and blue skin discoloration (10-31-2013) [online]. Available at: http://www.fda.gov/Drugs/DrugSafety/ucm372774.htm. Accessed December 15, 2014.
- 22.Garin Shkolnik T, Feuerman H, Didkovsky E, et al. . Blue-gray mucocutaneous discoloration: a new adverse effect of ezogabine. JAMA Dermatol 2014;150:984–989. [DOI] [PubMed] [Google Scholar]
- 23.Tompson DJ, Crean CS, Reeve R, Berry NS. Efficacy and tolerability exposure-response relationship of retigabine (ezogabine) immediate-release tablets in patients with partial-onset seizures. Clin Ther 2013;35:1174–1185.e4. [DOI] [PubMed] [Google Scholar]
- 24.Hunter J, Maljevic S, Shankar A, et al. . Subthreshold changes of voltage-dependent activation of the K(V)7.2 channel in neonatal epilepsy. Neurobiol Dis 2006;24:194–201. [DOI] [PubMed] [Google Scholar]
- 25.Ishii A, Fukuma G, Uehara A, et al. . A de novo KCNQ2 mutation detected in non-familial benign neonatal convulsions. Brain Dev 2009;31:27–33. [DOI] [PubMed] [Google Scholar]
- 26.Saadeldin IY, Milhem RM, Al-Gazali L, Ali BR. Novel KCNQ2 mutation in a large Emirati family with benign familial neonatal seizures. Pediatr Neurol 2013;48:63–66. [DOI] [PubMed] [Google Scholar]
- 27.Samanta D, Ramakrishnaiah R, Willis E, Frye RE. Myoclonic epilepsy evolved into West syndrome: a patient with a novel de novo KCNQ2 mutation. Acta Neurol Belg 2015;115:475–478. [DOI] [PubMed] [Google Scholar]
- 28.Peters HC, Hu H, Pongs O, Storm JF, Isbrandt D. Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci 2005;8:51–60. [DOI] [PubMed] [Google Scholar]
- 29.Soldovieri MV, Miceli F, Taglialatela M. Driving with no brakes: molecular pathophysiology of Kv7 potassium channels. J Physiol 2011;26:365–376. [DOI] [PubMed] [Google Scholar]
- 30.Milh M, Lacoste C, Cacciagli P, et al. . Variable clinical expression in patients with mosaicism for KCNQ2 mutations. Am J Med Genet A 2015;167A:2314–2318. [DOI] [PubMed] [Google Scholar]
- 31.Ronen GM, Rosales TO, Connolly M, Anderson VE, Leppert M. Seizure characteristics in chromosome 20 benign familial neonatal convulsions. Neurology 1993;43:1355–1360. [DOI] [PubMed] [Google Scholar]
- 32.Miceli F, Soldovieri MV, Ambrosino P, et al. . Early-onset epileptic encephalopathy caused by gain-of-function mutations in the voltage sensor of Kv7.2 and Kv7.3 potassium channel subunits. J Neurosci 2015;35:3782–3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Veerapandiyan A, Singh P, Mikati MA. Possible induction of West syndrome by oxcarbazepine therapy in a patient with complex partial seizures. Epileptic Disord 2012;14:99–103. [DOI] [PubMed] [Google Scholar]
- 34.Amato G, Roeloffs R, Rigdon GC, et al. . N-Pyridyl and Pyrimidine Benzamides as KCNQ2/Q3 potassium channel openers for the treatment of epilepsy. ACS Med Chem Lett 2011;2:481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gunthorpe MJ, Large CH, Sankar R. The mechanism of action of retigabine (ezogabine), a first-in-class K+ channel opener for the treatment of epilepsy. Epilepsia 2012;53:412–424. [DOI] [PubMed] [Google Scholar]
- 36.Qi J, Zhang F, Mi Y, et al. . Design, synthesis and biological activity of pyrazolo[1,5-a]pyrimidin-7(4H)-ones as novel Kv7/KCNQ potassium channel activators. Eur J Med Chem 2011;46:934–943. [DOI] [PubMed] [Google Scholar]
- 37.Xiong Q, Gao Z, Wang W, Li M. Activation of Kv7 (KCNQ) voltage-gated potassium channels by synthetic compounds. Trends Pharmacol Sci 2008;29:99–107. [DOI] [PubMed] [Google Scholar]
- 38.Kalappa BI, Soh H, Duignan KM, et al. . Potent KCNQ2/3-specific channel activator suppresses in vivo epileptic activity and prevents the development of tinnitus. J Neurosci 2015;35:8829–8842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar M, Reed N, Liu R, Aizenman E, Wipf P, Tzounopoulos T. Synthesis and evaluation of potent KCNQ2/3-specific channel activators. Mol Pharmacol 2016;89:667–677. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Supplement
Coinvestigators