The complete chemical structure of Saccharomyces cerevisiae rRNA: partial pseudouridylation of U2345 in 25S rRNA by snoRNA snR9 (original) (raw)
Abstract
We present the complete chemical structures of the rRNAs from the eukaryotic model organism, Saccharomyces cerevisiae. The final structures, as determined with mass spectrometry-based methodology that includes a stable isotope-labelled, non-modified reference RNA, contain 112 sites with 12 different post-transcriptional modifications, including a previously unidentified pseudouridine at position 2345 in 25S rRNA. Quantitative mass spectrometry-based stoichiometric analysis of the different modifications at each site indicated that 94 sites were almost fully modified, whereas the remaining 18 sites were modified to a lesser extent. Superimposed three-dimensional modification maps for S. cerevisiae and Schizosaccharomyces pombe rRNAs confirmed that most of the modified nucleotides are located in functionally important interior regions of the ribosomes. We identified snR9 as the snoRNA responsible for pseudouridylation of U2345 and showed that this pseudouridylation occurs co-transcriptionally and competitively with 2′-O-methylation of U2345. This study ends the uncertainty concerning whether all modified nucleotides in S. cerevisiae rRNAs have been identified and provides a resource for future structural, functional and biogenesis studies of the eukaryotic ribosome.
INTRODUCTION
The ribosome is the universal cellular machinery that catalytically translates the mRNA genetic code into proteins. The eukaryotic ribosome is composed of two distinct ribonucleoprotein subunits. The small subunit (SSU, 40S) contains the 18S rRNA and 33 ribosomal proteins (RPs), and the large subunit (LSU, 60S) contains the 5S, 5.8S, and 25S rRNAs and ∼47 RPs (1). In each subunit, the rRNAs form the core of the translation machinery, and the RPs appear to stabilize and fine-tune the structure and function of the ribosome (1–5).
Ribosome biogenesis involves de novo synthesis of its rRNAs and RPs and hundreds of _trans_-acting factors, which are responsible for post-transcriptional/translational modifications, assembly and transport (6). In the eukaryotic ribosome, >100 rRNA nucleotides (nt) are modified during biogenesis (7). These modifications include, but are not limited to, methylation of a nt base or the 2′-hydroxyl group of a ribose moiety (Nm), acetylation of a base, or conversion of uridine (U) to pseudouridine (Ψ) (8). In general, these modifications are thought to expand the naturally limited topological properties of RNAs and optimize the basic functions of the ribosome (i.e. decoding, peptidyl transfer and translocation) by stabilizing their structures via the local charge environment or hydrogen bonding and optimizing their interactions with ligands (i.e. tRNAs, mRNAs and translation factors) (9).
In eukaryotic ribosomes, ribose 2′-O methylation and pseudouridylation are mostly catalysed by box C/D or box H/ACA small nucleolar ribonucleoproteins, respectively (10,11), with the RNA components (small nucleolar (sno)RNAs) of the ribonucleoproteins determining the modification sites by base-paring with an rRNA sequence and the proteins catalysing the modification reactions (12–14). Conversely, base methylation, acetylation and other complex modifications are catalysed by snoRNA-independent enzymes, most of which have only recently been identified (15–24).
The ribosome of the eukaryotic budding yeast Saccharomyces cerevisiae (Sc) has been extensively studied. Its three-dimensional (3D) structure is known at molecular resolution (25) and its biogenesis has been studied in depth. The yeast SSU contains an 18S rRNA composed of 1800 nt, and the LSU contains 5S, 5.8S and 25S rRNAs composed of 121, 158 and 3394 nt, respectively (6). To date, more than 20 reports have been published concerning rRNA modifications as well as the enzymes and snoRNAs responsible for each modification (ref. 9 and references therein). A total of ∼112 modified nt have been identified in the Sc ribosomal rRNAs, but there is still uncertainty concerning whether all modifications are known.
We recently developed a mass spectrometry (MS)-based technology termed SILNAS (Stable Isotope–Labelled riboNucleic Acid as an internal Standard), which allows for the comprehensive determination of RNA modifications with the use of a stable isotope–labelled RNA synthesized in vitro that does not contain modified nt; using SILNAS, we presented the first complete map of modified rRNAs from the fission yeast Schizosaccharomyces pombe (Sp) (26). An important feature of SILNAS is that, at each site, it quantifies the stoichiometries of different nt, i.e. modified nt, allowing for assessment of RNA-modification dynamics dependent on changes in internal and external cellular conditions or accompanying biological events. We describe herein the results of a SILNAS-based structural analysis of Sc rRNAs that allowed us to identify and quantify each type of unmodified and modified nt at all positions. This study presents the complete chemical structure including all modifications in the mature Sc rRNAs.
MATERIALS AND METHODS
Reagents
Standard laboratory chemicals were obtained from Wako Pure Chemical Industries. Sodium guanosine-13C10 5′-triphosphate (98 atom% 13C) and RNase A were obtained from Sigma-Aldrich. Sodium cytidine-13C9 5′-triphosphate (98 atom% 13C) and sodium uridine-13C9 5′-triphosphate (98 atom% 13C) were purchased from Santa Cruz Biotechnology, Inc. Uracil-5-D (98 atom% 2H) was obtained from C/D/N Isotopes, Inc. RNase T1 was purchased from Worthington and purified with reversed-phase LC before use. Triethylammonium acetate (TEAA, 1.0 M) buffer, pH 7.0, was purchased from Glen Research. The chemically synthesized oligonucleotides were obtained from JBioS, given in Supplementary Table S1-1.
Yeast strains, genetic methods and media
Yeast strains and media used are listed in Supplementary Tables S1-2 and S1-3. To obtain U/C-5D-labelled rRNAs, yeast cells deficient in ura3 were cultured in U/C-5-D labelling medium containing 0.01% (w/v) uracil-5-D instead of U with the natural isotope distribution (Supplementary Table S1-2).
To construct strains deficient in a snoRNA gene, primers for the 60-bp upstream and downstream regions of the gene and the 20-bp upstream and downstream regions of his3 were used (Supplementary Table S1-1). The _his_3-related primers in conjunction with pCgHIS3 (provided by the National Bio-Resource Project of MEXT, Japan) were used to polymerase chain reaction (PCR)-amplify his3. Each isolated DNA carrying a snoRNA gene disrupted by his3 was used to transform the _his3_-deficient strain. Transformation of yeast was performed by the LiOAc-method (27). Stable his+ transformants were selected (termed ΔsnR9, ΔsnR9_2, ΔsnR33 and ΔsnR65; their genotypes given in Supplementary Table S1–3). The three transformants, ΔsnR9, ΔsnR33 and ΔsnR65, have same genetic background, but ΔsnR9_2 differs in the marker genes used during episomal expression (Figure 4 and Supplementary Figure S9). Proper replacement of each wild-type snoRNA gene with the corresponding disrupted construct was confirmed by PCR.
Figure 4.

Episomal expression of snR9 in ΔsnR9_2 restores U2345 pseudouridylation. EICs of fragments from the RNase A digestion of 25S rRNA containing U2345 are shown. 25S rRNA was purified from ΔsnR9_2 that had been transformed with pSEC, pSECR9-WT, pSECR9-A33G, pSECR9-G67A or pSECR9-G69A. Each strain was cultured at 30°C in U/C-5-D labelling medium. The extracted 25S rRNAs were individually digested with RNase A, and the resulting digests (50 fmol) were subjected to LC-MS. The sequence and m/z value of ΨmAΨp are indicated. A mass window of ±10 ppm was used the chromatograms. The most intense peak in the pSECR-WT spectrum was set to 100%. The MS signal of ΨmAΨp was detected only in the spectrum of ΔsnR9 that had been transformed with pSECR9-WT, which enabled expression of wild-type snR9.
snoRNA cDNAs were constructed by the four-primer overlapping PCR strategy (28) using the primers listed in Supplementary Table S1-1. The products were individually cloned into the SalI-PstI site in the multicopy expression vector pSEC (29) containing the GPD promoter, snoRNA-processing elements (an RNT1 cleavage site and an snR13 terminator) and leu2 using Ligation high kit reagents (TOYOBO). The resulting plasmids were designated pSECR9-WT, pSECR9-A33G, pSECR9-G67A and pSECR9-G69A.
Yeast cell culture and preparation of cellular RNAs
Yeast cells were cultivated at 30°C in one of the media described in Supplementary Table S1-2 and harvested during the logarithmic growth phase (∼1.0 × 107 cell/ml). Total RNA was prepared as reported (30).
The different types rRNAs were purified from total RNA by reversed-phase LC through a PLRP-S 4000Å column (4.6 × 150 mm, 10 μm, Agilent Technologies). After applying 20 μg total RNA to the column, the rRNAs were eluted with a 60-min linear gradient of 12–14% (v/v) acetonitrile in 100 mM TEAA, pH 7.0, 0.1 mM diammonium phosphate at a flow rate of 200 μl/min and 60°C while monitoring the eluate at A260 (26,31). The rRNA preparations were >95% pure.
In vitro transcription of internal standard RNAs for SILNAS
To construct the plasmids for in vitro transcription of the internal standard RNAs, the DNAs encoding Sc 5S, 5.8S, 18S and 25S rRNA were PCR-amplified using genomic DNA and the primers given in Supplementary Table S1-1. Each amplified DNA encoding an rRNA was individually inserted into the XhoI-HindIII site of plasmid pBluescript II KS (+) (Agilent Technologies). Before in vitro transcription, each plasmid was linearized with SpeI to terminate the product at the end of the rRNA. To synthesize the rRNAs, 2 μg of each template DNA was individually incubated in and transcribed by Megascript T3 kit reagents (Invitrogen). Guanosine-13C10 5′-triphosphate or cytidine-13C9 5′-triphosphate and uridine-13C9 5′-triphosphate were included in the synthesis mixture instead of the corresponding ribonucleoside 5′-triphosphate with a natural isotope distribution. The resulting 13C10 GTP labelled RNA or 13C9 CTP/UTP doubly labelled RNA was precipitated with ethanol, solubilized in nuclease-free water and then purified by reversed-phase LC as described above.
Sequence-specific RNase H cleavage of rRNAs
rRNA (2 pmol) was digested with 15 U RNase H (Takara Bio) at 42°C for 1 h, guided by a synthetic 20- to 28-mer RNA/DNA chimera complementary to the duplex cleavage site (4 pmol) in 100 μl of 40 mM Tris–HCl, pH 7.7, 4 mM MgCl2, 1 mM DTT (26). All RNA/DNA sequences are presented in Supplementary Table S1-1. Before adding the enzyme, the sample was denatured at 65°C for 10 min. The reaction was stopped by adding 8 μl of 0.5 M ethylenediaminetetraacetic acid, and the resulting fragments were separated through a PLRP-S 300Å column (2 mm i.d. × 100 mm, 3-μm particle size, Agilent Technologies) with a 60-min, 12.5–15% linear gradient of acetonitrile in 100 mM TEAA, pH 7.0, 0.1 mM diammonium phosphate at a flow rate of 50 μl/min at 60°C.
Preparation of RNase digests for SILNAS and Direct nanoflow LC-MS and MS2 of RNA fragments
RNA (∼100 fmol) from natural sources or cells grown in guanosine with natural isotope distribution was mixed with an equal amount of synthetic RNA transcribed in vitro with 13C-labelled guanosine. The mix was digested with RNase T1 in 10 mM sodium acetate buffer (pH 5.3) at 37°C for 60 min at an enzyme/substrate ratio of 1/5 (w/w). RNase A digested RNA fragments were produced by the same procedure using a reference RNA transcribed in vitro with 13C-labelled cytidine and uridine.
RNA fragments were separated using a direct nanoflow LC-MS system as described (32,33). Briefly, the RNase digests were first concentrated through a MonoCap C18 trap column (200 μm i.d. × 40 mm; GL Sciences) in 100 mM TEAA, pH 7.0, 0.1 mM diammonium phosphate. The samples were then injected onto a reversed-phase Develosil C30-UG tip column (150 μm i.d. × 120 mm, 3-μm particle size; Nomura Chemical Co., Ltd.) equilibrated with solvent A (10 mM TEAA, pH 7, in water:methanol, 9:1). Samples were eluted at 100 nl/min with a 60-min 0–24.5% linear gradient of solvent B (10 mM TEAA, pH 7:acetonitrile, 60:40). The column was subsequently washed with 70% B for 10 min and re-equilibrated with A.
Each LC eluate was sprayed online at −1.3 kV with the aid of a spray-assisting device (33) into a Q Exactive mass spectrometer (Thermo Fisher Scientific) operating in the negative ion mode and in the data-dependent mode to automatically switch between MS and tandem MS (MS2) acquisition. Full-scan mass spectra (m/z = 500–2000) were acquired at a mass resolution of 140 000. At most, the five most intense peaks, (>100 000 counts/s with a 60-ms maximum injection time), were isolated within a 3-m/z window for fragmentation. Precursors were fragmented by switching to a higher energy CID mode with a normalized collision energy of 20 or 50%. To retain mass resolution and to increase spectral quality, three tandem mass spectral micro-scans were acquired for each sample. A fixed starting value of m/z = 100 was set for each tandem mass spectrum. To detect the ions, including the characteristic Ψ signature ion at m/z 207.04, a mass resolution of 17 500 at m/z 200 for the tandem mass spectra was used (34).
Database searching and interpretation of the tandem mass spectra
Ariadne (35) (http://ariadne.riken.jp/) was used for database searches and assignment of the tandem mass spectral peaks in conjunction with the sequences in the Sc genome database (http://www.yeastgenome.org/download-data/sequence). The Ariadne default search parameters were: the maximum number of missed cleavages was one; two methylations per RNA fragment at any residue position were allowed; an RNA mass tolerance of ± 20 ppm and a tandem spectral tolerance of ± 50 ppm were allowed. For assignment of Ψ residues, the mass table and the variable modification parameters were changed from the default parameters to ‘5D_CU’ and ‘Ψ’, because cytosine and U can be deuterated when yeast was grown in U/C-5-D-labelling medium. In this condition, the pseudouridylation reaction results in an exchange of the 5-deuterium in uracil with a hydrogen in solvent, providing a −1 Da mass shift (36). The characteristic Ψ signature ion at m/z = 207.04 were also used for assignment of Ψ (34).
The stoichiometry of RNA modification at each site was estimated by Ariadne program designed for SILNAS (to be open to the public). In principle, the program first extracts the intensity of MS signals from raw data, assigns all pairs of light and heavy signal of RNA fragments produced by SILNAS, compares quantitatively the signal intensities of each pair, and determines the stoichiometry of modification as the ratio of the difference in signal intensities between the light (unmodified) and heavy (a sum of unmodified and modified) fragments to the signal intensity of the heavy isotope-labelled fragment (Supplementary Figure S1). Upon calculation, Ariadne program considers the isotopical impurities in commercial rNTPs (∼5%) and corrects the estimate by a correction factor obtained experimentally by the quantitative analysis of labelled and non-labelled RNA fragment pairs with known nucleotide sequences. Finally, we confirmed the results by manual inspection of the original MS spectrum to examine whether the estimates are based on ‘uncontaminated’ MS signals (Supplementary Figure S1).
Sucrose density gradient centrifugation
Ribosomes and polyribosomal particles were fractionated as described (30), except that Sc cells were disrupted by a multi-beads shocker (Yasui Kikai) in the presence of zirconium beads (5-mm diameter; 1.5 g/20 ml cell culture).
Other procedures
In-solution RNA digestion was performed as described (32). The oligonucleotide masses and a-, c-, w- and y-series ion masses were calculated by Ariadne. The most abundant isotopomer was calculated by Xcalibur 4.0 (Thermo Fisher Scientific). For calculation of the C/U-5D-labelled fragment masses, 98 atom% D was assumed. The secondary structures of H/ACA box snoRNAs were deduced by mFold (37). The predicted internal loop at the base of an upper stem and the 12–19 nt upstream of an H or ACA box were compared with the sequence around the target site. The snoRNA with the greatest sequence identity was selected as the responsible snoRNA. RNA tertiary structures defined by their PDB files were visualized and analysed with the Swiss-PdbViewer (http://spdbv.vital-it.ch/).
RESULTS
Identification and quantification of modified nt in Sc rRNAs
We isolated total RNA from logarithmic-growth phase Sc cells cultivated in YPD medium (Supplementary Table S1-2), and purified the rRNAs by reversed-phase liquid chromatography (LC) through a PLRP-S 4000-Å column (Methods). To chemically characterize the purified rRNAs, each was mixed with an in vitro transcribed 13G-labelled reference RNA having the same sequence with the exception that the incorporated nt had not been modified during transcription, and each mixture was then digested with RNase T1. Each digest was subjected to nanoflow LC-MS and MS2 followed by a database search to identify the chemical structure of each rRNA fragment. The oligonucleotides containing modified nt were identified based on shifts in their LC retention times and intensities of mass values in comparison with the corresponding in vitro transcribed fragments.
To align the RNase T1-digested oligonucleotides so as to determine the complete chemical structure of each rRNA, we also digested each rRNA with RNase A and performed LC-MS-Ariadne analysis on the RNase A digests. When the rRNA produced multiple fragments having the same nucleotide sequence, the RNA was systematically segmented with RNase H to avoid the production of such fragments and to distinguish the redundant sequences before RNase T1 or A digestion (detailed in Supplementary Figure S2). Where necessary, we also characterized rRNA prepared from Sc cells grown in the U/C-5-D-labelling medium to differentiate between U and Ψ in terms of the number of incorporated 2H atoms (Figure 1 and Supplementary Figures S2 and 3). These procedures allowed us to align all oligonucleotides, thereby completely determine the chemical structure of 5S, 5.8S, 18S and 25S rRNAs.
Figure 1.

Nucleotide heterogeneity at position 2345 in Saccharomyces cerevisiae 25S rRNA. LC-MS and MS2 of the RNase T1 digest of U/C-5D-labelled and reverse-phase LC-purified 25S rRNA (100 fmol). (A) Extracted ion monitoring of fragments containing a nt at position 2345 shows that nt2345 is a mixture containing 9% U, 74% Um, 4% Ψ and 13% Ψm (Table 2). The extracted ion masses have m/z values of 1421.688 (upper panel) and 1418.184 (lower panel) within a mass window of ±10 ppm. U, 2334UCmCCΨAUCUACUAΨCΨA2351Gp; Ψ, 2334UCmCCΨAUCUACΨAΨCΨA2351Gp; Um, 2334UCmCCΨAUCUACUmAΨCΨA2351Gp; Ψm, 2334UCmCCΨAUCUACΨmAΨCΨA2351Gp. (B) Mass spectra for the oligonucleotides containing U2345. The most abundant isotopomers are marked by asterisks. The m/z values of the most abundant signals coincide with the theoretical values within 5 ppm. (C) Tandem mass spectrum of 2334UCmCCΨAUCUACΨmAΨCΨA2351Gp. The blue arrows in the spectrum identify the major a, c, w and y ions. The cleavage positions of the assigned ions are mapped on the RNA sequence in the inset. Errors determined by Ariadne in the MS2 signals are plotted under the spectrum.
We identified 112 sites with 12 distinct modified nt in the Sc rRNAs (Table 1 and Supplementary Table S2). Based on the quantitative capability of the SILNAS procedure, we concluded that these represents all the modified nucleotides in the Sc rRNAs carrying the modification more than the level of ≥5%. Of the 112 sites, the 5S and 5.8S rRNAs each contained one modified nt, a Ψ, the 18S rRNA contained 37 modified nt (14 Ψ bases including a 1-methyl-3-(3-amino-3-carboxypropyl)Ψ base, 18 2′-O-methylated riboses, 2 acetylated bases, 2 dimethylatated bases and a monomethylated base) and the 25S rRNA contained 73 modified nt (31 Ψ bases, 37 2′-O-methylated riboses and 6 monomethylated bases) (Supplementary Table S2). Our results are consistent with those of many previous reports (references in ref. 9), with the following exceptions. This study found a formerly unknown Ψ at position 2345 in 25S rRNA, where we identified Ψ, U, Ψm or Um (with Um having been found previously (38)) (Figure 1 and Supplementary Figure S3). Conversely, we did not find the modified residues 5-methyluridine (m5U)954 (39), Gm1140 (40), Gm2393 (10) and m5U2919 (39) that have been reported by others (Supplementary Figure S4). Instead, we found only the corresponding unmodified nt at those positions. We assume that this discrepancy might simply be the results of incorrect assignments due to technical difficulties in identifying the modified nucleotides by the conventional technique, although we cannot exclude a possibility that those nucleotides are modified under specific cellular conditions or in response to specific stimuli to the cells.
Table 1. Position, type and stoichiometry of partially modified nucleotides found in Saccharomyces cerevisiae rRNAs.
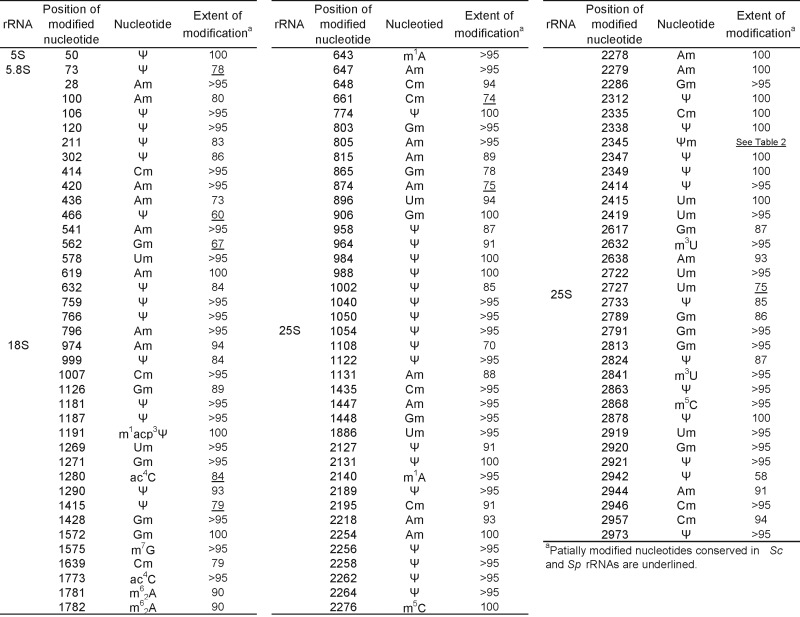
With the use of SILNAS, we quantified the stoichiometries of the modified nt and the original nt from which they were derived at each of the 112 positions. Of the 112 positions, 94 contained almost exclusively a modified nt (≥85%), whereas the nt at the other 18 sites had been modified to a lesser extent (∼5–85%) (Table 1 and Supplementary Table S2).
3D modification maps of the Sc rRNAs
We mapped the modified nt onto the 3D structural map of the Sc ribosome (Figure 2A). As noted previously for the Sp ribosome (26), most (∼90%) of the modified nt are located in functionally important, interior regions of the ribosome, i.e. the peptidyl transferase centre, the decoding centre, the A, P and E sites involved in tRNA and mRNA binding, the polypeptide exit tunnel and the SSU–LSU interface (Supplementary Table S2 and Figure 2). In the SSU, the modified nt are clustered on a plane that includes its head, neck, and body regions (Figure 2C). In the LSU, almost all modified nt are clustered as a funnel-shaped structure around the peptidyl transferase centre and the peptide exit tunnel. Modified nt are essentially absent on the exterior and at the periphery of each interior region of the ribosome, which mainly include RPs (Supplementary Figure S5). The 3D modification map and the published map of the Sc ribosome constructed from data accumulated by more than 20 previous reports (reviewed by Decatur and Fournier (41)), are basically the same.
Figure 2.

3D modification map of the Sc ribosome. The modified nt found in this study are mapped on the 3D structures of the Sc rRNAs (3U5B.pdb and 3U5D.pdb). The RNA backbones are shown as white wires and the colour codes for the modified nt (shown as balls) are: yellow, Ψ; red, 2′-O-methylated nt; blue, base modified nt found in Sc and Sp rRNAs; purple, modified nt unique to Sc rRNA; and white, modified nt unique to Sp rRNA. (A) Sc rRNAs. (B–D) Superimposed maps of modified nt found in Sc and Sp rRNAs; (B) SSU and LSU, (C) SSU and (D) LSU. Insets in C and D show the surface structures of the subunits. Domain names are in white and their positions are indicated by arrows.
The Sc 3D modification map compares well with that of the Sp ribosome (26) (Figure 2B–D). Of the 112 modifications located in the Sc rRNAs, 95 are found at equivalent positions in the Sp rRNAs, whereas others are located at positions unique to the Sc or Sp rRNAs (Figure 2B–D). Notably, of the 95 modification sites that are conserved in both yeast species, almost all are found centred in the aforementioned functionally important regions, whereas the remaining 17 nt are scattered outside of these regions (Figure 2B–D and Supplementary Figure S6). Also, ∼50% of the partially modified nt in the Sc rRNAs are located at positions corresponding to those found in Sp rRNAs (Supplementary Figure S7; discussed below).
Guide snoRNA for U2345 pseudouridylation
As noted above, in the 25S rRNA, U2345 is partially pseudouridylated. Because most pseudouridylations of yeast rRNAs are catalysed by snoRNA-directed box H/ACA complexes (9), we next identified the snoRNA responsible for U2345 pseudouridylation. We first analysed the nt sequences of the 29 known H/ACA box snoRNAs in Sc (42) to find a sequence (s) complementary to the sequence around U2345. Given that all box H/ACA snoRNAs have a 9–13-nt discontinuous sequence that is capable of base pairing with the targeted substrate RNA so as to form a pseudouridylation pocket 13–16 nt upstream of the 5′ end of the H or ACA box sequence (43), we predicted that snR9 or snR33 might be involved in the pseudouridylation of U2345 (Figure 3A and Supplementary Figure S8). We therefore prepared individual deletion mutants of snR9 and snR33 (designated ΔsnR9 and ΔsnR33, respectively), cultured them, purified their 25S rRNAs and analysed their structures around position 2345, 2345ΨmA2347Ψp, produced by RNase A digestion. In ΔsnR9 25S rRNA, only U and Um2345 was found (Table 2 and Figure 3B and Supplementary Figure S9), whereas in ΔsnR33 25S rRNA, Ψm2345 was present (Figure 3B), suggesting that snR9 is the snoRNA for U2345 pseudouridylation.
Figure 3.

Deletion of snR9 prevents pseudouridylation of U2345. (A) Potential base-pairing interactions between snR9 and Sc 25S rRNA. The upper sequence is that of the snoRNA snR9 with its hairpin shown as a solid line. The positions of A33, G67 and G69, which were mutated in our study, are indicted by the arrows. The lower sequence is that of the 25S rRNA around Ψ2345. The lower arrow points to Ψ2345. (B) Extracted ion chromatogram (EIC) of the fragments containing 2345ΨmA2347Ψp produced by the RNase A digestion of the 25S rRNA from wild-type (upper panel), ΔsnR9 (middle panel) and ΔsnR33 (lower panel) Sc strains. The strains were cultured in the U/C-5-D labelling medium at 30°C and purified 25S rRNA was digested with RNase A. Each digest (50 fmol) was subjected to LC-MS. The sequence and m/z value of ΨmAΨp are shown in the figure. The most intense signal in the ΔsnR33 spectrum was set to a relative intensity of 100%, and the peaks in the wild-type spectrum were scaled accordingly. The arrows in the left panels indicate the position of the MS signal for the Ψm2345-containing fragment, which was not detected in the ΔsnR9 chromatogram.
Table 2. Stoichiometries of original (U) and modified nucleotides at position 2345 in Saccharomyces cerevisiae 25S rRNA.
| Strain | U %a | Um %a | Ψ %a | Ψm %a | Ψ/Ψm %b | Um/Ψm %b |
|---|---|---|---|---|---|---|
| WT(BY5208) | 8.0 ± 1.7 | 76.2 ± 2.8 | 2.9 ± 1.0 | 12.9 ± 0.7 | 15.8 ± 1.5* | 89.1 ± 2.3** |
| ΔsnR9 | 6.1 ± 1.1 | 93.9 ± 1.1 | 0 | 0 | 0 | 93.9 ± 1.1** |
| ΔsnR65 | 57.8 ± 2.6 | 0 | 42.2 ± 2.6 | 0 | 42.2 ± 2.6* | 0 |
To confirm that snR9 mediates U2345 pseudouridylation and to rule out the presence of other mutations that might be cause the loss of U2345 pseudouridylation in a snR9 deficient mutant, we performed a complementation study by expressing snR9, carried by pSECR9, in the mutant and determining whether this episomal expression restored pseudouridylation of U2345. Subsequent LC-MS and MS2 of the RNase A or T1 fragments of the 25S rRNA obtained from these strains showed that Ψ2345 and Ψm2345 were present only when pSECR9-WT had been transformed into ΔsnR9_2 (Figure 4 and Supplementary Table S3). To reinforce this result, we introduced a point mutation at positions 33, 67 and 69, which are upstream of the snR9's H box and base-pair with the nt of 25S rRNA or within snR9. These mutations are in physical proximity of the U2345 pseudouridylation pocket (Figure 3A). Each point mutant completely abolished U2345 pseudouridylation (Figure 4 and Supplementary Table S3). Thus, the results clearly indicated that the snR9-directed box H/ACA RNP is responsible for U2345 pseudouridylation and that A33, G67 and G69 are essential for the reaction.
Because snR9 also participates in U2338 pseudouridylation of 25S rRNA in conjunction with slightly different anti-sense guide nt (42) (Supplementary Figure S8), we examined whether A33, G67 and/or G69, are involved in U2338 pseudouridylation. Interestingly, the mutations affected U2338 pseudouridylation only to the extent that the percentage of pseudouridylated U2338 was decreased in comparison with the wild-type snR9-supplemented strain (Supplementary Table S3), suggesting that the affinities of snoRNAs for their substrate RNAs affect the catalytic activities of the pseudouridylation reaction.
Interference between U2345 pseudouridylation and 2′-O-methylation
rRNA modifications have been found to occur during early ribosome biogenesis, i.e. co-transcriptionally, whereas other modifications, e.g. rRNA base methylation, take place at specific stages of ribosome biogenesis (6). Notably, most ribose 2′-O-methylations of Sc rRNA occur co-transcriptionally (44). Pseudouridylation has also been assumed to be a co-transcriptional event, but little experimental evidence supports this assumption because it has been difficult to assess the timing of the reaction (45). Given that each of U2345 and Ψ2345 may undergo 2′-O-methylation, we addressed this issue directly by MS. We prepared the snR65 deletion mutant ΔsnR65 in which U/Ψ 2345 cannot be 2′-O-methylated and compared the levels of U, Ψ, Um and Ψm at position 2345 with those found at the equivalent position in the 25S rRNA of ΔsnR9 and isogenic wild-type Sc. After isolating rRNAs from each strain, the levels of the aforementioned nt were quantified by LC-MS. In ΔsnR65, U2345 was not 2′-O-methylated and, in ΔsnR9, U2345 was not pseudouridylated. Interestingly, the amount of 2′-O-methylated U2345 was significantly greater in ΔsnR9 than in wild-type, and the amount of Ψ2345 in ΔsnR65 was significantly greater than in wild-type (Table 2), suggesting that the two reactions are competitive and occur during transcription of the 25S rRNA; namely, if a U is first 2′-O-methylated, then this partially inhibits subsequent pseudouridylation of the U and vice versa.
Loss of Ψ2345 has no detectable effect on cell growth and rRNA processing
Although U2345 is only partially pseudouridylated in the mature ribosome, we wondered whether complete inhibition of pseudouridylation might affect Sc cell growth. However, ΔsnR9 had a normal growth phenotype at 19, 30 and 37°C (Figure 5A), and ΔsnR9 and wild-type maintained the same steady-state levels for the 40S and 60S subunits as well as polysomes as monitored by sucrose density gradient centrifugation (Figure 5B).
Figure 5.

Loss of U2345 pseudouridylation causes no changes in cell growth and ribosome synthesis. (A) Cell growth study. Serial 10-fold dilutions of the isogenic wild-type BY5208 and ΔsnR9 strains grown at 19°C for 48 h or 30°C and 37°C for 24 h were spotted onto YPD-containing agar plates. (B) Ribosome profiles of BY5208 (upper panel) and ΔsnR9 (lower panel) strains. Ribosomal and polysomal fractions prepared from the wild-type and ΔsnR9 strains (each cultured at 30°C for 24 h in YPD medium) were subjected to sucrose density gradient centrifugation. A254 was monitored.
DISCUSSION
We present herein the complete chemical structures of the rRNAs in the eukaryotic model organism, Sc. These rRNAs contain 112 sites with 12 different types of modified nt. Although all modified nt, except that at position 2345 in 25S rRNA, had been reported before, we believe that our study, which used a synthetic heavy isotope-labelled internal standard and SILNAS-based MS methodology, is the first to directly identify all modified nt. Our study, therefore, ends any uncertainty concerning whether all modified nt in Sc rRNAs have been identified. We also showed that snR9, a component of the box H/ACA snoRNP particle, is responsible for U2345 pseudouridylation. By doing so, the complete set of snoRNAs for pseudouridylation of Sc rRNAs has been determined (Supplementary Table S2).
Our SILNAS-based analysis also provides the first stoichiometric determination of each site modification in the Sc rRNAs (Table 1 and Supplementary Table S2). We found that a part of the sites were only partially modified (∼5–85%) and, interestingly, that these partially modified nt are located at positions similar to those found in Sp rRNAs (Supplementary Figure S7). We previously identified a number of partially modified nt at specific locations in rRNAs of Sp cells that were altered in response to the culture temperature (26), which suggested that the cells might coordinately regulate the modification levels of nt at specific sites in their rRNAs; however, we could not correlate the locations of those partially modified nt with ribosome function. We have also examined various culture conditions that might alter the stoichiometries of the modifications at specific sites of Sp ribosome, but the cells maintained approximately the same steady-state levels of rRNA modification regardless of the logarithmic and stationary growth phases, the ploidy state, or the nutritive condition (starvation of carbon and nitrogen sources) (our unpublished results). Thus, why the nt of rRNAs are only partially modified remains unknown. It should be noted, however, that the rRNAs used in our studies were mostly derived from mature ribosomes because exponentially growing yeast cells were the source. Given that rRNA modifications occur throughout ribosome biogenesis, i.e. from the initial steps in the nucleolus to the final steps in the cytoplasm, studies concerning rRNA modification in pre-ribosome intermediates may provide additional insight into the roles of rRNA modification during eukaryotic ribosome biogenesis.
We found that position 2345 in Sc 25S rRNA may contain any one of four different nt, i.e. 9% U, 74% Um, 4% Ψ and 13% Ψm (Table 2). This nt is located within Helix 61, which contacts the ‘basic thumb’ domain of RPL3, and this protein is part of the aminoacyl-tRNA binding site (46). Mutation of Sc RPL3 affects yeast growth rate. However, why this heterogeneity exists is not clear, particularly because the complete loss of Ψ/Ψm2345 apparently does not affect cell growth and ribosome synthesis (Figure 5).
The superimposed 3D modification maps of the Sc and Sp rRNAs (Figure 2) completely support a previous conclusion that rRNA modifications are mostly located in functionally important regions (41). Given that modified nt are also found in functionally important regions of bacterial (47) and human rRNAs (7) (also Taoka et al., unpublished results), rRNA modifications are most likely highly conserved in all organisms, implying that their fundamental biological functions are universally beneficial. It has been reported that pseudouridylation of a U in the Escherichia coli ribosome alters the orientation of the base, thereby optimizing the local ribosomal structure by positioning the base properly with respect to the rRNA backbone (47); however, a mutant E. coli strain lacking all pseudouridylation enzymes grew normally (48). Conversely, in the eukaryotic ribosome, alterations in rRNA pseudouridylation appear to negatively affect translation of certain mRNAs containing internal ribosome entry sequences (IRESs) (49), a structural element positioned at the 5′ UTR of these mRNAs that directly recruits the ribosome to the initiation region of the mRNAs and thereby reduces the requirement for canonical initiation factors (50). In mice that are hypomorphic for dyskerin, an enzyme that pseudouridylates U in rRNAs, translation of IRES-containing mRNAs, including those of the tumour suppressors p27 and p53, is perturbed (51,52), and consequently these mice are more likely to develop cancer (53). Notably, eukaryotic ribosomes lacking the Ψ modification have impaired IRES-element binding (49). In addition, in human cells, defects in rRNA modification, including snoRNA-dependent 2′-O-methylation/pseudouridylation and base modifications have frequently been associated with genetic diseases and cancers, although the molecular mechanisms of pathology are largely unknown (53–56). Because our SILNAS-based MS methodology enables the study of rRNA modification dynamics accompanying cellular events, we expect that its use in future studies will improve our understanding of the physiological roles of rRNA modifications and how defects in rRNA modifications affect human diseases.
Supplementary Material
SUPPLEMENTARY DATA
Acknowledgments
We thank Prof Yi-Tao Yu and Dr Hironori Adachi of the University of Rochester, Rochester, NY for the expression vector pSEC and Ms C. Fujita and Ms M. Koike for technical assistance.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Core Research for Evolutional Science and Technology, Japan Science and Technology Agency. Funding for open access charge: Core Research for Evolutional Science and Technology, Japan Science and Technology Agency.
Conflict of interest statement. None declared.
REFERENCES
- 1.Wilson D.N., Doudna Cate J.H. The structure and function of the eukaryotic ribosome. Cold Spring Harb. Perspect. Biol. 2012;4:a011536. doi: 10.1101/cshperspect.a011536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yusupova G., Yusupov M. High-resolution structure of the eukaryotic 80S ribosome. Annu. Rev. Biochem. 2014;83:467–486. doi: 10.1146/annurev-biochem-060713-035445. [DOI] [PubMed] [Google Scholar]
- 3.Voigts-Hoffmann F., Klinge S., Ban N. Structural insights into eukaryotic ribosomes and the initiation of translation. Curr. Opin. Struct. Biol. 2012;22:768–777. doi: 10.1016/j.sbi.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 4.Jenner L., Melnikov S., Garreau de Loubresse N., Ben-Shem A., Iskakova M., Urzhumtsev A., Meskauskas A., Dinman J., Yusupova G., Yusupov M. Crystal structure of the 80S yeast ribosome. Curr. Opin. Struct. Biol. 2012;22:759–767. doi: 10.1016/j.sbi.2012.07.013. [DOI] [PubMed] [Google Scholar]
- 5.Klinge S., Voigts-Hoffmann F., Leibundgut M., Ban N. Atomic structures of the eukaryotic ribosome. Trends Biochem. Sci. 2012;37:189–198. doi: 10.1016/j.tibs.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 6.Woolford J.L., Jr, Baserga S.J. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics. 2013;195:643–681. doi: 10.1534/genetics.113.153197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Piekna-Przybylska D., Decatur W.A., Fournier M.J. The 3D rRNA modification maps database: with interactive tools for ribosome analysis. Nucleic Acids Res. 2008;36:D178–D183. doi: 10.1093/nar/gkm855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Machnicka M.A., Milanowska K., Osman Oglou O., Purta E., Kurkowska M., Olchowik A., Januszewski W., Kalinowski S., Dunin-Horkawicz S., Rother K.M., et al. MODOMICS: a database of RNA modification pathways–2013 update. Nucleic Acids Res. 2012;41:D262–D267. doi: 10.1093/nar/gks1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharma S., Lafontaine D.L. 'View From A Bridge': a new perspective on eukaryotic rRNA base modification. Trends Biochem. Sci. 2015;40:560–575. doi: 10.1016/j.tibs.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 10.Kiss-Laszlo Z., Henry Y., Bachellerie J.P., Caizergues-Ferrer M., Kiss T. Site-specific ribose methylation of preribosomal RNA: a novel function for small nucleolar RNAs. Cell. 1996;85:1077–1088. doi: 10.1016/s0092-8674(00)81308-2. [DOI] [PubMed] [Google Scholar]
- 11.Ganot P., Bortolin M.L., Kiss T. Site-specific pseudouridine formation in preribosomal RNA is guided by small nucleolar RNAs. Cell. 1997;89:799–809. doi: 10.1016/s0092-8674(00)80263-9. [DOI] [PubMed] [Google Scholar]
- 12.Tollervey D., Lehtonen H., Jansen R., Kern H., Hurt E.C. Temperature-sensitive mutations demonstrate roles for yeast fibrillarin in pre-rRNA processing, pre-rRNA methylation, and ribosome assembly. Cell. 1993;72:443–457. doi: 10.1016/0092-8674(93)90120-f. [DOI] [PubMed] [Google Scholar]
- 13.Lafontaine D.L., Bousquet-Antonelli C., Henry Y., Caizergues-Ferrer M., Tollervey D. The box H + ACA snoRNAs carry Cbf5p, the putative rRNA pseudouridine synthase. Genes Dev. 1998;12:527–537. doi: 10.1101/gad.12.4.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lapinaite A., Simon B., Skjaerven L., Rakwalska-Bange M., Gabel F., Carlomagno T. The structure of the box C/D enzyme reveals regulation of RNA methylation. Nature. 2013;502:519–523. doi: 10.1038/nature12581. [DOI] [PubMed] [Google Scholar]
- 15.Figaro S., Wacheul L., Schillewaert S., Graille M., Huvelle E., Mongeard R., Zorbas C., Lafontaine D.L., Heurgue-Hamard V. Trm112 is required for Bud23-mediated methylation of the 18S rRNA at position G1575. Mol. Cell. Biol. 2012;32:2254–2267. doi: 10.1128/MCB.06623-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lafontaine D., Delcour J., Glasser A.L., Desgres J., Vandenhaute J. The DIM1 gene responsible for the conserved m6(2)Am6(2)A dimethylation in the 3′-terminal loop of 18 S rRNA is essential in yeast. J. Mol. Biol. 1994;241:492–497. doi: 10.1006/jmbi.1994.1525. [DOI] [PubMed] [Google Scholar]
- 17.Peifer C., Sharma S., Watzinger P., Lamberth S., Kotter P., Entian K.D. Yeast Rrp8p, a novel methyltransferase responsible for m1A 645 base modification of 25S rRNA. Nucleic Acids Res. 2012;41:1151–1163. doi: 10.1093/nar/gks1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharma S., Watzinger P., Kotter P., Entian K.D. Identification of a novel methyltransferase, Bmt2, responsible for the N-1-methyl-adenosine base modification of 25S rRNA in Saccharomyces cerevisiae. Nucleic Acids Res. 2013;41:5428–5443. doi: 10.1093/nar/gkt195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharma S., Yang J., Watzinger P., Kotter P., Entian K.D. Yeast Nop2 and Rcm1 methylate C2870 and C2278 of the 25S rRNA, respectively. Nucleic Acids Res. 2013;41:9062–9076. doi: 10.1093/nar/gkt679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharma S., Yang J., Duttmann S., Watzinger P., Kotter P., Entian K.D. Identification of novel methyltransferases, Bmt5 and Bmt6, responsible for the m3U methylations of 25S rRNA in Saccharomyces cerevisiae. Nucleic Acids Res. 2013;42:3246–3260. doi: 10.1093/nar/gkt1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharma S., Langhendries J.L., Watzinger P., Kotter P., Entian K.D., Lafontaine D.L. Yeast Kre33 and human NAT10 are conserved 18S rRNA cytosine acetyltransferases that modify tRNAs assisted by the adaptor Tan1/THUMPD1. Nucleic Acids Res. 2015;43:2242–2258. doi: 10.1093/nar/gkv075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ito S., Akamatsu Y., Noma A., Kimura S., Miyauchi K., Ikeuchi Y., Suzuki T. A single acetylation of 18 S rRNA is essential for biogenesis of the small ribosomal subunit in Saccharomyces cerevisiae. J. Biol. Chem. 2014;289:26201–26212. doi: 10.1074/jbc.M114.593996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leulliot N., Bohnsack M.T., Graille M., Tollervey D., Van Tilbeurgh H. The yeast ribosome synthesis factor Emg1 is a novel member of the superfamily of alpha/beta knot fold methyltransferases. Nucleic Acids Res. 2008;36:629–639. doi: 10.1093/nar/gkm1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meyer B., Wurm J.P., Sharma S., Immer C., Pogoryelov D., Kotter P., Lafontaine D.L., Wohnert J., Entian K.D. Ribosome biogenesis factor Tsr3 is the aminocarboxypropyl transferase responsible for 18S rRNA hypermodification in yeast and humans. Nucleic Acids Res. 2016;44:4304–4314. doi: 10.1093/nar/gkw244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garreau de Loubresse N., Prokhorova I., Holtkamp W., Rodnina M.V., Yusupova G., Yusupov M. Structural basis for the inhibition of the eukaryotic ribosome. Nature. 2014;513:517–522. doi: 10.1038/nature13737. [DOI] [PubMed] [Google Scholar]
- 26.Taoka M., Nobe Y., Hori M., Takeuchi A., Masaki S., Yamauchi Y., Nakayama H., Takahashi N., Isobe T. A mass spectrometry-based method for comprehensive quantitative determination of post-transcriptional RNA modifications: the complete chemical structure of Schizosaccharomyces pombe ribosomal RNAs. Nucleic Acids Res. 2015;43:e115. doi: 10.1093/nar/gkv560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ito H., Fukuda Y., Murata K., Kimura A. Transformation of intact yeast cells treated with alkali cations. J. Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang C., Wu G., Yu Y.T. Inducing nonsense suppression by targeted pseudouridylation. Nat. Protoc. 2012;7:789–800. doi: 10.1038/nprot.2012.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang C., Karijolich J., Yu Y.T. Post-transcriptional modification of RNAs by artificial Box H/ACA and Box C/D RNPs. Methods Mol. Biol. 2011;718:227–244. doi: 10.1007/978-1-61779-018-8_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taoka M., Ishikawa D., Nobe Y., Ishikawa H., Yamauchi Y., Terukina G., Nakayama H., Hirota K., Takahashi N., Isobe T. RNA cytidine acetyltransferase of small-subunit ribosomal RNA: identification of acetylation sites and the responsible acetyltransferase in fission yeast, Schizosaccharomyces pombe. PLoS One. 2014;9:e112156. doi: 10.1371/journal.pone.0112156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamauchi Y., Taoka M., Nobe Y., Izumikawa K., Takahashi N., Nakayama H., Isobe T. Denaturing reversed phase liquid chromatographic separation of non-coding ribonucleic acids on macro-porous polystyrene-divinylbenzene resins. J. Chromatogr. A. 2013;1312:87–92. doi: 10.1016/j.chroma.2013.09.021. [DOI] [PubMed] [Google Scholar]
- 32.Taoka M., Yamauchi Y., Nobe Y., Masaki S., Nakayama H., Ishikawa H., Takahashi N., Isobe T. An analytical platform for mass spectrometry-based identification and chemical analysis of RNA in ribonucleoprotein complexes. Nucleic Acids Res. 2009;37:e140. doi: 10.1093/nar/gkp732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakayama H., Yamauchi Y., Taoka M., Isobe T. Direct identification of human cellular microRNAs by nanoflow liquid chromatography-high-resolution tandem mass spectrometry and database searching. Anal. Chem. 2015;87:2884–2891. doi: 10.1021/ac504378s. [DOI] [PubMed] [Google Scholar]
- 34.Yamauchi Y., Nobe Y., Izumikawa K., Higo D., Yamagishi Y., Takahashi N., Nakayama H., Isobe T., Taoka M. A mass spectrometry-based method for direct determination of pseudouridine in RNA. Nucleic Acids Res. 2015;44:e59. doi: 10.1093/nar/gkv1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakayama H., Akiyama M., Taoka M., Yamauchi Y., Nobe Y., Ishikawa H., Takahashi N., Isobe T. Ariadne: a database search engine for identification and chemical analysis of RNA using tandem mass spectrometry data. Nucleic Acids Res. 2009;37:e47. doi: 10.1093/nar/gkp099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Popova A.M., Williamson J.R. Quantitative analysis of rRNA modifications using stable isotope labeling and mass spectrometry. J. Am. Chem. Soc. 2014;136:2058–2069. doi: 10.1021/ja412084b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lowe T.M., Eddy S.R. A computational screen for methylation guide snoRNAs in yeast. Science. 1999;283:1168–1171. doi: 10.1126/science.283.5405.1168. [DOI] [PubMed] [Google Scholar]
- 39.Bakin A., Lane B.G., Ofengand J. Clustering of pseudouridine residues around the peptidyltransferase center of yeast cytoplasmic and mitochondrial ribosomes. Biochemistry. 1994;33:13475–13483. doi: 10.1021/bi00249a036. [DOI] [PubMed] [Google Scholar]
- 40.Birkedal U., Christensen-Dalsgaard M., Krogh N., Sabarinathan R., Gorodkin J., Nielsen H. Profiling of ribose methylations in RNA by high-throughput sequencing. Angew. Chem. Int. Ed. Engl. 2014;54:451–455. doi: 10.1002/anie.201408362. [DOI] [PubMed] [Google Scholar]
- 41.Decatur W.A., Fournier M.J. rRNA modifications and ribosome function. Trends Biochem. Sci. 2002;27:344–351. doi: 10.1016/s0968-0004(02)02109-6. [DOI] [PubMed] [Google Scholar]
- 42.Piekna-Przybylska D., Decatur W.A., Fournier M.J. New bioinformatic tools for analysis of nucleotide modifications in eukaryotic rRNA. RNA. 2007;13:305–312. doi: 10.1261/rna.373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reichow S.L., Hamma T., Ferre-D'Amare A.R., Varani G. The structure and function of small nucleolar ribonucleoproteins. Nucleic Acids Res. 2007;35:1452–1464. doi: 10.1093/nar/gkl1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kos M., Tollervey D. Yeast pre-rRNA processing and modification occur cotranscriptionally. Mol. Cell. 2010;37:809–820. doi: 10.1016/j.molcel.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Turowski T.W., Tollervey D. Cotranscriptional events in eukaryotic ribosome synthesis. Wiley Interdiscip. Rev. RNA. 2014;6:129–139. doi: 10.1002/wrna.1263. [DOI] [PubMed] [Google Scholar]
- 46.Meskauskas A., Dinman J.D. A molecular clamp ensures allosteric coordination of peptidyltransfer and ligand binding to the ribosomal A-site. Nucleic Acids Res. 2010;38:7800–7813. doi: 10.1093/nar/gkq641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Noeske J., Wasserman M.R., Terry D.S., Altman R.B., Blanchard S.C., Cate J.H. High-resolution structure of the Escherichia coli ribosome. Nat. Struct. Mol. Biol. 2015;22:336–341. doi: 10.1038/nsmb.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O'Connor M., Gregory S.T. Inactivation of the RluD pseudouridine synthase has minimal effects on growth and ribosome function in wild-type Escherichia coli and Salmonella enterica. J. Bacteriol. 2011;193:154–162. doi: 10.1128/JB.00970-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jack K., Bellodi C., Landry D.M., Niederer R.O., Meskauskas A., Musalgaonkar S., Kopmar N., Krasnykh O., Dean A.M., Thompson S.R., et al. rRNA pseudouridylation defects affect ribosomal ligand binding and translational fidelity from yeast to human cells. Mol. Cell. 2011;44:660–666. doi: 10.1016/j.molcel.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sonenberg N., Hinnebusch A.G. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bellodi C., Kopmar N., Ruggero D. Deregulation of oncogene-induced senescence and p53 translational control in X-linked dyskeratosis congenita. EMBO J. 2010;29:1865–1876. doi: 10.1038/emboj.2010.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yoon A., Peng G., Brandenburger Y., Zollo O., Xu W., Rego E., Ruggero D. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science. 2006;312:902–906. doi: 10.1126/science.1123835. [DOI] [PubMed] [Google Scholar]
- 53.Buchhaupt M., Sharma S., Kellner S., Oswald S., Paetzold M., Peifer C., Watzinger P., Schrader J., Helm M., Entian K.D. Partial methylation at Am100 in 18S rRNA of baker's yeast reveals ribosome heterogeneity on the level of eukaryotic rRNA modification. PLoS One. 2014;9:e89640. doi: 10.1371/journal.pone.0089640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lafontaine D.L. Noncoding RNAs in eukaryotic ribosome biogenesis and function. Nat. Struct. Mol. Biol. 2015;22:11–19. doi: 10.1038/nsmb.2939. [DOI] [PubMed] [Google Scholar]
- 55.McMahon M., Contreras A., Ruggero D. Small RNAs with big implications: new insights into H/ACA snoRNA function and their role in human disease. Wiley Interdiscip. Rev. RNA. 2014;6:173–189. doi: 10.1002/wrna.1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Williams G.T., Farzaneh F. Are snoRNAs and snoRNA host genes new players in cancer? Nat. Rev. Cancer. 2012;12:84–88. doi: 10.1038/nrc3195. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY DATA